磁性纳米微粒的制作方法
本申请要求于2010年5月26日提交的美国临时申请号No.61/348,561的优先权,其内容通过引用整体并入本申请。
联邦政府资助的研究和开发
本发明是在政府支持下通过美国国立卫生研究院授予的基金号UO1 HL080731-02而完成,政府在本发明中拥有某些权利。
技术领域
本公开涉及磁性纳米微粒。
背景技术:
生物可相容性的和/或可降解的磁性纳米微粒在生物技术和医学里有着广阔的应用。尤其是,考虑到生物样本和组织有着固有的低磁化感受性,磁性纳米微粒可以为高度选择性检测提供一种有效的对比机制。举例来说,磁性纳米微粒可用于应用范围包括但不限于生物分离,药物传递,和基因导入。
技术实现要素:
本公开涉及磁性纳米微粒以及它们的合成方法和使用。每个磁性纳米微粒包括一个磁芯和一个生物相容性的外壳,这种外壳既保护磁芯不受氧化且增强纳米微粒的磁性。增强的磁性可包括增加磁化强度和减少磁芯的矫顽力,这使得高灵敏度检测和减弱纳米微粒的非特异性聚集成为可能。通过形成具有增强磁特性的生物可相容性纳米微粒,特定靶分子的检测,例如蛋白质和单细胞,可以得到提高。
一般来说,本说明书所描述主体的一方面可以体现在包括一个铁磁芯和一个包被磁芯的超顺磁外壳的纳米微粒。
在某些实施方案中,纳米微粒的直径大于或等于2nm。在某些例子中,
铁磁内芯的内径在大约1nm到15nm的范围内。在某些实施例中,超顺磁性外壳的厚度大于或等于0.1nm。铁磁芯可包括Fe,Co,Ni,FePt或SmCo。
在某些实施方案中,超顺磁性外壳包括一种磁性材料的氧化物。在某些情况下,超顺磁性外壳包括一种掺杂剂。掺杂剂可包括选择于一组包含Mn,Co,Ni,Zn和ZnMn的一种金属。
在某些实施方案中,纳米微粒包括超顺磁性外壳上的一层涂层,其中形成涂层以增加纳米微粒的水溶性。涂层可包括2,3-二巯基琥珀酸(DMSA)。
在某些情况下,纳米微粒包括超顺磁性外壳上的一层涂层,其中形成涂层使纳米微粒与靶分子结合。
在某些实施方案中,纳米微粒包括超顺磁性外壳上的葡聚糖聚合物涂层。
在某些实施方案中,超顺磁外壳包括铁氧化物。
另一方面,构建纳米微粒的方法,该方法包括构建一个或多个铁磁性纳米微粒核心;和在每个一个或多个铁磁性纳米微粒核心上构成一个超顺磁外壳。构建铁磁性纳米微粒包括在第一种溶液中合并一种金属复合物和表面活性剂,并且热处理第一种溶液以热降解金属复合物从而在第一种溶液中形成一个或多个铁磁性纳米微粒核心。
在某些实施方案中,在一个或多个铁磁性纳米微粒核心上构建超顺磁外壳包括,在热处理第一种溶液后,将一种或多种金属油酸酯复合物与第一种溶液结合以形成第二种溶液,并且热处理第二种溶液以在一个或多个铁磁性纳米微粒外壳上形成超顺磁外壳。
另一方面,检测个体中目标分子存在的方法包括给个体摄入纳米微粒,其中纳米微粒包括一个铁磁芯,一个超顺磁外壳,以及超顺磁性外壳上一个与目标分子特异结合的靶向部分,为纳米微粒结合靶分子提供充分的时间,并且产生个体的核磁共振成像,其中成像中的信号表示存在目标分子。
另一方面,一种治疗的方法包括给个体摄入纳米微粒,其中纳米微粒包括一个铁磁芯,一个超顺磁外壳,以及超顺磁性外壳上一个与个体中靶细胞结合的涂层,提供充分的时间使纳米微粒结合靶细胞,并且给个体形成一个交替的电磁场来处理靶细胞。
交联聚合物是指一聚合物链上和/或支链上的功能基团与另一聚合物上的功能基团反应而形成的聚合物网。
非交联聚合物是指很少或没有单独的聚合物链与另一聚合物链的功能基团反应以形成一种相互联系的聚合物网。
磁矩是指磁体趋向磁场的趋势。
磁矫顽力是指铁磁性材料对消磁的抗力。
磁化率是指物质应答外加磁场的磁化程度。
超顺磁性指的是材料在没有外加磁场的情况下不能展现出磁性,但在有磁场的情况下表现出与磁体相似。
本发明提供了多个优点。例如,在某些实施方案中,磁性纳米微粒的外壳可防止纳米微粒核心的逐步氧化,转而防止纳米微粒整个磁化的减少。在某些情况下,在磁性纳米微粒的外壳中掺入金属可提高纳米微粒的整体磁化。磁性纳米微粒有着稳定的外壳的优势,因为外壳的厚度随着时间的推移总体上是恒定的。在特定的实施方案中,磁性纳米微粒的外壳提供了靶分子或其他结合基团可以附着上的生物可相容性表层。由于磁性纳米微粒的高度磁化,它们可以用于对此类靶分子和/或基团高灵敏检测。此外,由于超顺磁外壳,磁性纳米微粒核心在低磁场几乎没有矫顽力。因此,在没有外加磁场的存在时多个磁性纳米微粒的非特异性聚集可以避免。相反的,当加入一个超过阈值的外部磁场时,可诱导磁性纳米微粒特异聚集。这种诱导性聚集可用于收集和/或分离附着于磁性纳米微粒的特定靶分子。
除非另外定义,这里用到的所有技术和科学用语有着如通常为本发明所属领域内普通技术人员所知的相同意思。尽管与那些描述于此的相似或相当的方法和材料可用于本发明的实践或测试,合适的方法和材料描述如下。所有这里提到的发表的论文,专利说明书,专利和其他参考文献都通过引用结合到全文。万一存在矛盾,以本说明书包括定义为准。另外,材料,方法和实例只是说明性的而不作为限定。
其他的特点和优势可以在以下详细的描述、图表和权利要求中显而易见。
附图说明
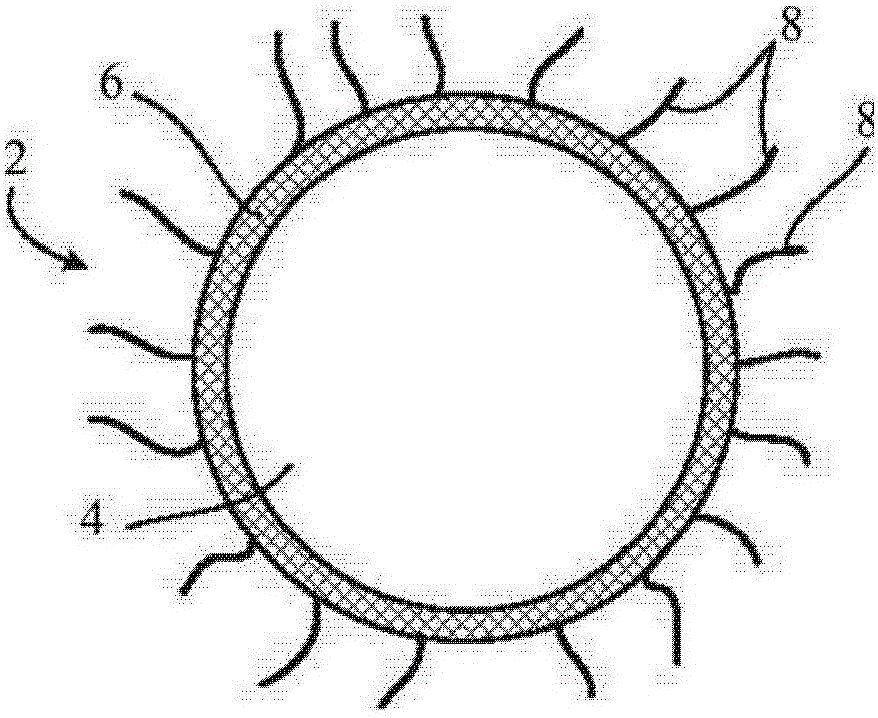
图1A和1B是示范性的磁性纳米微粒横截面。
图2是描绘制备一个磁性纳米微粒典型的进程的流程图。
图3A到3D是用金属复合物:表面活性剂溶液制备的纳米微粒核心的图像,每种都包括不同的金属复合物对表面活性剂摩尔比。
图4A到4F是在不同的反应温度下从金属复合物:表面活性剂溶液制作的纳米微粒核心的图像。
图4G是纳米微粒直径对应反应温度的图表。
图5是FeMFe 2O 4(M=Fe,Co,Mn)磁性纳米微粒(MNPs)标准化的磁化强度对应温度的图表。
图6a-6b是FeFeO MNPs和FeMFe 2O 4(M=Fe,Co,Mn)磁性纳米微粒的磁化曲线。
图7是不同磁性纳米微粒的横向弛豫值(r 2)的比较图表。
图8是FeMnFe 2O 4磁性纳米微粒在涂上二巯基琥珀酸(DMSA)之前和之后以结合靶基团的示意图。
图9是几种类型的纳米微粒的标准化ΔT2与亲和素浓度的关系图。
图10A是几种类型的磁性纳米微粒弛豫率相对细胞浓度的图。
图10B是显示FeMnFe 2O 4MNPs的标准化ΔT2与细胞浓度的关系图。
图11A到11D是不同磁性纳米微粒的MRI成像图。
具体实施方式
更好的,磁性纳米微粒有着一个高的磁矩为检测提高了纳米微粒的选择性。尽管增大纳米微粒的大小可导致更高的磁矩,但是过大的纳米微粒难以用于检测小分子和细胞。此外,在某些情况下,磁性微粒易于氧化,而随时间减少磁化。而且,由于微粒的磁力特性,自发聚集可发生,使得纳米微粒难以应用以及使得分子与纳米微粒难以选择性结合。
本公开涉及磁性纳米微粒以及它们的合成和使用的方法。每个磁性纳米微粒包括一个磁芯和一个生物相容性外壳,这种外壳既保护磁芯不受氧化且增强纳米微粒的磁性。增强的磁性可包括增加磁化强度和减少磁芯的矫顽力,达到高敏感度检测并减弱纳米微粒的非特异性聚集。通过有增强的磁性的成型的生物可相容性纳米微粒,特定靶分子的检测,例如蛋白质和单细胞,可以得到提高。
磁性纳米微粒
图1A表示一个典型的磁性纳米微粒2的横截面。磁性纳米微粒2包括一个由外壳6包被的纳米微粒核心4。尽管显示为一个圆截面(三维球体),但磁性纳米微粒2可以有各种形状。例如,磁性纳米微粒2可以形成类似棒状/圆柱体,电线,或晶须。纳米微粒也可有其它的形状。磁性纳米微粒2也可有不同的规格。在某些实施例中,磁性纳米微粒可有任何一个范围在1nm到20nm之间的平均最大尺寸,包括,例如,大约2nm,5nm,10nm,15nm,或16nm。
在某些实施方案中,纳米微粒核心4由结构为晶体、多晶的或非结晶的铁磁性材料构成。例如,纳米微粒核心4可包括材料比如但不限于,Fe,Co,Ni,FeOFe 2O 3,NiOFe 2O 3,CuOFe 2O 3,MgOFe 2O 3,MnBi,MnSb,MnOFe 2O 3,Y3Fe 5O 12,CrO 2,MnAs,SmCo,FePt,或者它们的组合。纳米微粒核心4可以与整个磁性纳米微粒2形状一样。例如,纳米微粒核心4可以形状类似棒状/圆柱体、电线、或晶须。纳米微粒核心4也可有其它形状。纳米微粒核心4的平均最大尺寸可从1nm到20nm不等包括例如大约2nm,5nm,10nm,或15nm。
磁性纳米微粒2的外壳6部分或全部围绕纳米核心4。在某些实施方案中,外壳6是由结构为结晶的、多晶的或非结晶的超顺磁材料构成。在某些实施方案中,通常构成外壳的材料是生物可相容性的,也就是,外壳材料在有机体中几乎不会引起不利的生物/免疫反应和/或对细胞和器官无毒。可用于外壳6的典型材料包括但不限于,金属氧化物,例如,铁氧体(Fe 3O 4)、FeO、Fe2O3、CoFe 2O 4、MnFe 2O 4、NiFe 2O 4、ZnMnFe 2O 4,或者它们的组合物。外壳6可以有范围从大约0.5nm到3nm,包括,例如,大约1nm、1.5nm、2nm、或2.5nm的厚度。
磁性纳米微粒2的外壳6可以提供多种功能。例如,当没有外壳的孤立的纳米微粒核心暴露在一种氧化剂(如空气中的氧气)时,全部或部分的核心材料可被氧化导致纳米微粒的磁特性退化。此外,形成的氧化物可能有相对于核心材料弱磁性。在核心4上形成外壳6,可以防止不受欢迎的核心材料的氧化因此保护核心4的磁性能。另外,在某些实施方案中,外壳6提高了纳米微粒核心4的磁性能。例如,有外壳6的磁性纳米微粒可显示出与仅仅由外壳材料组成的纳米微粒核心大约10倍,100倍或1000倍的磁矩。
磁性纳米微粒2的磁化强度(每单位微粒体积的磁矩)在某种程度上依赖核心4的平均最大尺寸,外壳6的厚度,以及核心4和外壳6各自的磁化强度。例如,FeFe3O4微粒总的磁化强度(M 0)由Fe核心和铁氧体外壳分别贡献,给出体积-加权平均值如下,
其中M Fe和M Ferrite是磁化强度,V Fe and V Ferrite分别是核心与外壳的体积。
在某些情况下,外壳材料相当于核心材料的氧化物,其中氧化物是在一个有控制的程序中包被在纳米微粒核心4表面上的,没有使核心4进一步氧化,而不是由核心材料自己形成。这种可控的外壳形成过程可随时间产生一个有固定厚度的外壳(也就是,外壳厚度几乎不增加)而且有着强的磁性能。此外,通过在纳米微粒核心4的表面形成外壳6,纳米微粒核心的大小也不会减小。
由于外壳6的超顺磁特性,当没有外加磁场施加于纳米微粒2时,磁性纳米微粒2几乎不显示出磁特性。相反,当阈值之上的外加磁场施加于纳米微粒2时,纳米微粒显示出上述增大的磁矩。换言之,外壳6改变了纳米微粒2的磁特性以至于整个纳米微粒2表现出超顺磁的。显示出增大的磁矩的阈值是基于外壳6的材料和结构。
在无外加磁场存在下,磁矩的减弱和/或缺失在某些实施方案中能减少和/或阻止成群的纳米微粒2由于它们的磁特性而发生的非特异性/自发的聚集。相反,通过施加一个外部磁场给多个上述有核心/外壳结构的纳米微粒,在某些实施方案中,很可能会引起纳米微粒的聚集。这种引起的聚集在试图检测结合到磁性纳米微粒的分析物或其它分子时是有用的。
在某些实施方案中,纳米微粒2的磁矩通过包含掺杂剂进外壳材料甚至可进一步增大。例如,外壳6可以参杂金属包括但不限于,Mn、Co、Ni、Zn、ZnMn,以及它们的组合。纳米微粒外壳6中掺杂物的存在也可以在有一个外加磁场存在时影响纳米微粒2显示出磁特性的临界点。磁性纳米微粒在掺杂剂离子组成为铁水平的一半,例如MnFe2O4,CoFe2O4,NiFe2O4,ZnFe2O4,ZnMnFe2O4时可有最高的磁矩。
在某些实施方案中,纳米微粒2也包括表面涂层。例如,图1B展示的是纳米微粒2其中在超顺磁外壳6的表面加工形成一层表面涂层8。表面涂层8完全或部分包被外壳6。涂层8的至少一个目的是提供一个生物相容性表面可以容易地使靶分子,结合基团等等功能化。在某些情况下,表面涂层8使磁性纳米微粒实质上亲水或者疏水。表面涂层可由聚合物加工形成,包括但不限于,合成聚合物比如聚乙二醇或硅烷,天然聚合物,合成或天然聚合物的衍生物,以及它们的组合。天然聚合物是当纯聚合物比如多聚糖通过微生物、植物或动物合成并且提纯而获得。合成聚合物是由非生物合成,通过采用标准的为领域内技术人员所知的高分子化学技术将单体融合成聚合物而获得。聚合物可以是同聚物,即由单一类型的单体合成,或者是共聚物,即由两种或多种单体合成。聚合物可以使交联或非交联的。交联聚合物是以热稳定和在生物系统中抗分解为特征的。交联聚合物分子量显著高于原始聚合物。
在某些实施方案中,表面涂层8不是包围纳米微粒2的连续膜,而是依附于并且围绕磁性纳米微粒2的扩展的聚合物链形成的“网状物”或“云状物”。典型的高聚物包括但不限于多糖及衍生物,比如葡聚糖、pullanan、羧基右旋糖苷、羧甲基右旋糖苷、和/或还原羧甲基右旋糖苷、聚甲基丙烯酸甲酯聚合物和聚乙烯醇聚合物。在某些实施方案中,这些聚合物涂层提供了比外壳材料更容易让靶向部分和/或结合基团结合的表面。
例如,可以制备葡聚糖包被的纳米微粒并与表氯醇交联。添加氨与环氧基反应以形成氨基。这种材料被认为是交联氧化铁或“CLIO”并且当被氨基功能化后被称为氨基-CLIO或NH 2-CLIO。羧基修饰的纳米微粒可以通过用水溶性的碳化二亚胺和二元胺如乙二胺或己烷二胺转换成氨基-功能化的磁性微粒。抗生物素(Avidin)或链霉亲和素(streptavidin)可以附着于纳米微粒以供生物素化的结合基团,比如寡核苷酸或多肽使用。同样的,生物素可以附着于纳米微粒以供抗生物素示踪的结合基团使用。
在某些实施方案中,纳米微粒与非聚合物功能基团组合在一起。已知合成稳定的、功能化的无聚合物的纳米微粒的方法,也在本发明的范围之内。该方法描述于例如Halbreich et al.,Biochimie,80(5-6):379-90,1998和Caroline R.A.Valois et al.,Biomaterials,31(2):366-374。
一般而言,结合基团是指能特异性地结合或者链接,举例来说,共价或非共价结合或杂交到一个靶分子或其它结合基团(或者在特定的实施方案中,聚合诱导分子)上的合成的或天然的分子。例如,结合基团可以是合成的寡核苷酸以杂交一个特定互补的靶核酸。结合基团也可以是针对抗原或者任何蛋白-蛋白间的相互作用的抗体。此外,结合基团可以是结合对应靶标的多糖。在特定的实施方案中,结合基团在结合到另一结合基团时,可以设计或挑选以适合靶分子比如溶液中的酶的底物。结合基团包括,例如,寡核苷酸结合基团、多肽结合基团、抗体结合基团以及多糖结合基团。
磁性纳米微的合成
图2表示描述制备磁性纳米微粒典型过程的流程图。如图2所示,磁性纳米微粒的制备始于纳米微粒核心的形成(步骤200)。为了制备纳米核心,金属复合物在包含表面活性剂的溶液中加热分解。在某些情况下,热分解发生在无氧环境中(例如,在真空或氮气环境下)以避免反应中发生纳米微粒核心的氧化。在一个例子中,1-十八烯(ODE)和油酰胺(OY)表面活性剂的溶液加热到250℃。当溶液的温度稳定时,金属复合物比如Fe(CO)5加入溶液并搅拌。Fe磁性纳米微粒(Fe MNP)核心形成并从溶液中析出。然后溶液冷却到室温。其他金属复合物的例子包括但不限于Fe(acac)2(acac是乙酰丙酮化合物)、Fe(acac)3、钴络合物、镍络合物,其它表面活性剂的例子包括但不限于油胺、十八烯酸和其它包括胺或羧酸化学官能团的化学物质。
纳米微粒核心大小在某种程度上取决于金属源和表面活性剂的摩尔比率。在某些实施方案中,降低表面活性剂相对于金属源的水平可导致微粒大小增加。例如,图3显示从包含OY和铁金属复合物的溶液制备的铁纳米微粒核心图像,其中摩尔比率([Fe]:[OY])在一个固定温度下从5:1到35:1不等。当比率从5:1变为12:1时,微粒直径才增加约2nm。一旦比率超过12:1,微粒大小稳定在2nm。
纳米微粒核心大小也取决于纳米微粒核心形成期间的温度。在某些实施方案中,提高反应发生时的温度导致微粒的大小的增加。例如,图4A到4F显示从包含OY和铁金属复合物的溶液制备的铁纳米微粒核心图像,其中纳米微粒核心形成期间的反应温度在固定摩尔比率([Fe]:[OY])下从140℃到260℃变化。当反应温度逐步从140℃升到260℃时,纳米微粒核心的直径从4.4nm增大到14.5nm。
在如图3和图4所示的每一个示范纳米微粒核心的图像中都可见到,随后可见纳米微粒核心材料暴露于空气中而后氧化而形成的天然非晶形的铁酸盐外壳(FeO)。天然非晶形外壳有弱的或没有磁性并且可随着时间逐步加厚以至于将铁核心改变为非晶形氧化物。前面提到的典型纳米微粒的直径通过调整铁酸盐外壳体积而估算。图4G表示的是有自然生成氧化物外壳的纳米微粒核心直径和估算的没有自然生成氧化物外壳的纳米微粒核心直径相对于反应温度。
再次参考图2,随着纳米微粒核心的制备,合成的外壳在核心微粒的表面形成(步骤202)。为了制备外壳,包含金属复合物的独立的溶液需制备并且加入到包含磁性纳米微粒核心的溶液中。为防止天然非晶形铁酸盐外壳先于合成外壳的形成,包含纳米微粒核的溶液可以保持在一个无氧的环境中(例如,在真空或主要为氮气的环境下)。然后合并的混合物可退火以在纳米微粒核心的表面形成纳米微粒外壳。
在一个例子中,铁-油酸盐复合物的溶液由上述包含Fe MNP核的溶液单独制备。特别是,包含Fe(CO)5、十八烯酸(OA)、和ODE的溶液最好在无氧环境下制备和退火。冷却后,新的溶液转移到包含Fe MNP核心的溶液中。然后包含Fe MNP核心和金属复合物的混合物温度升高以在Fe MNP核心周围形成合成外壳。特别是,反应温度须缓缓加热到一个最佳的退火温度(如300℃)以形成具有铁核心和合成的多晶铁酸盐外壳的Fe MNPs,其中合成外壳展现出超顺磁性。合成外壳起着屏障的作用以防止核心的氧化并且通常是稳定的,表现出厚度不随时间变化。混合物冷却后,通过离心收集包含超顺磁外壳的磁性纳米微粒。
在某些实施方案中,磁性纳米微粒的合成外壳的形成步骤包括加入更多不同的金属复合物到包含MNP核心的溶液中。所添加的不同金属复合物可提供一种或多种掺杂剂到随后形成的磁性纳米微粒的合成外壳中,其中掺杂剂可提高MNPs的磁性(例如磁矩)。不同金属复合物的一个或多个的组合(如,Ni(CO)4,Co 2(CO)8)可添加到溶液中以在合成外壳中形成一个或多个掺杂物。在一个例子中,在加热混合物之前,Mn 2(CO)10添加到上述包含Fe MNP核心和铁金属复合物的混合物中。如前一样,反应温度须缓缓加热到一个最佳的退火温度(如300℃)以形成具有铁核和合成的掺杂Mn的多晶铁酸盐(Fe 2O 4)外壳(FeMnFe 2O 4)的Fe MNPs。
在某些实施方案中,MNPs可以进一步加工在合成外壳上形成一层表面涂层(步骤204),其中表面涂层包括功能团以连接MNP到结合配基。如前所述,表面涂层可包括一个功能性的聚合或非聚合涂层。在某些情况下,涂层可以提高或降低MNP的润湿性。聚合的或非聚合的涂层可以提供一个暴露的官能团用以结合特异或非特异配基。例如,官能团可包括氨基、羧基、或其它结合特异或非特异配基的反应基团。在某些实施方案中,配基可以包括具有末端氨基、巯基、或磷酸基等等能结合氨基或羧基的寡核苷酸。或者说,结合配基可以通过形成于合成MNP外壳上的功能性聚合物附着于磁性纳米微粒上。合成功能化涂层的纳米微粒方法的各种非限制性例子描述如下。
在一个例子中,非聚合物的DMSA涂层可以在合成的外壳的表面形成(例见Albrecht et al.,Biochimie,80(5-6):379-90,1998)。DMSA偶联到人工合成的铁酸盐外壳,提供一个暴露的官能团。在另一个例子中,羧基功能化表面涂层可以根据Gorman的方法合成于MNP之上(见WO00/61191)。该方法中,还原的羧甲基(CM)葡聚糖由商用葡聚糖合成。CM-葡聚糖和铁盐混合在一起然后用氢氧化铵中和。最终的羧基功能化的纳米微粒可用于偶联氨基功能化的寡核苷酸。
在另一个例子中,羧基功能化的MNPs也可以用多糖包被的MNPs通过与溴或氯乙酸在强碱中反应以结合羧基来制成。此外,羧基功能化微粒可由氨基功能化的MNPs用诸如琥珀酐或顺丁烯二酸酐等试剂将氨基转换为羧基来制备。
在另一个例子中,可以制备葡聚糖涂层的MNPs并与表氯醇交联。添加胺与环氧基反应而形成氨基(例见U.S.Patent App.Pub.No.20030124194and U.S.Patent App.Pub.20030092029,incorporated herein by reference,Hogemann,D.,et al.,“Improvement of MRI probes to allow efficient detection of gene expression,”Bioconjug.Chem.2000.11(6):941-6,and Josephson et al.,“High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates,”Bioconjug.Chem.,1999,10(2):186-91)。该材料被认为是交联氧化铁或“CLIO”并且当用氨基功能化时被称为氨基-CLIO或NH 2-CLIO。
在另一个例子中,羧基功能化MNPs可以通过水溶性碳化二亚胺和二元胺如乙二胺或己二胺转化成氨基功能化磁性微粒。在某些实施方案中,亲和素或链亲和素可以连接到纳米微粒以供与生物素化的结合配基如寡核苷酸或多肽使用(例见.,Shen et al.,“Magnetically labeled secretin retains receptor affinity to pancreas acinar cells,”Biocojug.Chem.,1996,7(3):311-6)。同样的,生物素也可连接到纳米微粒以供与亲和素标记的结合配基使用。
MNPs的特性
MNPs的特性可通过多种技术来鉴定。例如,MNPs磁特性可以通过测定零场冷却或场冷却磁化曲线评估。在一个例子中,制备FeMnFe 2O4MNPs并用扩展的Stoner-Wohlfarth磁化模型评估。该模型被开发来解释一组均一的单畴纳米微粒的整体磁性行为,而这些纳米微粒随机排列并且彼此互不影响。我们已经进一步扩展了该模型以反映实际的FeMFe2O 4微粒物理系统总结如下。
·为适应铁和铁酸盐的立方磁晶各向异性,我们采用Joffe和Heuberger建立的架构。
·包括磁化的热激活影响,通过允许磁矩的热激活逆转。
·我们用TEM分析获得的粒径分布函数来说明微粒系综里的大小差异。
使用该方法,已经确定合成外壳遵循典型的超顺磁曲线而纳米微粒核心具有非零矫顽力(H c=350Oe)呈现一个稳定的单畴特性。然而,MNP的总磁化强度(也就是核心和外壳磁化强度的体积加权平均数)相比于纳米微粒核心有一个相当小的矫顽力(H c=40Oe)。在这个例子中,合成外壳超顺磁的贡献在低磁场下饱和而超过相对较慢的纳米微粒核心磁化。相反的,核心对磁化的贡献在高磁场下在增加MNP总的磁化强度成为主导。其他技术,比如微磁学模拟和核磁弛豫色散可用于进一步测定MNP特征。
MNPs的用途
磁性纳米微粒可用于各种应用中,包括医药如生物兼容的和环境敏感的传感器和/或分子显像剂。例如,MNPs可被用做基于磁共振传感器,其中MNPs用来做探测水溶性(即含水)样品中各种分析物的遥感器以及用于对水溶性样品中分析物水平变化的持续监控。MNPs可以悬浮于水相中,并有一个或多个配基以共价或非共价连接或者固定其上,此配基能够根据溶液中分析物的存在和浓度而改变MNPs聚集状态(例见美国专利申请公开号20060269965,以引用的方式并入此).
在另一个例子中,MNPs可用于聚集形成试验以检测靶分子(例见美国专利申请公开号20030092029,通过引用合并于此)。在聚集形成试验中,一种缀合体(或对应一种靶分子或一类型靶分子的两种或多种带不同结合配基的缀合体混合物)加到样品溶液。每个缀合体包括一个或多个结合配基(例如,寡核苷酸,核酸,多肽,或多糖),例如,共价或非共价地连接到一种磁性例如超顺磁纳米微粒。结合配基与靶分子(或在某些实施方案中,引起聚集的分子如亲和素)发生特异性相互作用。结合配基特异地结合选择的靶分子,可以是例如核酸、多肽或多糖。因此,缀合体的分散状态转换到聚合状态,从而减少了水相溶液中相邻水质子的自旋-自旋弛豫时间(T2)。在某些情况下,MNPs可用于靶分子如蛋白的磁力分离。
在另一个例子中,MNPs可在聚集色散分析中用于检测靶分子(例见美国专利申请公开号20030092029,通过引用合并于此)。在聚集色散分析中,缀合体用来制备小的聚集物,然后聚集物加到样品溶液中。在该分析系统中,设计结合配基使它们能互相结合(或结合引起聚合的分子如亲和素)以形成所述聚集物,成为(或形成相互间结合或结合到引起聚集的分子上)特异性靶向分子裂解的底物。如果样品溶液含有靶分子,结合配基形成的底物则裂解,导致聚集物的分散。
该分析系统中的聚集物可以在体外例如小瓶或列阵里如2-D或3-D列阵,也可在体内例如摄入缀合体或聚集物后的磁共振成像观察和探测。在某些例子中,MNPs可用于成像而不要求多个纳米微粒的聚集。前述用途的进一步说明例见美国专利号6,818,199,6,203,778,和5,766,572,Jae-Hyun Lee et al.,Nature Medicine13:95-99;Daniel L.J.Thorek Annals of Biomedical Engineering2006,23-38.
在另一个例子中,MNPs可以结合靶细胞,比如癌症细胞,适用于磁热疗。特别是,缀合MNPs可用于癌细胞或组织,体外或体内,其中缀合MNPs结合癌细胞/组织。在某些实施方案中,MNPs包含使MNPs结合特定癌细胞/组织或当全身注射时引起MNPs转移到个体的特定部位的靶向配基。例见Maite Lewin et al.,Nature Biotechnology18:410-414;PCT No.PCT/KR07/00961。然后,给结合的MNPs施加一个外加的交变磁场(如100kHz)以致MNPs在对外加磁场应答中的运动产生增加的热能来治疗靶向细胞/组织。特别是,热能的增加可导致癌症细胞/组织的破坏。
在另一个例子中,MNPs可用于磁转染。在磁转染中,缀合的MNPs结合到靶向分子比如核酸上,然后给结合MNPs的分子施加一个磁场以刻意地引进和集中纳米微粒到一个或多个靶向细胞中。而后核酸通过多种不同的机制被释放到细胞质中比如,例如:1)由包被在MNPs的阳离子聚合物引起的质子海绵效应促进胞内体渗透膨胀,胞膜的破坏以及核酸的胞内释放;或2))由包被在MNPs的阳离子脂质体引起胞内体的不稳定以通过细胞阴性脂质体的转变和电荷中和释放核酸到细胞。例见美国专利号7,635,734and Eric M Pridgen et al.,Nanomedicine2(5):669-680.
实施例
本发明在以下实施例中做了进一步描述,但不限制权利要求里所描述的本发明的范围。
例1:铁磁芯/超顺磁氧化铁外壳纳米微粒的合成与鉴定
我们首先制备Fe MNPs核心。往250mL的3颈圆底玻璃烧瓶里加入20mL ODE和0.3mL OY(0.64mmol)。冷凝器和电热偶连接到烧瓶不同的颈部。混合物真空下加热到60℃1小时,再充入氮气彻底排除氧气。该步骤重复两次。然后将混合物加热到要求的温度(例如,对于16nmFe MNPs核心260℃)。当温度稳定时,Fe(CO)5(1.4mL,10mmol)小心的加入反应器中同时大力搅拌;随着羰基分解溶液迅速变黑,纳米微粒开始形成。溶液维持在这种高温和氮气中1个小时,之后室温冷却。
当FeMNPs核心成型时,分别制备锰和铁-油酸复合物。Mn 2(CO)10(156mg,0.8mmol),OA(2.3mL,7.26mmol)和10mL ODE加到100mL的3颈圆底玻璃烧瓶里。真空下升温到60℃一个小时,通入氮气,然后再重复两次。无氧溶液加热到120℃后加入Fe(CO)5(0.21mL,1.61mmol)。混合物搅拌30min。期间溶液会变黄。但是由于温度较低(120℃)而不会形成微粒。包含金属-油酸盐复合物的溶液室温下冷却,然后用双向针管小心的转移到仅含FeMNP核心的溶液中。仅含FeMNP核心和金属-油酸盐复合物的混合物室温下搅拌30分钟。然后反应温度以5℃/分钟缓慢升高。我们监测温度上升期间纳米微粒的结晶度,测定铁酸盐外壳形成的最佳热处理温度(300℃)。当温度稳定后,混合物搅拌一小时以上。随着反应的完成,溶液在室温下冷却并加入150mL异丙醇溶液(ODE/异丙醇=0.2v/v)。FeMnFe 2O 4MNPs通过离心(3,000rpm,15分钟)收集再分散在10mL己烷中。为了清洗微粒,加入50mL乙醇到微粒溶液中,离心分离混合物,再将微粒分散到10mL己烷中。清洗步骤重复3次以确保除去多余的化学物质。
例2:纳米微粒核心/外壳的磁特性
制备纳米微粒的大小是用动态光散射法(Zetasizer Nano-ZS,Malvern)鉴定的。形状、结构、组成进一步分别用透射电子显微镜描述(TEM;JEOL2100,JOEL USA)、X射线粉末衍射仪(XRD;RU300,Rigaku)、电感耦合等离子体原子发射光谱仪(ICP-AES;Activa-S,HORIBA Jobin Yvon)鉴定。磁特性用震动样品磁强计(EV-5,ADE Magnetics)和超导量子干涉(SQUID)磁强计(MPMS-5,Quantum Design)分析。
我们鉴定了FeMFe 2O 4MNPs的磁特性。为了比较分析,我们也合成了不同大小和组成成分的铁酸盐MNPs。所有类型的微粒Fe核心(FeFeO and FeMFe 2O 4,M=Fe,Co,Mn)的Ms大于铁酸盐MNPs FeMnFe 2O 4所假设的最大Ms。铁酸盐外壳与疏松物质相比有低的磁化强度,可能由于他们的多晶特性。图5表示FeMFe 2O 4(M=Fe,Co,Mn)MNPs的零场冷却和场冷却磁场强度,其中峰值在两个不同的温度(TB1>TB2)出现。这两个峰分别表示Fe核心(TB1)和铁酸盐外壳(TB2)中超顺磁性的发生。Fe核心的TB1≈290℃,表现出室温下核心的稳定的,强磁特性。
图6a-6b表示FeFeO MNPs和FeMFe2O4(M=Fe,Co,Mn)MNPs的磁化曲线。磁化曲线用振动样品磁强计在T=300K下获得的。所有的测量都是在合成2小时后以MNPs的粉末形式进行的。样品中MNPs的数量用电感耦合等离子体原子发射光谱仪(ICPAES)定量。首先,我们分析FeFeO MNPs的磁特性(图6a)。磁滞曲线证实了铁核心直径在11nm以上的微粒的超顺磁特性。总的来说,观测到的Ms在同比于Fe核心部分大的大微粒中增加得多。用仅有外壳的MNPs测得的FeO外壳的Ms相当小(8emu/g[Fe])。在补偿外壳的作用后,预计Fe核心的Ms为206emu/g[Fe](对于11nm的核心),接近于疏松材料的Ms(210emu/g[Fe])。
FeMFe 2O 4(M=Fe,Co,Mn)MNPs的磁化曲线每个都表现出异常的磁滞损失特点同时伴随可忽略不计的剩磁(图6b)。在同样大小Fe核心的MNPs中,总磁化强度随外壳材料的Ms排序(MnFe 2O 4>Fe 3O 4>CoFe 2O 4)。该趋势因此可验证我们的方法通过铁酸盐外壳的金属掺杂来提高整个微粒的Ms。表1列出不同类型MNPs的总Ms、氧化物外壳的Ms、以及外壳对总Ms的贡献。正如仅有铁酸盐的MNPs没有核心/外壳结构,对于这些纳米微粒总的Ms和铁氧化物的Ms没有差异。每种类型的MNP的Ms是在温度为300K外加磁场强度为H ext=10kOe下测得。测得纳米微粒的外直径约为16nm。铁酸盐外壳估计的Ms小于那些仅有铁酸盐的MNPs,可能由于外壳的多畴性。
表1
为了进一步分析,用扩展的Stoner-Wohlfarth磁化模型计算FeMFe2O4MNPs的磁性,包括非零温度和立方磁晶各向异性的影响。根据模拟结果,外壳磁化强度遵循典型的超顺磁的曲线,相反核心部分对于非零矫顽力(Hc=350Oe)呈现出稳定的单畴反应。微粒的总磁化强度(M tot),核心的磁化强度和外壳的磁化强度的加权平均值,然而有一个相当小的矫顽力(Hc=40Oe):超顺磁的贡献(来自M shell)在低磁场下迅速饱和而超过相对较慢的核心磁化。而在高磁场下核心的贡献(M core)对增加总M tot成为主导。
例3:磁化的数值模拟
FeMnFe 2O 4微磁学模拟进一步证实所假设的磁化机制。在无外加磁场(H ext)存在以及T=300K下,铁磁芯的磁化矢量形成一个涡旋状态,表明强烈的交换耦合以抵抗磁化的热反转。在微粒外壳中,磁化矢量是随机定向的,显示出超顺磁特性。这组成结构定性上讲与零场冷却和场冷却磁化强度测量值一致,显示在室温下铁核心高TB1和外壳低的多的TB2。在非零磁场强度下,磁矩的调整是路径依赖的并且导致磁滞回线。铁酸盐MNPs的相似模拟显示无磁滞现象并且磁化矢量在H ext=0时为热能随机。
我们随后鉴定了铁核心MNPs的横向弛豫率(r 2)。为了交叉检测Ms和MNPs的大小(d)对r 2的影响,我们也制备了不同直径的铁酸盐(Fe 3O 4)MNPs。图7是r 2值与不同MNPs的关系,r 2随着微粒大小的增加而上升。对于一个固定的微粒大小(d=16nm),r 2值正比于Ms,相应的,铁核心MNPs相比于基于铁酸盐的微粒有更高的r 2值。FeMnFe 2O 4MNPs获得最高的r 2值由于其最高的Ms;r 2值增大超过Fe 3O 4或交联氧化铁(CLIO)纳米微粒分别为2倍和300倍。我们用一个考虑r 2值计算中水扩散的外球面模型进一步分析MNPs的r 2值。总之,对于足够小的微粒,水分子的扩散运动足够快而使MNPs的磁场达到平衡,而体系中的r 2等于d2·Ms。其实,观测到的r 2值高度符合理论值,1)通过提高微粒大小和磁化强度,验证提高r 2值的方法;2)证实MNPs分散性好而在分子运动体系中无聚合产生(FeMnFe 2O 4d小于23nm)。
为理解FeMFe 2O 4MNPs的异常磁性,我们演示了两种不同层次的磁模拟:
·整体MNPs所测得磁滞回线,包括探测热激活的反磁化效果。
·单一微粒的磁化矢量的构型通过微磁学模拟分析。
结果显示FeMFe 2O 4MNPs磁化的新机制。该机制中,Fe核心(直径=12nm)表现出是单畴的,对很大矫顽力有铁磁反应,然而铁酸盐外壳有超顺磁特性并且在很低的外加磁场(Hext)下易磁化。当这两者(核心与外壳)结合在一起时,矫顽力减小因为外壳在低Hext促使初始磁化,但是与核心相关的磁滞损耗在高Hext下保留。该模型因此解释了FeMFe 2O 4MNPs中的在温度和外部磁场依赖磁化中的磁行为。
例4:表面修饰以及与生物素和抗体的缀合
FeMnFe 2O 4MNPs悬浮在10mL氯仿中,加入50μL三乙胺。10mL含DMSA(50mg)的DMSO注入纳米微粒溶液。混合物40℃摇6小时直到逐步变为两相,并且通过离心(3,000rpm,10minutes)沉淀。用乙醇小心清洗沉淀物以除去多余的DMSA,再用匀质器打散于10mL乙醇中。再次向纳米微粒-乙醇溶液加入10mL含DMSA(50mg)的DMSO,并且重复整个过程。最终的沉淀物散布于50mL水中。DMSA处理的纳米微粒最后用巯基(-SH)和未结合的羧酸作为末端;两者化学官能团可用于结合特定的靶向配体。通过Ellman’s试剂(Pierce Biotechnology)测得每个纳米微粒的巯基数约为50个。为了将DMSE处理的FeMnFe 2O 4MNPs和(+)生物素-酰肼(Aldrich)缀合,用羧酸在FeMnFe 2O 4MNPs上形成酰胺键,用NHS/EDC的化学反应在生物素上形成氨基。
DMSA处理的FeMnFe 2O 4MNPs(25mg)加到10mL水中,然后添加NHS(3.5mg),,EDC(5mg),以及生物素(1mg)。混合物在室温下摇匀3小时。结合的FeMnFe 2O 4MNPs随后沉淀下来(12,000rpm,20minutes),再用水洗3次。为了结合抗体,抗体首先要通过附加马来酰亚胺官能团呈现硫醇活性。然后抗体(anti-HER2/neu:Herceptin,6mgmL)溶于1mLPBS缓冲液中并且通过加入0.1M NaHCO 3,和1mg sulfo-SMCC调整PH到8.2。混合物在37℃孵育30分钟。然后有马来酰亚胺活性的抗体用PD-10去盐柱(GE Healthcare Bio-Sciences)纯化并且立刻与DMSA处理的纳米微粒(5mgmL)结合。混合物在4℃摇晃6小时,然后用Sephadex G-100(DNA grade,GE Healthcare Bio-Sciences)纯化。用二喹啉甲酸法(BCA protein assay kit,Pierce Biotechnology)测得每个纳米微粒结合抗体数约为10。
例5:MNPs的表面改质和细胞毒性试验
为了转移所合成的疏水性的FeMnFe 2O 4MNPs到水相缓冲液中,我们通过配基的交换在微粒的表面进行了2,3-二巯基琥珀酸(DMSA)修饰。DMSA包括两个羧基(-COOH)和两个巯基(-SH)。当与MNPs混合时,羧酸先与FeMnFe 2O 4MNP表面形成直接螯合带。图8是FeMnFe 2O 4MNP涂上DMSA以与目标配体结合的示意图。DMSA涂层是通过DMSA分子间的二硫交联而稳定。采用这种方法,我们确实能使FeMnFe 2O 4MNPs溶于水。
为了结合目标配体到微粒上,我们利用末端的巯基或羧酸官能团。例如,我们可运用NHS/EDC(N羟基琥珀酸亚胺,NHS;乙基二甲基胺丙基碳化二亚胺,EDC)化学作用结合生物素分子到DMSA的羧酸基上。抗体分别用马来酰亚胺官能团修饰,并通过双硫键结合到巯基上。然后我们检测与DMSA包被的FeMnFe 2O 4MNPs相关的潜在细胞毒性。正常细胞(3T3纤维原细胞)和癌症细胞(HCT116)在加入不同数量的MNPs之后培养24小时。然后用3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT)法分析细胞活力(见第7单元)。未发现严重的细胞毒性效应,从而证实了DMSA涂层和磁性材料两者的生物可相容性。
例6:生物学应用的演示包括亲和素滴定法和细胞试验
为了演示FeMFe 2O 4MNPs的生物学应用,我们通过基于NMR的诊断平台(DMR,诊断磁共振成像)使用微粒来检测生物标记。DMR检测是基于当样品中的待测目标被MNPs识别的时候横截面的弛豫时间(ΔT2)的改变。对于分子检测(如蛋白),DMR检测借助磁性弛豫转换现象,其中MNPs与靶向分子交联以形成纳米簇。对更大的靶标(如细菌,哺乳动物细胞),靶标由MNPs标记,并且自由微粒在测量之前转移。对于两者试验,检测的灵敏度与MNPs的r2是相称的。
首先,我们用生物素-亲和素作为模型评估了分子检测中FeMFe 2O 4MNPs的传感容量。生物素化MNPs的滴定实验显示亲和素剂量与T2的改变相关。图9表示的是对于几种类型的纳米微粒,亲和素浓度对应的标准ΔT2的图表。在其他类型的MNPs中,FeMnFe 2O 4通过检测约1.5pM的亲和素显示出最高的灵敏性。DMR试验与ELISA一样灵敏但是具有样品量需求更少(约1μl)和测定时间更短(小于30min)等额外的优势。
亲和素滴定法:亲和素(ImmunoPure Avidin#21121;Pierce Biotechnology)溶于PBS。样品通过混合各种大量亲和素到生物素化的FeMnFe 2O 4MNPs而制备。37℃孵育15分钟后,样品的T2值用先前报导于“Chip-NMR biosensor for detection and molecular analysis of cells,”(Lee et al.,Nat.Med.14,pp.869-874(2008))的微型核磁共振(NMR)系统以1μL样品测得。Carr-Purcell-Meiboom-Gill脉冲序列与下列参数一同使用:回声时间(TE),4msec;重复时间(TR),6sec;每次扫描180°脉冲数,500;扫描次数,8;作为参照,根据无亲和素样品的T2值计算出ΔT2。所有测量都测试3次并且数据显示平均值±标准差。
对于细胞检测,我们用MNPs结合的以肿瘤标记物(HER2)为目标的抗体标记癌症细胞(SkBr3)。随着MNP孵育,细胞松弛,定义为每单位细胞浓度的弛豫速率(1/T2)被发现与MNPs的r2成正比。图10A表示几种类型MNPs的每单位细胞浓度的弛豫速率。结果是,FeMnFe 2O 4产生最显著的ΔT2,而允许目标检测接近于单细胞水平。图10B表示FeMnFe 2O 4MNPs.标准的ΔT2与细胞浓度关系。
人类的SkBr3乳腺癌细胞在供应商推荐的培养基里培养并且维持在37℃,含5%CO2的湿润空气中。在细胞聚合时,用FeMnFe 2O4MNPs结合的HER2/neu抗体37℃孵育10分钟。参照样本在没有靶蛋白下制备。所有的样本通过离心(1,000rpm,5分钟)洗涤3次,重悬于PBS,并且梯度稀释。样本中的细胞浓度用细胞计数器测定。相同的微型NMR系统和脉冲序列(之前所描述的)用来获得T2测定值。
例7:核磁共振成像(MRI)
FeMFe2O4MNPs已评估作为MRI显像剂使用。影像的对比研究证实使用FeMFe2O4MNPs可以增加对比对。例如,与广泛使用的CLIO纳米微粒相比,FeMFe2O4MNPs在低10倍剂量下能产生一样的信号变化。我们也在体内影像学研究中进行初步实践。我们给小鼠注射各种MNPs[交联氧化铁(CLIO),Fe 3O 4,和FeMnFe 2O 4],并保持一样的金属量(10mg[metal]/kg)。然后我们用7T MRI仪在不同的时间点得到T2加权影像。注射3小时候的影像证实对比增强的确与每种类型的MNPs的r2值相当。
图11A是一个在注射MNPs前的器官MRI影像。图11B到11D是已经注射不同MNPs.的器官的MRI影像。影像中的字母“L”识别肝的位置而影像中的字母“K”则识别肾的位置。FeMnFe 2O 4MNPs(图11D)导致最显著变黑,从而支持了FeMnFe 2O 4MNPs作为一种有效的显像剂的潜在效用。在包含吞噬细胞的器官中比如肝,脾,或者骨髓中,静脉注射约3小时后发生峰值信号的改变,与一小时长的血液半衰期一致。在早期的时间点,大多器官包括肾脏由于血管灌注而显示相当暗。这种改变在24小时内都恢复到基准线。
影像学研究使用鸟笼设计模型中的4.7T和7.0T扫描仪(Bruker)以及一卷体积线圈来进行(Rapid Biomedical,Germany)。为了确定r2,准备由不同金属浓度的FeMnFe 2O 4MNPs组成的影像,而T2值用以下自旋回波参数测得:TE=10msec;TR=2000msec;矩阵256×256;每次扫描180°脉冲数=16。对于小鼠成像,FeMnFe 2O 4MNPs(在PBS里)通过尾静脉静脉注射10mg[metal]/kg的金属剂量。小鼠用N 2O和O 2(7:3)混合气体里的异氟烷(5%诱导麻醉,1.5%维持麻醉)麻醉。T2加权像用以下参数获得:TE=36msec;TR=2420msec;翻转角位90°;矩阵128×128;视野4×4cm2,层厚1mm。
其它实施方案
应该了解虽然本发明在它的详细说明书中已经做了描述,但前述说明目的是说明而不是限制由所附权利要求范围定义的本发明的范围。另有其它方面、优势、以及修饰在以下权利要求范围之内。
- 还没有人留言评论。精彩留言会获得点赞!