与多个分子的抗原反复结合的抗原结合分子的制作方法
本申请是国际申请号pct/jp2009/057309,国际申请日为2009年4月10日,进入中国国家阶段日期为2010年12月10日,中国申请号为200980122466.6,发明名称为“与多个分子的抗原反复结合抗原结合分子”的分案申请。本发明涉及提高抗原结合分子的药物动力学的方法、增加抗原结合分子与抗原的结合次数的方法、药物动力学提高的抗原结合分子、抗原结合分子与抗原的结合次数增加的抗原结合分子、以及它们的制备方法等。
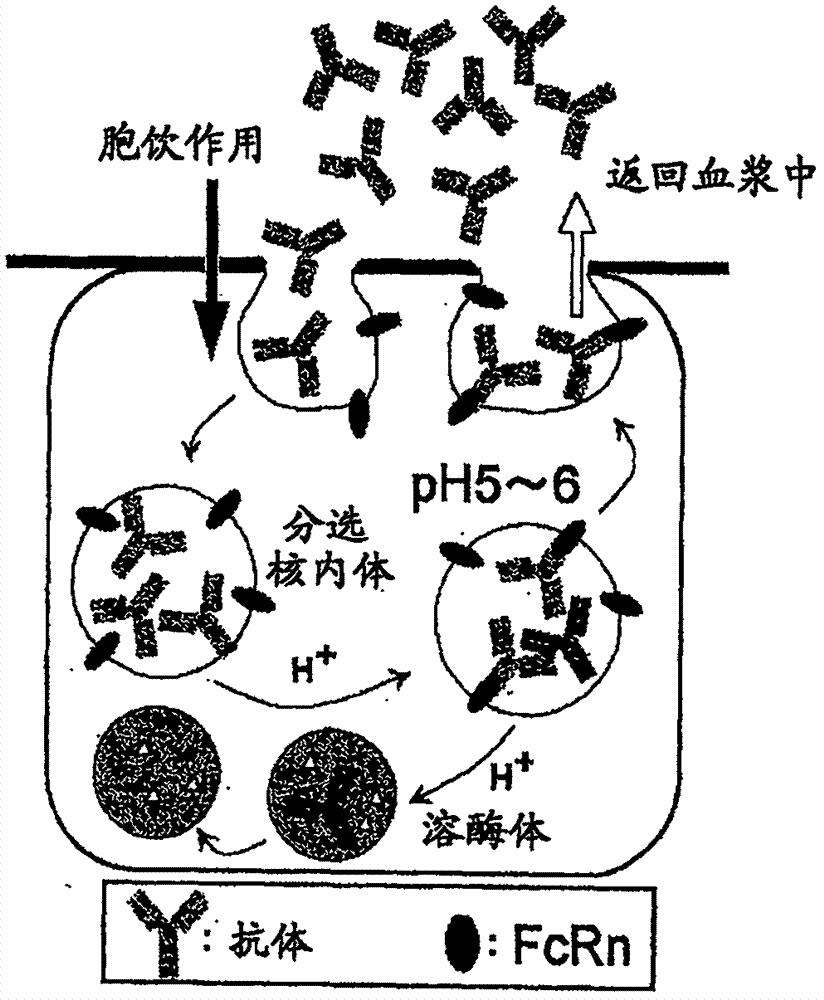
背景技术:
:抗体在血浆中的稳定性高、副作用也少,因此作为药品而受到关注。其中,有多种igg型抗体药物已上市,目前还有多种抗体药物正在开发中(非专利文献1、非专利文献2)。另一方面,作为可适用于第2代抗体药物的技术,人们开发了各种技术,有人报道了提高效应子功能、抗原结合能力、药物动力学、稳定性的技术或降低免疫原性风险的技术等(非专利文献3)。认为问题在于:由于抗体药物通常给药量非常大,所以难以制备皮下给药制剂;以及制造成本高等。作为减少抗体药物的给药量的方法,人们在考虑提高抗体的药物动力学的方法和提高抗体与抗原的亲和性(亲和力)的方法。作为提高抗体的药物动力学的方法,有人报道了恒定区的人工氨基酸取代(非专利文献4、5)。作为增强抗原结合能力、抗原中和能力的技术,有人报道了亲和力成熟技术(非专利文献6),通过向可变区的cdr区等的氨基酸中导入突变,可以增强与抗原的结合活性。通过增强抗原结合能力,可以提高在体外的生物活性、或者减少给药量,还可以进一步提高在体内的药效(非专利文献7)。另一方面,每1分子的抗体能够中和的抗原量依赖于亲和力,通过增强亲和力,可以以少的抗体量中和抗原,可以通过各种方法来增强抗体的亲和力。并且,只要与抗原进行共价键结合、使亲和力无限大,就可以用1分子的抗体中和1分子的抗原(抗体为二价时中和两分子的抗原)。但在现有方法中,用1分子的抗体中和1分子的抗原(抗体为二价时中和两分子的抗原)的化学量论的中和反应是界限,以抗原量以下的抗体量无法完全中和抗原。即,在增强亲和力的效果方面存在界限(非专利文献9)。当抗体为中和抗体时,为了使其中和效果持续一定期间,必需给予该期间内在体内产生的抗原量以上的抗体量,仅凭提高上述抗体的药物动力学或亲和力成熟技术,在减少必需的抗体给药量方面存在界限。因此,为了以抗原量以下的抗体量目标期间持续抗原的中和效果,必需用1个抗体中和多个抗原。作为用1个抗体中和多个抗原的方法,有利用赋予抗体催化功能的催化抗体进行的抗原的灭活。当为蛋白抗原时,认为通过水解抗原的肽键可以将抗原灭活,通过抗体催化该水解反应,可以反复中和(失活)抗原(非专利文献8)。迄今为止,报道了许多有关催化抗体和催化抗体制作技术,但作为药品具有足够的催化活性的催化抗体则未见报道。即,在抗某抗原的抗体的体内试验中,与普通的不具有催化功能的中和抗体相比,以低剂量发挥同等以上的效果、或者以相同的给药量可以更持续地发挥效果的催化抗体迄今为止未见报道。这样,没有关于用1分子的抗体中和多个抗原、可以发挥优于普通中和抗体的体内效果的抗体的报道,为了减少给药量以及延长持续性,人们希望开发用1个抗体中和多个抗原、在体内较普通的中和抗体更能发挥效果的新型抗体制作技术。本发明的现有技术文献如下所示。现有技术文献非专利文献非专利文献1:monoclonalantibodysuccessesintheclinic,janicemreichert,clarkjrosensweig,laurabfaden&matthewcdewitz,naturebiotechnology23,1073-1078(2005).非专利文献2:pavlouak,belseymj.thetherapeuticantibodiesmarketto2008.eurjpharmbiopharm.2005apr;59(3):389-96.非专利文献3:kimsj,parky,honghj.antibodyengineeringforthedevelopmentoftherapeuticantibodies.molcells.2005aug31;20(1):17-29.review.非专利文献4:hintonpr,xiongjm,johlfsmg,tangmt,kellers,tsurushitan.anengineeredhumanigg1antibodywithlongerserumhalf-life.jimmunol.2006jan1;176(1):346-56.非专利文献5:ghetiev,popovs,borvakj,raduc,matesoid,medesanc,oberrj,wardes.increasingtheserumpersistenceofaniggfragmentbyrandommutagenesis.natbiotechnol.1997jul;15(7):637-40.非专利文献6:procnatlacadsciusa.2005jun14;102(24):8466-71.epub2005jun6.ageneralmethodforgreatlyimprovingtheaffinityofantibodiesbyusingcombinatoriallibraries.rajpala,beyazn,haberl,cappuccillig,yeeh,bhattrr,takeuchit,lernerra,crear.非专利文献7:wuh,pfarrds,johnsons,brewahya,woodsrm,patelnk,whitewi,youngjf,kienerpa.developmentofmotavizumab,anultra-potentantibodyforthepreventionofrespiratorysyncytialvirusinfectionintheupperandlowerrespiratorytract.jmolbiol.2007,368,652-665.非专利文献8:hansoncv,nishiyamay,pauls.catalyticantibodiesandtheirapplications.curropinbiotechnol.2005dec;16(6):631-6.非专利文献9:rathanaswamip,roalstads,roskosl,suqj,lackies,babcookj.demonstrationofaninvivogeneratedsub-picomolaraffinityfullyhumanmonoclonalantibodytointerleukin-8.biochembiophysrescommun.2005sep9;334(4):1004-13.技术实现要素:发明所要解决的课题本发明鉴于上述状况而设,其目的在于提供:抗原结合分子与抗原多次结合的方法、提高抗原结合分子的药物动力学的方法、可以多次与抗原结合的抗原结合分子、药物动力学得到改善的抗原结合分子、含有该抗原结合分子的药物组合物、以及它们的制备方法。解决课题的方法本发明人等对抗原结合分子等具有抗原结合能力的多肽与抗原多次结合的方法、改善血浆中半衰期(血中半衰期)(提高药物动力学)的方法进行了深入研究。其结果,本发明人等发现:与在血浆中(血中)的ph下的抗原结合活性相比,在早期核内体内的ph下的抗原结合活性弱的抗原结合分子与抗原多次结合,血浆中半衰期长。本发明涉及抗原结合分子与抗原多次结合的方法、提高抗原结合分子的药物动力学的方法、能够多次与抗原结合的抗原结合分子、药物动力学提高的抗原结合分子、药物动力学提高的抗原结合分子的制备方法等,更具体而言,本发明提供下述[1]-[50]。[1]抗原结合分子,其对于抗原的在ph5.8下的kd与在ph7.4下的kd之比、即kd(ph5.8)/kd(ph7.4)的值为2以上。[2][1]所述的抗原结合分子,其中kd(ph5.8)/kd(ph7.4)的值为10以上。[3][1]所述的抗原结合分子,其中kd(ph5.8)/kd(ph7.4)的值为40以上。[4][1]-[3]中任一项所述的抗原结合分子,其特征在于:至少1个氨基酸被组氨酸取代、或者插入至少1个组氨酸。[5][1]-[4]中任一项所述的抗原结合分子,其特征在于:该抗原结合分子具有拮抗活性。[6][1]-[5]中任一项所述的抗原结合分子,其特征在于:该抗原结合分子与膜抗原或可溶型抗原结合。[7][1]-[6]中任一项所述的抗原结合分子,其特征在于:抗原结合分子为抗体。[8]药物组合物,其中含有[1]-[7]中任一项所述的抗原结合分子。[9]提高抗原结合分子的药物动力学的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[10]增加抗原结合分子与抗原的结合次数的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[11]增加抗原结合分子所能结合的抗原数目的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[12]使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[13]使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[14]增加抗原结合分子的血浆中抗原消除能力的方法,该方法通过使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性来进行。[15][9]-[14]中任一项所述的方法,其特征在于:对于抗原的在ph5.8下的kd与在ph7.4下的kd之比、即kd(ph5.8)/kd(ph7.4)的值为2以上。[16][9]-[14]中任一项所述的方法,其特征在于:使kd(ph5.8)/kd(ph7.4)的值为10以上。[17][9]-[14]中任一项所述的方法,其特征在于:使kd(ph5.8)/kd(ph7.4)的值为40以上。[18]提高药物动力学的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[19]增加抗原结合分子与抗原的结合次数的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[20]增加抗原结合分子所能结合的抗原数目的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[21]使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[22]使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[23]增加抗原结合分子的血浆中抗原消除能力的方法,该方法通过用组氨酸取代抗原结合分子的至少1个氨基酸或插入至少1个组氨酸来进行。[24][18]-[23]中任一项所述的方法,其特征在于:通过取代成组氨酸或插入组氨酸,使ph5.8下的抗原结合活性与在ph7.4下的抗原结合活性之比即kd(ph5.8)/kd(ph7.4)的值与组氨酸取代或插入前相比变大。[25][9]-[24]中任一项所述的方法,其特征在于:抗原结合分子具有拮抗活性。[26][9]-[25]中任一项所述的方法,其特征在于:抗原结合分子与膜抗原或可溶型抗原结合。[27][9]-[26]中任一项所述的方法,其特征在于:抗原结合分子为抗体。[28]抗原结合分子的筛选方法,该方法包括以下步骤:(a)获得ph6.7-ph10.0下的抗原结合分子的抗原结合活性的步骤;(b)获得ph4.0-ph6.5下的抗原结合分子的抗原结合活性的步骤;(c)选择ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤。[29][28]所述的筛选方法,其特征在于:选择ph6.7-ph10.0下的抗原结合活性为ph4.0-ph6.5下的抗原结合活性的两倍以上的抗体。[30]抗原结合分子的筛选方法,该方法包括以下步骤:(a)在ph6.7-ph10.0的条件下使抗原结合分子与抗原结合的步骤;(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤;(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤。[31]第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的筛选方法,该方法包括以下步骤:(a)在第一ph条件下使抗原结合分子与固定有抗原的柱结合的步骤;(b)在第二ph条件下从柱上洗脱在第一ph条件下与柱结合的抗原结合分子的步骤;(c)获得被洗脱的抗原结合分子的步骤。[32]第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的筛选方法,该方法包括以下步骤:(a)在第一ph条件下使抗原结合分子文库与固定有抗原的柱结合的步骤;(b)在第二ph条件下从柱上洗脱抗原结合分子的步骤;(c)扩增编码被洗脱的抗原结合分子的基因的步骤;(d)获得被洗脱的抗原结合分子的步骤。[33][31]或[32]所述的筛选方法,其特征在于:第一ph为ph6.7-ph10.0,第二ph为ph4.0-ph6.5。[34][28]-[33]中任一项所述的筛选方法,其中抗原结合分子是抗原结合分子中的至少1个以上的氨基酸被组氨酸取代或插入至少1个组氨酸的抗原结合分子。[35][28]-[33]中任一项所述的筛选方法,该方法以获得血浆中滞留性优异的抗原结合分子为目的。[36][28]-[33]中任一项所述的筛选方法,该方法以获得能够与抗原进行两次以上结合的抗原结合分子为目的。[37][28]-[33]中任一项所述的筛选方法,该方法以获得可结合的抗原数多于抗原结合位点的抗原结合分子为目的。[38][28]-[33]中任一项所述的筛选方法,该方法以获得在细胞内解离在细胞外结合的抗原的抗原结合分子为目的。[39][28]-[33]中任一项所述的筛选方法,该方法以获得以与抗原结合的状态摄入细胞内、以未与抗原结合的状态被释放到细胞外的抗原结合分子为目的。[40][28]-[33]中任一项所述的筛选方法,该方法以获得血浆中抗原消除能力增加的抗原结合分子为目的。[41][28]-[40]中任一项所述的筛选方法,其中抗原结合分子是用作药物组合物的抗原结合分子。[42][28]-[41]中任一项所述的筛选方法,其特征在于:抗原结合分子为抗体。[43]抗原结合分子的制备方法,该制备方法包括以下步骤:(a)获得ph6.7-ph10.0下的抗原结合分子的抗原结合活性的步骤;(b)获得ph4.0-ph6.5下的抗原结合分子的抗原结合活性的步骤;(c)选择ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤;(d)获得编码(c)中所选择的抗原结合分子的基因的步骤;(e)使用(d)中得到的基因制备抗原结合分子的步骤。[44]抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在ph6.7-ph10.0的条件下使抗原结合分子与抗原结合的步骤;(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤;(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤;(d)获得编码(c)中取得的抗原结合分子的基因的步骤;(e)使用(d)中得到的基因制备抗原结合分子的步骤。[45]第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在第一ph条件下使抗原结合分子与固定有抗原的柱结合的步骤;(b)在第二ph条件下从柱上洗脱在第一ph条件下与柱结合的抗原结合分子的步骤;(c)获得被洗脱的抗原结合分子的步骤;(d)获得编码(c)中取得的抗原结合分子的基因的步骤;(e)使用(d)中得到的基因制备抗原结合分子的步骤。[46]第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的制备方法,该方法包括以下步骤:(a)在第一ph条件下使抗原结合分子文库与固定有抗原的柱结合的步骤;(b)在第二ph条件下从柱上洗脱抗原结合分子的步骤;(c)扩增编码被洗脱的抗原结合分子的基因的步骤;(d)获得被洗脱的抗原结合分子的步骤;(e)获得编码(d)中取得的抗原结合分子的基因的步骤;(f)使用(e)中得到的基因制备抗原结合分子的步骤。[47][45]或[46]所述的制备方法,其特征在于:第一ph为ph6.7-ph10.0,第二ph为ph4.0-ph6.5。[48][43]-[47]中任一项所述的制备方法,该制备方法进一步包括:用组氨酸取代抗原结合分子中的至少1个以上的氨基酸或插入至少1个组氨酸的步骤。[49][43]-[48]中任一项所述的制备方法,其特征在于:抗原结合分子为抗体。[50]药物组合物,其中含有利用[43]-[49]中任一项所述的制备方法制备的抗原结合分子。发明效果根据本发明,提供使1分子的抗原结合分子与多个抗原反复结合的方法。1分子的抗原结合分子通过与多个抗原结合,可以提高抗原结合分子的药物动力学,且可以在体内发挥优于普通的抗原结合分子的效果。附图说明图1是显示与膜型抗原结合的抗体的分解路径的图。图2是显示基于fcrn的igg分子的补救机制的图。图3是显示igg分子在核内体内从膜型抗原上解离,从而再次与新的抗原结合的示意图。图4是显示igg分子在核内体内从可溶型抗原上解离,从而再次与新的抗原结合的示意图。图5是显示固定有抗原的柱淘选的图。图6是显示由柱淘选获得的克隆的噬菌体elisa的结果的曲线图。上段是wt,下段是cl5。图7是显示ph依赖性结合抗il-6受体抗体的生物学中和活性的曲线图。图8是显示ph依赖性结合抗il-6受体抗体在ph7.4下与可溶型il-6受体结合的biacore传感图的结果的图。最上的是wt,上起第2位是h3pi/l73,上起第3位是h170/l82,最下的是clh5/l73。图9是显示ph依赖性结合抗il-6受体抗体在ph5.8下与可溶型il-6受体结合的biacore传感图的结果的图。最上的是wt,上起第2位是h3pi/l73,上起第3位是h170/l82,最下的是clh5/l73。图10是显示ph依赖性结合抗il-6受体抗体与膜型il-6受体结合(ph7.4)和解离(ph5.8)的biacore传感图的结果的图。最上的是wt,上起第2位是h3pi/l73,上起第3位是h170/l82,最下的是clh5/l73。图11是显示ph依赖性结合抗il-6受体抗体与sr344反复结合的biacore的传感图。图12是显示在ph依赖性结合抗il-6受体抗体与sr344反复结合试验中的总抗原结合量的曲线图。图13是显示ph依赖性结合抗il-6受体抗体在人il-6受体转基因小鼠体内的血浆中抗体浓度变化的曲线图。图14是显示ph依赖性结合抗il-6受体抗体在食蟹猴体内的血浆中抗体浓度变化的曲线图。图15是显示ph依赖性结合抗il-6受体抗体在食蟹猴体内的crp浓度变化的曲线图。图16是显示ph依赖性结合抗il-6受体抗体在食蟹猴体内的非结合型食蟹猴il-6受体浓度变化的曲线图。图17是显示ph依赖性结合抗il-6受体抗体与膜型il-6受体结合(ph7.4)和解离(ph5.8)的biacore传感图的结果的图。上起依次为wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1。图18是显示ph依赖性结合抗il-6受体抗体在人il-6受体转基因小鼠体内的wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1的血浆中抗体浓度变化的曲线图。图19是显示ph依赖性结合抗il-6受体抗体与膜型il-6受体的结合(ph7.4)和解离(ph5.8)的biacore传感图的结果的图。上起依次为wt、fv4-igg1、fv4-igg2、fv4-m58。图20是显示ph依赖性结合抗il-6受体抗体在人il-6受体转基因小鼠体内的wt、fv4-igg1、fv4-igg2、fv4-m58的血浆中抗体浓度变化的曲线图。图21是显示ph依赖性结合抗il-6受体抗体与膜型il-6受体的结合(ph7.4)和解离(ph5.8)的biacore传感图的结果的图。上起依次为fv1-m71、fv1-m73、fv3-m71、fv3-m73。图22是显示在食蟹猴体内以0.5mg/kg给予h3pi/l73-igg1、fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73和以1.0mg/kg给予高亲和性抗体时,ph依赖性结合抗il-6受体抗体的血浆中抗体浓度变化的曲线图。图23是显示在食蟹猴体内ph依赖性结合抗il-6受体抗体的h3pi/l73-igg1、fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73、高亲和性ab给药组的crp浓度变化的曲线图。图24是显示在食蟹猴体内ph依赖性结合抗il-6受体抗体的h3pi/l73-igg1、fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73、高亲和性ab给药组的非结合型食蟹猴il-6受体浓度变化的曲线图。图25是显示重链(vh1、vh2、vh3、vh4)和轻链(vl1、vl2、vl3)的fr1、fr2、fr3、fr4和cdr1、cdr2、cdr3的图。星号显示排比序列中存在氨基酸突变的位点。图26是显示作为ph依赖性结合抗il-6抗体的抗il6克隆2在ph7.4和ph5.5下与il-6结合的biacore传感图的结果的图。ph7.4的图从上起依次显示100、50、25、12.5、6.25ng/ml的il-6。图27是显示作为ph依赖性结合抗il-31受体抗体的抗il31r克隆1在ph7.4和ph5.5下与il-31受体结合的biacore传感图的结果的图。ph5.5的图从上起依次显示100、50、25、12.5ng/ml的il-31受体。图28是显示对小鼠静脉内给予sr344和抗人il-6受体抗体的混合溶液后的血浆中抗体浓度变化的图。图29是显示对小鼠静脉内给予sr344和抗人il-6受体抗体的混合溶液后的血浆中sr344浓度变化的图。具体实施方式本发明提供增加抗原结合分子与抗原的结合次数的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来增加抗原结合分子与抗原的结合次数的方法。本发明还提供增加抗原结合分子与抗原的结合次数的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供增加抗原结合分子与抗原的结合次数的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供增加抗原结合分子所能结合的抗原数的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来增加抗原结合分子所能结合的抗原数的方法。本发明还提供增加抗原结合分子所能结合的抗原数的方法,其特征在于:将抗原结合分子中的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供增加抗原结合分子所能结合的抗原数的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法。本发明还提供使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法。本发明还提供使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法。更具体而言,本发明提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来增加抗原结合分子的血浆中抗原消除能力的方法。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供提高抗原结合分子的药物动力学的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来提高抗原结合分子的药物动力学(延长血浆中滞留性)的方法。本发明还提供提高药物动力学的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供提高药物动力学的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法。更具体而言,提供通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来增加抗原结合分子的血浆中抗原消除能力的方法。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法,其特征在于:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸。本发明还提供增加抗原结合分子的血浆中抗原消除能力的方法,其特征在于:取代、缺失、添加和/或插入抗原结合分子所含的抗体恒定区中的氨基酸。本发明中,“药物动力学的提高”、“药物动力学的改善”或“优异的药物动力学”可以改称“血浆中(血中)滞留性的提高”、“血浆中(血中)滞留性的改善”、“优异的血浆中(血中)滞留性”,这些语句以相同的意义使用。本发明中,使酸性ph下的抗原结合能力弱于中性ph下的抗原结合能力,是指使抗原结合分子在ph4.0-ph6.5下的抗原结合活性弱于在ph6.7-ph10.0下的抗原结合活性。优选使抗原结合分子在ph5.5-ph6.5下的抗原结合活性弱于在ph7.0-ph8.0下的抗原结合活性,特别优选使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性。因此,本发明中酸性ph通常是指ph4.0-ph6.5,优选为ph5.5-ph6.5,特别优选为ph5.8。本发明中,中性ph通常是指ph6.7-ph10.0,优选为ph7.0-ph8.0,特别优选为ph7.4。本发明中,“使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力”的描述还可以描述成使抗原结合分子在中性下的抗原结合能力高于在酸性ph下的抗原结合能力。即,本发明中,只要增大抗原结合分子在酸性ph下的抗原结合能力与在中性ph下的抗原结合能力之差即可(例如象后述那样,只要增大kd(ph5.8)/kd(ph7.4)的值即可)。为了增大抗原结合分子在酸性ph下的抗原结合能力与在中性ph下的抗原结合能力之差,例如可以降低酸性ph下的抗原结合能力,也可以提高中性ph下的抗原结合能力,或者可以是两者。测定抗原的结合活性时,除ph以外的条件可以由本领域技术人员适当选择,没有特别限定,例如,如实施例所述,可以在mes缓冲液中、37℃的条件下进行测定。抗原结合分子的抗原结合活性的测定可以利用本领域技术人员所公知的方法进行,例如,如实施例所述,可以使用biacore(gehealthcare)等进行测定。当抗原为可溶型抗原时,通过使抗原以分析物的形式流经固定有抗原结合分子的芯片(chip),可以评价抗原结合分子与可溶型抗原的结合能力;当抗原为膜型抗原时,通过使抗原结合分子以分析物的形式流经固定有抗原的芯片,可以评价抗原结合分子与膜型抗原的结合能力。本发明中,只要酸性ph下的抗原结合活性弱于中性ph下的抗原结合活性,对酸性ph下的抗原结合活性与中性ph下的抗原结合活性之差没有特别限定,优选对于抗原的在ph5.8下的kd与在ph7.4下的kd(dissociationconstant:解离常数)之比即kd(ph5.8)/kd(ph7.4)的值为2以上,进一步优选kd(ph5.8)/kd(ph7.4)的值为10以上,更进一步优选kd(ph5.8)/kd(ph7.4)的值为40以上。对kd(ph5.8)/kd(ph7.4)的值的上限没有特别限定,只要在本领域技术人员的技术中能够制作,可以是400、1000、10000等任何值。作为抗原结合活性的值,当抗原为可溶型抗原时,可以使用kd(解离常数);当抗原为膜型抗原时,可以使用表观kd(apparentdissociationconstant:表观解离常数)。kd(解离常数)和表观kd(表观解离常数)可以按照本领域技术人员公知的方法进行测定,例如可以使用biacore(gehealthcare)、斯卡恰特作图法(scatchardplot)、facs等。本发明中,作为表示酸性ph下的抗原结合活性与中性ph下的抗原结合活性之差的其他指标,例如还可以使用解离速度常数kd(dissociationrateconstant:解离速度常数)。作为表示结合活性之差的指标,当使用kd(解离速度常数)来代替kd(解离常数)时,对于抗原的在ph5.8下的kd(解离速度常数)与在ph7.4下的kd(解离速度常数)之比即kd(ph5.8)/kd(ph7.4)的值优选为2以上,进一步优选为5以上,更进一步优选为10以上,更优选为30以上。对kd(ph5.8)/kd(ph7.4)的值的上限没有特别限定,只要在本领域技术人员的技术常识内能够制作,可以是50、100、200等任何值。作为抗原结合活性的值,当抗原为可溶型抗原时,可以使用kd(解离速度常数);当抗原为膜型抗原时,可以使用表观kd(apparentdissociationrateconstant:表观解离速度常数)。kd(解离速度常数)和表观kd(表观解离速度常数)可以按照本领域技术人员公知的方法进行测定,例如可以使用biacore(gehealthcare)、facs等。本发明中,在不同ph下测定抗原结合分子的抗原结合活性时,优选除ph以外的条件相同。对使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性的方法(赋予ph依赖性结合能力的方法)没有特别限定,可以通过任何方法来进行。可以列举:例如通过将抗原结合分子中的氨基酸取代成组氨酸、或在抗原结合分子中插入组氨酸,使ph5.8下的抗原结合活性弱于ph7.4下的抗原结合活性的方法。已知通过用组氨酸取代抗体中的氨基酸,可以赋予抗体ph依赖性的抗原结合活性(febsletter,309(1),85-88,(1992))。对导入(进行)组氨酸突变(取代)或插入的位点没有特别限定,只要与突变或插入前相比在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性(kd(ph5.8)/kd(ph7.4)的值变大),可以是任何位点。例如,当抗原结合分子为抗体时,可以列举抗体的可变区等。导入(进行)组氨酸突变或插入的数可以由本领域技术人员适当确定,可以用组氨酸只取代1个位点,或者只在1个位点插入组氨酸,也可以用组氨酸取代2个位点以上的多个位点,或者在2个位点以上的多个位点插入组氨酸。还可以同时导入除组氨酸突变以外的突变(突变成组氨酸以外的氨基酸)。还可以同时进行组氨酸突变和组氨酸插入。组氨酸取代或组氨酸插入可以利用本领域技术人员所公知的、将丙氨酸分区(scanning)的丙氨酸取代成组氨酸的组氨酸分区等方法随机进行,可以从随机导入组氨酸突变或插入的抗原结合分子文库中选择与突变前相比kd(ph5.8)/kd(ph7.4)的值变大的抗原结合分子。将抗原结合分子的氨基酸取代成组氨酸或在抗原结合分子的氨基酸中插入组氨酸时,虽然没有特别限定,但优选组氨酸取代或插入后的抗原结合分子在ph7.4下的抗原结合活性与组氨酸取代或插入前的抗原结合分子在ph7.4下的抗原结合活性同等。这里,组氨酸取代或插入后的抗原结合分子在ph7.4下的抗原结合活性与组氨酸取代或插入前的抗原结合分子在ph7.4下的抗原结合活性同等,是指组氨酸取代或插入后的抗原结合分子维持着组氨酸取代或插入前的抗原结合分子所具有的抗原结合活性的10%以上、优选50%以上、进一步优选80%以上、更优选90%以上。通过组氨酸取代或插入,抗原结合分子的抗原结合活性变低时,通过抗原结合分子中的1或多个氨基酸的取代、缺失、添加和/或插入等,可以使抗原结合活性与组氨酸取代或插入前的抗原结合活性同等。本发明中,还包括这样的在组氨酸取代或插入后通过进行1个或多个氨基酸的取代、缺失、添加和/或插入使结合活性同等的抗原结合分子。作为使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性的其他方法,可以列举:将抗原结合分子中的氨基酸取代成非天然型氨基酸或在抗原结合分子中的氨基酸中插入非天然型氨基酸的方法。已知可以人为地控制非天然氨基酸的pka(angew.chem.int.ed.2005,44,34、chemsocrev.2004sep10;33(7):422-30、aminoacids.1999;16(3-4):345-79)。因此,在本发明中,可以使用非天然型氨基酸来代替上述组氨酸。另外,上述组氨酸取代和/或插入以及非天然型氨基酸的取代和/或插入可以同时进行。本发明中使用的非天然型氨基酸可以是任何非天然型氨基酸,可以使用本领域技术人员所公知的非天然型氨基酸等。并且,当抗原结合分子是包含抗体恒定区的物质时,作为使抗原结合分子在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性的其他方法,可以列举:修饰抗原结合分子所含的抗体恒定区的方法。这样的抗体恒定区的修饰的具体例子有:例如实施例中所述的取代成恒定区的方法。作为抗体恒定区的修饰方法,可以列举:例如研究恒定区的多个同种型(igg1、igg2、igg3、igg4),选择在ph5.8下的抗原结合活性降低(在ph5.8下的解离速度变快)的同种型的方法。还可以列举:通过向野生型同种型的氨基酸序列(野生型igg1、igg2、igg3、igg4氨基酸序列)中导入氨基酸取代,使在ph5.8下的抗原结合活性降低(在ph5.8下的解离速度变快)的方法。根据同种型(igg1、igg2、igg3、igg4),抗体恒定区的铰链区的序列大不相同,由于铰链区的氨基酸序列的不同对抗原结合活性影响很大,所以通过根据抗原或表位的种类来选择适当的同种型,可以选择ph5.8下抗原结合活性降低的(ph5.8下的解离速度加快)同种型。由于铰链区的氨基酸序列的不同对抗原结合活性影响很大,所以作为野生型同种型的氨基酸序列的氨基酸取代位点,认为优选铰链区。利用上述方法等使抗原结合物质在ph5.8下的抗原结合活性弱于在ph7.4下的抗原结合活性(kd(ph5.8)/kd(ph7.4)的值变大)时,虽然没有特别限定,但优选与原始抗体相比,kd(ph5.8)/kd(ph7.4)的值通常为2倍以上,优选5倍以上,进一步优选10倍以上。本发明中,“药物动力学提高”不仅包括抗原结合分子从对人、小鼠、大鼠、猴、兔、狗等动物给药到从血浆中消除(例如,直至形成抗原结合分子在细胞内被分解等而无法返回到血浆中的状态)的时间变长,还包含抗原结合分子从给药起到从血浆中消除这一期间以能够与抗原结合的状态(例如,抗原结合分子未与抗原结合的状态)滞留在血浆中的时间变长。即使抗原结合分子存在于血浆中,当抗原已经与该抗原结合分子结合时,该抗原结合分子也无法与新的抗原结合。因此,若抗原结合分子未与抗原结合的时间变长,则能够与新的抗原结合的时间变长(能够与新的抗原结合的机会增多),可以减少在体内抗原未与抗原结合分子结合的时间(换言之,可以延长抗原结合分子与抗原结合的时间)。例如,相对于存在于血浆中等体内的抗原(与抗原结合分子结合的分子和未与抗原结合分子结合的抗原的总量),与抗原结合分子结合的抗原的比例,通常在给予抗原结合分子后经过一定时间即减少。但是,若抗原结合分子以能够与抗原结合的状态滞留的时间变长,则可以抑制(例如减少其减少的程度等)其减少,结果,自给予抗体起经过一定期间后,相对于体内存在的抗原,与抗原结合分子结合的抗原的比例变高。即,本发明的“药物动力学的提高”,未必是指从给予抗原结合分子到抗原结合分子消除的时间延长(变长)。下述情形也均包含在本发明的“药物动力学的提高”中:即使抗原结合分子从给药起到消除的时间没有变化,但抗原结合分子以能够与抗原结合的状态(例如抗原结合分子未与抗原结合的状态)滞留于血浆中的时间变长的情形;以及体内的抗原未与抗原结合分子结合的时间减少(换言之,抗原结合分子与抗原结合的时间变长)的情形;或者相对于体内存在的抗原,与抗原结合分子结合的抗原的比例变高的情形。因此,本发明的“药物动力学的提高”至少包含以下(1)-(4)。(1)从给予抗原结合分子到抗原结合分子从血浆中消除的时间延长。(2)给予抗原结合分子后,抗原结合分子以能够与抗原结合的状态存在于血浆中的时间延长。(3)给予抗原结合分子后,体内的抗原未与抗原结合分子结合的时间减少(抗原结合分子与体内的抗原结合的时间延长)。(4)相对于体内存在的抗原,与抗原结合分子结合的抗原的比例增加。当抗原为存在于血浆中的可溶型抗原时,即使抗原结合分子的药物动力学(从血浆中消除的速度)同等,有时抗原结合分子所结合的抗原的消除也变快。这与通过降低抗原的药物动力学(从血浆中的消除变快),使相对于抗原的相对的抗原结合分子的药物动力学提高有关,即、与抗原结合分子以可与抗原结合的状态存在于血浆中的时间延长有关。因此,作为本发明的抗原结合分子的“药物动力学提高”的一种方式,还包含给予抗原结合分子后可溶型抗原从血浆中消除的速度(抗原结合分子的血浆中抗原消除能力)加快。本发明中,1分子的抗原结合分子是否与多个抗原结合,当抗原为膜型抗原时,可以根据抗原结合分子的药物动力学是否提高来判断。是否“药物动力学提高了”可以如下判断。例如,从给予抗原结合分子到抗原结合分子消除的时间是否延长,这可以通过测定抗原结合分子的血浆中半衰期、平均血浆中滞留时间、血浆中清除率等任意一个参数(フア一マコキネテイクス演習によゐ理解(南山堂))来判断。例如,对小鼠、大鼠、猴、兔、狗、人等给予抗原结合分子时,当为血浆中半衰期变长或平均血浆中滞留时间变长的情况等时,可以说抗原结合分子的药物动力学提高。上述参数可以利用本领域技术人员所公知的方法来测定,例如使用药物动力学分析软件winnonlin(pharsight),按照附录的操作指南进行无区室分析(noncompartmentalanalysis),由此可以适当评价。抗原结合分子从给药到消除这一期间以可与抗原结合的状态存在于血浆中的时间是否被延长,这可以通过测定未与抗原结合的抗原结合分子的血浆中浓度,并测定未与抗原结合的抗原结合分子的血浆中半衰期、平均血浆中滞留时间、血浆中清除率等任一个参数来判断。未与抗原结合的抗原结合分子的血浆中浓度的测定可以按照本领域技术人员公知的方法进行,例如按照clinpharmacol.2008apr;48(4):406-17中所述的方法进行测定。给予抗原结合分子后体内的抗原未与抗原结合分子结合的时间是否减少(抗原结合分子与抗原结合的时间变长),这可以通过测定抗原结合分子未结合的非结合型抗原的血浆中浓度,根据非结合型抗原的血浆中浓度、或非结合型抗原量相对于总抗原量的比例维持在低水平的期间来判断。非结合型抗原的血浆中浓度或非结合型抗原的抗原量相对于总抗原量的比例的测定可以按照本领域技术人员公知的方法进行,例如按照pharmres.2006jan;23(1):95-103中所述的方法进行测定。当抗原在体内显示出任何功能时,抗原是否被中和抗原的功能的抗原结合分子(拮抗分子,antagonisticmolecule)结合,这还可以根据该抗原的功能是否被中和来评价。抗原的功能是否被中和,这可以通过测定反映抗原的功能的任何体内的标志物来评价。抗原是否被激活抗原的功能的抗原结合分子(激动分子,agonisticmolecule)结合,这可以通过测定反映抗原的功能的任何体内的标志物来评价。对非结合型抗原的血浆中浓度的测定、非结合型抗原的抗原量相对于总抗原量的比例的测定、体内标志物的测定等测定没有特别限定,优选自给予抗原结合物质起经过一定时间后进行测定。本发明中,对自给予抗原结合物质起经过一定时间后没有特别限定,根据所给予的抗原结合物质的性质等,本领域技术人员可以适时确定,例如自给予抗原结合物质起经过1天后、自给予抗原结合物质起经过3天后、自给予抗原结合物质起经过7天后、自给予抗原结合物质起经过14天后、自给予抗原结合物质起经过28天后等。本发明中,优选在人体内的药物动力学提高。当难以测定在人体内的血浆中滞留性时,可以根据在小鼠(例如正常小鼠、人抗原表达转基因小鼠、人fcrn表达转基因小鼠等)或猴(例如食蟹猴等)体内的血浆中滞留性,预测在人体内的血浆中滞留性。对血浆中滞留性的测定方法没有特别限定,例如可以按照实施例所述的方法进行。抗原结合分子能否与抗原多次结合,这可以通过测定在与血浆中相同的中性条件下与抗原结合分子结合的抗原是否在与核内体内相同的酸性条件下解离、再次在中性条件下能够与多少抗原结合来评价。具体而言,使用biacore这样的评价抗原结合分子-抗原反应的仪器,在中性条件下使抗原结合分子-抗原复合体作用,之后暴露于酸性条件下一定时间,之后再次在中性条件下测定抗原结合分子能否与抗原结合,由此可以进行评价。与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的抗原结合量提高两倍时,可以说与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的结合次数提高两倍。当抗原为膜型抗原,与抗原结合的抗原结合分子经由抗原被摄入并被溶酶体分解,从而从血浆中消除时,通过评价与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的药物动力学或与抗原的结合期间是否提高或提高了多少,可以评价与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的结合次数是否增加。例如,与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子与抗原的结合期间提高两倍时,可以说与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的结合次数提高两倍。另外,测定抗原结合分子未结合的非结合型抗原的血浆中浓度,当非结合型抗原的血浆中浓度、或非结合型抗原的抗原量相对于总抗原量的比例维持在低水平的时间延长两倍时,可以说与修饰前的抗原结合分子相比,赋予了ph依赖性结合能力的抗原结合分子的结合次数提高两倍。当抗原为可溶型抗原时,在血浆中的中性条件下与抗原结合分子结合的抗原在核内体内解离,抗原结合分子返回血浆中时,抗原结合分子可以在血浆中的中性条件下再次与抗原结合,所以具有在核内体内的酸性条件下解离抗原的性质的抗原结合分子可以与抗原多次结合。和与抗原结合分子结合的抗原在核内体内未解离的情形(抗原维持着与抗原结合分子结合的状态返回血浆中)相比,与抗原结合分子结合的抗原在核内体内解离时,抗原被运送到溶酶体中被分解,所以抗原从血浆中消除的速度增加。即,以抗原从血浆中消除的速度为指标,还可以判断抗原结合分子能否与抗原多次结合。抗原从血浆中消除的速度的测定,例如还可以通过对体内给予抗原(例如膜抗原)和抗原结合分子,测定给药后血浆中的抗原浓度来进行。当抗原(例如膜抗原)在体内产生(分泌)时,若抗原从血浆中的消除速度增加,则血浆中抗原浓度降低,因此也可以以血浆中抗原浓度为指标,判断抗原结合分子能否与抗原多次结合。本发明中,“增加抗原结合分子与抗原的结合次数”是指,对人、小鼠、猴等给予抗原结合分子时,以抗原结合分子与抗原结合并摄入细胞内的步骤为1次,增加该步骤。即,在本发明中,“抗原结合分子两次与抗原结合”是指,抗原结合分子以与抗原结合的状态摄入细胞内,之后以解离了抗原的状态被释放到细胞外,被释放的抗原结合分子再次与抗原结合并摄入细胞内。抗原结合分子摄入细胞内时,抗原结合分子可以以结合1个抗原的状态摄入,也可以以结合2个或2个以上的抗原的状态摄入。本发明中,“抗原结合分子与抗原的结合次数增加”是指,不必所有抗原结合分子的抗原结合次数都增加,例如可以是抗原结合分子组合物所含的抗原结合分子中,两次以上与抗原结合的抗原结合分子的比例上升、或抗原结合分子组合物所含的抗原结合分子的结合次数的平均值上升等。本发明中,优选对人给予抗原结合分子时抗原结合分子与抗原的结合次数增加,但在难以测定在人体内的抗原结合次数时,根据体外的测定结果、在小鼠(例如表达抗原的转基因小鼠、表达人fcrn的转基因小鼠等)或猴(例如食蟹猴等)等体内的测定结果,可以预测在人体内的抗原结合次数。本发明中,优选抗原结合分子两次以上与抗原结合,例如优选抗原结合分子组合物中所含的抗原结合分子的至少10%以上、优选30%以上、进一步优选50%以上、更优选80%以上(例如90%以上、95%以上等)的抗原结合分子两次以上与抗原结合。本发明中,“增加抗原结合分子所能结合的抗原数”是指,在对人、小鼠、猴等动物给予抗原结合分子起到抗原结合分子被细胞内的溶酶体分解这一期间,增加抗原结合分子所能结合的抗原数。通常,igg等抗体具有2个结合位点,所以1个抗体最多与2个抗原结合,与抗原结合的抗体摄入细胞内,与抗原一同被溶酶体分解。因此,通常igg等抗体最多可以与2个抗原结合。利用本发明的方法,通过使抗体等抗原结合分子在核内体内的ph下的抗原结合活性弱于在血浆中的ph下的抗原结合活性,摄入细胞内的抗体等抗原结合分子在细胞内解离抗原,被释放到细胞外,能够再次与抗原结合。即,利用本发明的方法,可以与多于抗原结合分子的抗原结合位点数的数目的抗原结合。具体而言,例如当为具有2个结合位点的igg时,通过采用本发明的方法,在给予抗体到抗体被分解这一期间可以与3个以上、优选4个以上的抗原结合。例如,当抗体为中和抗体时,“增加抗原结合分子所能结合的抗原数”还可以是指增加抗原结合分子所能中和的抗原数。因此,当抗体为中和抗体时,还可以将“结合”与“中和”互换。本发明中,“增加抗原结合分子所能结合的抗原数”是指,不必所有抗原结合分子所能结合的抗原数都增加,例如可以是抗原结合分子组合物中所含的抗原结合分子所能结合的抗原数的平均值增加,还可以是能够与多于抗原结合分子的抗原结合位点数的抗原结合的抗原结合分子的比例增加等。本发明中,优选将抗原结合分子给予人时抗原结合分子所能结合的抗原数增加,当难以测定人体内的抗原数时,根据体外的测定结果、小鼠(例如表达抗原的转基因小鼠、表达人fcrn的转基因小鼠等)或猴(例如食蟹猴等)等的测定结果,可以预测人体内的可结合的抗原数。通常,当抗体为中和抗体时,认为上述抗原结合分子与抗原的结合次数与抗原结合分子所能中和的抗原数相关,因此抗原结合分子所能中和的抗原数的测定可以和上述抗原结合分子与抗原的结合次数的测定同样进行。本发明还提供:通过给予酸性ph下的抗原结合活性低于中性ph下的抗原结合活性的抗原结合分子,使抗原结合分子在体内两次以上与抗原结合的方法。本发明还涉及:在具有中和活性的抗原结合分子中,通过给予酸性ph下的抗原结合活性低于中性ph下的抗原结合活性的抗原结合分子,中和多于抗原结合分子的抗原结合位点数的数目的抗原的方法。优选涉及通过给予酸性ph下的抗原结合活性低于中性ph下的抗原结合活性的igg,中和3个以上、优选4个以上的抗原的方法。本发明还涉及:通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法。本发明中,抗原从抗原结合分子上解离的位置只要是细胞内即可,可以是任何位置,优选为早期核内体内。本发明中,“在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离”是指,未必与抗原结合分子结合摄入细胞内的抗原全部在细胞内从抗原结合分子上解离,只要与使抗原结合分子在酸性ph下的抗原结合能力低于在中性ph下的抗原结合能力之前相比,在细胞内从抗原结合分子上解离的抗原的比例增加即可。本发明还涉及:通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,促进细胞内的未与抗原结合的抗原结合分子与fcrn的结合的方法。通常,fcrn在核内体内与抗原结合分子结合,当抗原结合分子与膜型抗原结合时,认为其无法与fcrn结合,因此,作为本发明的优选方案,当抗原为膜型抗原时,可以列举下述方法:通过使抗原结合分子在核内体内的ph(酸性ph)下的抗原结合能力弱于在血浆中的ph(中性ph)下的抗原结合能力,促进核内体内的抗原结合分子从抗原上解离,促进抗原结合分子与fcrn的结合。当抗原为可溶型抗原时,可以列举下述方法:无论是否存在抗原的结合,抗原结合分子可以与fcrn结合,但若通过使抗原结合分子在核内体内的ph(酸性ph)下的抗原结合能力弱于在血浆中的ph(中性ph)下的抗原结合能力,在核内体内可以促进抗原从抗原结合分子上解离,则会促进“未与抗原结合”的抗原结合分子与fcrn的结合。无论抗原是膜型还是可溶型,只要未与抗原结合的抗原结合分子可以通过fcrn返回到血浆中,就可以再次与抗原结合,因此通过重复此过程,抗原结合分子可以多次与抗原结合。本发明中,“促进细胞内的抗原结合分子与fcrn的结合”是指,不必所有抗原结合分子都与fcrn结合,只要与使抗原结合分子在核内体内的ph下的抗原结合能力弱于在血浆中的ph下的抗原结合能力之前相比,细胞内与fcrn结合而未与抗原结合的抗原结合分子的比例增加即可。在本发明的促进细胞内的抗原结合分子与fcrn的结合的方法中,优选的抗原结合分子的例子有:与膜蛋白等膜型抗原(膜抗原)结合的抗原结合分子。其他优选的抗原结合分子的例子有:与可溶型蛋白等可溶型抗原结合的抗原结合分子。促进细胞内的抗原结合分子与fcrn的结合的方法,还可称作是增强抗原结合分子与细胞内(例如核内体内)的fcrn的结合活性的方法。本发明还涉及:通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法。本发明中,“使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外”是指,不必所有以与抗原结合的状态摄入细胞内的抗原结合分子都以未与抗原结合的状态被释放到细胞外,只要与使抗原结合分子在酸性ph下的抗原结合能力低于在中性ph下的抗原结合能力之前相比,被释放到细胞外的抗原结合分子的比例增加即可。优选被释放到细胞外的抗原结合分子维持抗原结合能力。使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法,在抗原结合分子与抗原结合摄入细胞内的情况下,也可说成是赋予抗原结合分子以未与抗原结合的状态容易被释放到细胞外的性质的方法。本发明还涉及:通过使抗原结合分子在酸性ph下的抗原结合能力弱于在中性ph下的抗原结合能力,来增加抗原结合分子的血浆中抗原消除能力的方法。本发明中,“血浆中抗原消除能力”是指,对体内给予抗原结合分子或机体分泌时,使存在于血浆中的抗原从血浆中消除的能力。因此,本发明中,“抗原结合分子的血浆中抗原消除能力增加”是指,对体内给予抗原结合分子时,只要与使抗原结合分子在酸性ph下的抗原结合能力低于在中性ph下的抗原结合能力之前相比,抗原从血浆中消除的速度加快即可。抗原结合分子的血浆中抗原消除能力是否增加,例如可以通过对体内给予可溶型抗原和抗原结合分子,测定给药后的可溶型抗原的血浆中浓度来判断。通过使抗原结合分子在酸性ph下的抗原结合能力低于在中性ph下的抗原结合能力,给予可溶型抗原和抗原结合分子后血浆中的可溶型抗原的浓度降低时,可以判断抗原结合分子的血浆中抗原消除能力增加。本发明还涉及:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸来提高抗原结合分子的药物动力学的方法。本发明还提供:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸来增加抗原结合分子与抗原的结合次数的方法。本发明还涉及:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸来增加抗原结合分子所能结合的抗原数的方法。本发明还提供:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸,使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法。本发明还提供:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸,使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法。本发明还提供:通过用组氨酸或非天然氨基酸取代抗原结合分子的至少1个氨基酸、或插入组氨酸或非天然氨基酸,使抗原结合分子的血浆中抗原消除能力增加的方法。对导入组氨酸或非天然氨基酸突变(取代、插入等)的位点没有特别限定,任何位点均可取代成组氨酸或非天然氨基酸,或者可以在任何位点插入组氨酸或非天然氨基酸。取代成组氨酸或非天然氨基酸或插入组氨酸或非天然氨基酸的位点的优选例子有:影响抗原结合分子的抗原结合能力的区。例如,当抗原结合分子为抗体时,可以列举抗体的可变区或cdr等。对导入组氨酸或非天然氨基酸突变的数没有特别限定,可以用组氨酸或非天然氨基酸只取代1个位点,或只在1个位点插入组氨酸或非天然氨基酸。或者,可以用组氨酸或非天然氨基酸取代2个位点以上的多个位点,或者在多个位点插入组氨酸或非天然氨基酸。除了用组氨酸或非天然氨基酸取代或插入以外,可以同时进行其他氨基酸的缺失、添加、插入和/或取代等。本发明中,作为取代成组氨酸或非天然氨基酸的位点的例子,当抗原结合分子为抗体时,考虑抗体的cdr序列或决定cdr的结构的序列作为修饰位点,可以列举例如以下位点。氨基酸位点以kabat编号(kabatea等人.1991.sequencesofproteinsofimmunologicalinterest.nih)表示。重链:h27、h31、h32、h33、h35、h50、h58、h59、h61、h62、h63、h64、h65、h99、h100b、h102轻链:l24、l27、l28、l32、l53、l54、l56、l90、l92、l94这些修饰位点中,认为h32、h61、l53、l90、l94是普遍性高的修饰位点。虽然没有特别限定,但在作为抗原为il-6受体(例如人il-6受体)时的优选的修饰位点,可以列举以下位点。重链:h27、h31、h32、h35、h50、h58、h61、h62、h63、h64、h65、h100b、h102轻链:l24、l27、l28、l32、l53、l56、l90、l92、l94组合多个位点并取代成组氨酸或非天然氨基酸时,优选的组合的具体例子有:例如h27、h31、h35的组合;h27、h31、h32、h35、h58、h62、h102的组合;l32、l53的组合;l28、l32、l53的组合等。并且,作为重链与轻链的取代位点的优选组合的例子,可以列举:h27、h31、l32、l53的组合。虽然没有特别限定,但在抗原为il-6(例如人il-6)的情况下,作为优选的修饰位点,可以列举以下位点。重链:h32、h59、h61、h99轻链:l53、l54、l90、l94虽然没有特别限定,但在抗原为il-31受体(例如人il-31受体)的情况下,优选的修饰位点有h33。这些位点中,可以只将1个位点用组氨酸或非天然氨基酸取代,也可以将多个位点用组氨酸或非天然氨基酸取代。本发明的方法可适用于不依赖靶抗原的种类的任意抗原结合分子。本发明中,抗原结合分子只要是与作为对象的抗原具有特异性的结合活性的物质即可,没有特别限定,抗原结合分子的优选的例子有:具有抗体的抗原结合区的物质。抗体的抗原结合区的例子有:cdr或可变区。当抗体的抗原结合区为cdr时,可以包含全长抗体所含的全部6个cdr,也可以包含1个或2个以上的cdr。当包含cdr作为抗体的结合区时,所含的cdr可以进行氨基酸的缺失、取代、添加和/或插入等,或者可以是cdr的一部分。当抗原结合分子中包含抗体恒定区时,本发明还涉及:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,来提高抗原结合分子的药物动力学的方法。当抗原结合分子中包含抗体恒定区时,本发明还提供:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,来增加抗原结合分子与抗原的结合次数的方法。当抗原结合分子中包含抗体恒定区时,本发明还涉及:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,来增加抗原结合分子所能结合的抗原数的方法。当抗原结合分子中包含抗体恒定区时,本发明还涉及:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,使在细胞外与抗原结合分子结合的抗原在细胞内从抗原结合分子上解离的方法。当抗原结合分子中包含抗体恒定区时,本发明还涉及:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外的方法。当抗原结合分子中包含抗体恒定区时,本发明还涉及:通过修饰(氨基酸的取代、缺失、添加和/或插入等)抗原结合分子中所含的抗体恒定区,使抗原结合分子的血浆中抗原消除能力增加的方法。作为本发明的抗原结合物质的优选方案,可以列举包含fcrn结合区的抗原结合物质。包含fcrn结合区的抗原结合物质可以通过fcrn的补救路径摄入细胞内,之后再次返回到血浆中。fcrn结合区优选为直接与fcrn结合的区。fcrn结合区的优选的例子有:抗体的fc区。但是,自蛋白或igg等的能够和与fcrn具有结合能力的多肽结合的区,可以经由白蛋白或igg等间接地与fcrn结合,因此,本发明中的fcrn结合区可以是和与这样的fcrn具有结合能力的多肽结合的区。对作为本发明的方法的对象的抗体等的抗原结合分子所识别的抗原没有特别限定,可以以识别任何抗原的抗体作为对象。利用本发明的方法提高药物动力学的抗体的例子有:例如识别受体蛋白(膜结合型受体、可溶型受体)或细胞表面标志物等膜抗原的抗体、识别细胞因子等可溶型抗原的抗体等。本发明中,膜抗原的优选的例子有:膜蛋白。本发明中,可溶型抗原的例子有:可溶型蛋白。利用本发明的方法使药物动力学提高的抗体所识别的抗原的具体例子有:例如il-1、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-10、il-11、il-12、il-15、il-31、il-23、il-2受体、il-6受体、osm受体、gp130、il-5受体、cd40、cd4、fas、骨桥蛋白、crth2、cd26、pdgf-d、cd20、单核细胞趋化活化因子、cd23、tnf-α、hmgb-1、α4整合素、icam-1、ccr2、cd11a、cd3、ifnγ、blys、hla-dr、tgf-β、cd52、il-31受体等。特别优选的抗原有il-6受体。作为本发明的方法的对象的抗原结合分子,可以列举:具有拮抗活性的抗原结合分子(拮抗抗原结合分子)、具有激动活性的抗原结合分子(激动抗原结合分子)等,作为优选方案,可以列举:拮抗抗原结合分子、特别是识别受体等的膜抗原或细胞因子等的可溶型抗原的拮抗抗原结合分子。例如,识别受体的拮抗抗原结合分子是与受体结合,阻碍受体与其配体的结合,抑制由受体介导的信号传递的抗原结合分子。本发明中,对作为对象的抗原结合分子没有特别限定,可以是任何的抗原结合分子。本发明中使用的抗原结合分子优选具有抗原结合活性(抗原结合区)和fcrn结合区。本发明中,特别优选为包含人fcrn结合区的抗原结合分子。具有抗原结合活性和fcrn结合区的抗原结合分子的例子有抗体。本发明的抗体的优选例子有igg抗体。使用igg抗体作为抗体时,对其种类没有限定,可以使用igg1、igg2、igg3、igg4等同种型(亚纲)的igg。对于这些同种型的igg的恒定区,可以象m73那样,向恒定区部分导入氨基酸突变。导入的氨基酸突变可以列举:例如增大或减少与fcγ受体的结合的氨基酸突变(procnatlacadsciusa.2006mar14;103(11):4005-10)、增大或减少与fcrn的结合的氨基酸突变(jbiolchem.2001mar2;276(9):6591-604)等,但并不限于这些。另外,通过选择igg2等的适当的恒定区,也可以改变ph依赖性结合。当作为本发明的对象的抗原结合分子为抗体时,抗体可以是小鼠抗体、人抗体、大鼠抗体、兔抗体、山羊抗体、骆驼抗体等来源于任一种动物的抗体。而且,可以是例如嵌合抗体、其中人源化抗体等取代了氨基酸序列的修饰抗体。还可以是双重特异性抗体、结合有各种分子的抗体修饰物、包含抗体片段的多肽等。“嵌合抗体”是指将来源于不同动物的序列组合而制作的抗体。嵌合抗体的具体例子有:例如包括小鼠抗体的重链、轻链的可变(v)区和人抗体的重链、轻链的恒定(c)区的抗体。“人源化抗体”也称作重构(reshaped)人抗体,是将来源于人以外的哺乳动物的抗体、例如小鼠抗体的互补性决定区(cdr,complementaritydeterminingregion)移植到人抗体的cdr中而得到的抗体。用于鉴定cdr的方法是公知的(kabat等人,sequenceofproteinsofimmunologicalinterest(1987),nationalinstituteofhealth,bethesda,md.;chothia等人,nature(1989)342:877)。另外,其通常的基因重组方法也是公知的(参照欧洲专利申请公开号ep125023号公报、wo96/02576号公报)。双重特异性抗体是指在同一抗体分子内具有识别不同表位的可变区的抗体。双重特异性抗体可以是识别2个以上不同抗原的抗体,也可以是识别同一抗原上的不同的2个以上表位的抗体。作为包含抗体片段的多肽,例如有fab片段、f(ab’)2片段、scfv(natbiotechnol.2005sep;23(9):1126-36)结构域抗体(dab)(wo2004/058821,wo2003/002609)、scfv-fc(wo2005037989)、dab-fc、fc融合蛋白等。这些分子中,特别是包含fc区的分子具有与fcrn的结合活性,所以适于采用本发明中发现的方法。并且,本发明可以使用的抗原结合分子可以是抗体样分子。抗体样分子是指通过与靶分子结合来发挥作用的分子(currentopinioninbiotechnology2006,17:653-658、currentopinioninbiotechnology2007,18:1-10、currentopinioninstructuralbiology1997,7:463-469、proteinscience2006,15:14-27),例如有darpins(wo2002/020565)、affibody(亲和体)(wo1995/001937)、avimer(wo2004/044011,wo2005/040229)、adnectin(wo2002/032925)等。即使是这些抗体样分子,只要其能够与靶分子进行ph依赖性结合,就可以以1分子与多个靶分子结合。抗原结合分子可以是进行靶向结合的受体蛋白和受体fc融合蛋白,例如有tnfr-fc融合蛋白、il1r-fc融合蛋白、vegfr-fc融合蛋白、ctla4-fc融合蛋白等(natmed.2003jan;9(1):47-52、biodrugs.2006;20(3):151-60)。即使是这些受体蛋白和受体fc融合蛋白,只要其能够与靶分子进行ph依赖性结合,就可以以1分子与多个靶分子结合。抗原结合分子还可以是与靶向结合具有中和效果的人工配体蛋白和人工配体融合蛋白,例如有突变il-6(emboj.1994dec15;13(24):5863-70)等。即使是这些人工配体蛋白和人工配体融合蛋白,只要其能够与靶分子进行ph依赖性结合,就可以以1分子与多个靶分子结合。并且,本发明的抗体可以是糖链被修饰的抗体。糖链被修饰的抗体的例子有:例如糖苷化修饰的抗体(wo99/54342等)、糖链中添加的岩藻糖缺失的抗体(wo00/61739、wo02/31140、wo2006/067847、wo2006/067913等)、具有含二等分(bisecting)glcnac的糖链的抗体(wo02/79255等)等。本发明的方法不受特定理论的限定,例如使酸性ph下的抗原结合能力弱于中性ph下的抗原结合能力、提高药物动力学、以及多次与抗原结合的关系可以如下说明。例如,当抗体是与膜抗原结合的抗体时,给予体内的抗体与抗原结合,之后抗体保持着与抗原结合的状态和抗原一起通过内化作用摄入细胞内的核内体中。之后,抗体保持着与抗原结合的状态向溶酶体移动,抗体与抗原一起被溶酶体分解。由内化作用介导的从血浆中的消除被称作抗原依赖性消除,在大多数抗体分子中有报道(drugdiscovtoday.2006jan;11(1-2):81-8)。当1分子的igg抗体以二价与抗原结合时,以1分子的抗体与2分子的抗原结合的状态被内化,直接被溶酶体分解。因此,当为普通抗体时,1分子的igg抗体无法与3分子以上的抗原结合。例如,当为具有中和活性的1分子的igg抗体时,无法中和3分子以上的抗原。igg分子的血浆中滞留性较长(消除慢),这是由于作为igg分子的补救受体而已知的fcrn在起作用。通过胞饮作用摄入核内体中的igg分子,其在核内体内的酸性条件下与在核内体内表达的fcrn结合。无法与fcrn结合的igg分子摄入溶酶体内,被溶酶体分解,但与fcrn结合的igg分子向细胞表面移动,在血浆中的中性条件下自fcrn上解离,又返回到血浆中。当抗体是与可溶型抗原结合的抗体时,给予体内的抗体与抗原结合,之后抗体保持着与抗原结合的状态摄入细胞内。摄入细胞内的抗体多半通过fcrn被释放到细胞外,但由于是保持着与抗原结合的状态被释放到细胞外,因此无法再次与抗原结合。因此,和与膜抗原结合的抗体一样,当为普通抗体时,1分子的igg抗体无法与3分子以上的抗原结合。本发明人等认为:通过内化作用与膜抗原等抗原结合的抗体摄入细胞内的核内体中时,与抗原结合着的抗体移动到溶酶体中被分解,相对于此,在核内体内抗原已解离的igg抗体可以与在核内体内表达的fcrn结合。即,本发明人等发现:在血浆中与抗原强力结合、在核内体内与抗原微弱结合的抗体,在血浆中与抗原结合,与抗原形成复合体,在此状态下通过内化作用摄入细胞内的核内体内,在核内体内与抗原解离后,与fcrn结合并向细胞表面移动,以未与抗原结合的状态再次返回到血浆中,可以中和多个膜型抗原。进一步发现:具有在血浆中与抗原强力结合、在核内体内与抗原微弱结合的性质的抗体,即使在与可溶型抗原等抗原结合的情况下,在核内体内也与抗原解离,因此以未与抗原结合的状态再次被释放到血浆中,可以中和多个可溶型抗原。特别是本发明人等着眼于血浆中的ph与核内体内的ph不同,发现在血浆中的ph条件下与抗原强力结合、在核内体内的ph条件下与抗原微弱结合的抗体,1个抗体分子可以与多个抗原结合,血浆中滞留性优异。核内体是膜小胞之一,在真核细胞的细胞质内形成网络,在从细胞膜到溶酶体的过程中掌管高分子的代谢。据报道,核内体内的ph通常为ph5.5-ph6.0的酸性(natrevmolcellbiol.2004feb;5(2):121-32),又已知血浆中的ph几乎为中性(通常为ph7.4)。因此,酸性ph下的抗原结合活性弱于中性ph下的抗原结合活性的抗原结合分子在中性ph的血浆中与抗原结合摄入细胞内,之后在酸性ph的核内体内与抗原解离。与抗原解离的抗原结合分子与fcrn结合并向细胞表面移动,以未与抗原结合的状态再次返回到血浆中,结果,可以与抗原多次结合,药物动力学提高。<抗原结合分子物质>本发明还提供在ph4.0-ph6.5下的抗原结合活性低于在ph6.7-ph10.0下的抗原结合活性的抗原结合分子、优选在ph5.0-ph6.0下的抗原结合活性低于在ph7.0-8.0下的抗原结合活性的抗原结合分子。在ph4.0-ph6.5下的抗原结合活性低于在ph6.7-10.0下的抗原结合活性的抗原结合分子的具体例子有:ph5.8下的抗原结合活性低于ph7.4下的抗原结合活性的抗原结合分子。ph5.8下的抗原结合活性低于ph7.4下的抗原结合活性的抗原结合分子还可称作ph7.4下的抗原结合活性高于ph5.8下的抗原结合活性的抗原结合分子。本发明的ph5.8下的抗原结合活性低于ph7.4下的抗原结合活性的抗原结合分子,只要其在ph5.8下的抗原结合活性低于在ph7.4下的结合即可,对其结合活性之差没有限定,即使差别甚微但只要ph5.8下的抗原结合活性低即可。作为本发明的ph5.8下的抗原结合活性低于ph7.4下的抗原结合活性的抗原结合分子的优选方案,可以列举ph7.4下的抗原结合活性为ph5.8下的抗原结合活性的两倍以上的抗原结合分子;作为进一步优选的方案,可以列举ph7.4下的抗原结合活性为ph5.8下的抗原结合活性的10倍以上的抗原结合分子;作为更优选的方案,可以列举ph7.4下的抗原结合活性为ph5.8下的抗原结合活性的40倍以上的抗原结合分子。具体而言,作为本发明的ph5.8下的抗原结合活性低于ph7.4下的抗原结合活性的抗原结合分子的优选方案,对于抗原的在ph5.8下的kd与在ph7.4下的kd之比、即kd(ph5.8)/kd(ph7.4)的值为2以上,进一步优选kd(ph5.8)/kd(ph7.4)的值为10以上,更进一步优选kd(ph5.8)/kd(ph7.4)的值为40以上。对kd(ph5.8)/kd(ph7.4)的值的上限没有特别限定,只要在本领域技术人员的技术中可以制作,可以是400、1000、10000等任何值。并且,作为本发明的在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性的抗原结合分子的其他优选方案,对于抗原在ph5.8下的kd与在ph7.4下的kd之比、即kd(ph5.8)/kd(ph7.4)的值为2以上,优选kd(ph5.8)/kd(ph7.4)的值为5以上,进一步优选kd(ph5.8)/kd(ph7.4)的值为10以上,更进一步优选kd(ph5.8)/kd(ph7.4)的值为30以上。对kd(ph5.8)/kd(ph7.4)的值的上限没有特别限定,只要在本领域技术人员的技术中可以制作,可以是50、100、200等任何值。测定抗原的结合活性时,除ph以外的条件可以由本领域技术人员适当选择,没有特别限定,例如,如实施例所述,可以在mes缓冲液中、37℃的条件下进行测定。另外,抗原结合分子的抗原结合活性的测定可以利用本领域技术人员所公知的方法进行,例如,如实施例所述,可以使用biacoret100(gehealthcare)等进行测定。认为这样的在酸性ph下与抗原微弱结合的抗原结合分子在核内体内的酸性条件下容易自抗原上解离,认为其在细胞内被内化后与fcrn结合,容易被释放到细胞外。在细胞内未被分解而被释放到细胞外的抗原结合分子可以再次与抗原结合。因此,例如当抗原结合分子为中和抗原结合分子时,在核内体内的酸性条件下容易自抗原上解离的抗原结合分子可以多次与抗原结合并中和抗原。结果,ph4.0-ph6.5下的抗原结合活性低于ph6.7-ph10.0下的抗原结合活性的抗原结合分子成为血浆中滞留性优异的抗原结合分子。作为在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性的抗原结合分子的优选方案,可以列举抗原结合分子中的至少1个氨基酸被取代成组氨酸或非天然氨基酸、或插入有至少1个组氨酸或非天然氨基酸的抗原结合分子。对导入组氨酸或非天然氨基酸突变的位点没有特别限定,只要与取代前相比ph5.8下的抗原结合活性弱于ph7.4下的抗原结合活性(kd(ph5.8)/kd(ph7.4)的值变大、或kd(ph5.8)/kd(ph7.4)的值变大),可以是任何位点。例如,当抗原结合分子为抗体时,可以是抗体的可变区或cdr等。被取代成组氨酸或非天然氨基酸的氨基酸的数或插入的氨基酸的数可以由本领域技术人员适当确定,可以用组氨酸或非天然氨基酸取代1个氨基酸,也可以插入1个氨基酸,还可以用组氨酸或非天然氨基酸取代2个以上的多个氨基酸,也可以插入2个以上的氨基酸。另外,除取代成组氨酸或非天然氨基酸、或者插入组氨酸或非天然氨基酸以外,还可以同时进行其他氨基酸的缺失、添加、插入和/或取代等。取代成组氨酸或非天然氨基酸、或者插入组氨酸或非天然氨基酸,这可以利用本领域技术人员公知的将丙氨酸分区的丙氨酸取代成组氨酸的组氨酸分区等方法随机进行,也可以从随机导入了组氨酸或非天然氨基酸突变的抗原结合分子中选择与突变前相比kd(ph5.8)/kd(ph7.4)或kd(ph5.8)/kd(ph7.4)的值变大的抗原结合分子。作为如此地突变成组氨酸或非天然氨基酸、且在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性的抗原结合分子的优选例子,可以列举如:突变成组氨酸或非天然氨基酸后在ph7.4下的抗原结合活性与突变成组氨酸或非天然氨基酸之前在ph7.4下的抗原结合活性同等的抗原结合分子。本发明中,组氨酸或非天然氨基酸突变后的抗原结合分子与组氨酸或非天然氨基酸突变前的抗原结合分子具有同等的抗原结合活性,是指以组氨酸或非天然氨基酸突变前的抗原结合分子的抗原结合活性为100%时,组氨酸或非天然氨基酸突变后的抗原结合分子的抗原结合活性至少为10%以上、优选50%以上、进一步优选80%以上、更优选为90%以上。组氨酸或非天然氨基酸突变后在ph7.4下的抗原结合活性可以高于组氨酸或非天然氨基酸突变前在ph7.4下的抗原结合活性。通过组氨酸或非天然氨基酸的取代或插入,抗原结合分子的抗原结合活性变低时,可以通过取代、缺失、添加和/或插入抗原结合分子中的1个或多个氨基酸等,使抗原结合活性与组氨酸取代或插入前的抗原结合活性同等。本发明还包含:在这样的组氨酸取代或插入后,通过进行1个或多个氨基酸的取代、缺失、添加和/或插入,使结合活性同等的抗原结合分子。并且,当抗原结合分子为包含抗体恒定区的物质时,在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性的抗原结合分子的其他优选方案有:修饰抗原结合分子中所含的抗体恒定区的方法。修饰后的抗体恒定区的具体例子有:例如实施例中记载的恒定区。利用上述方法等使抗原结合物质的ph5.8下的抗原结合活性弱于ph7.4下的抗原结合活性(kd(ph5.8)/kd(ph7.4)的值变大)时,虽然没有特别限定,但优选与原始抗体相比,kd(ph5.8)/kd(ph7.4)的值通常为2倍以上、优选5倍以上、进一步优选10倍以上。本发明的抗原结合分子,只要其在ph4.0-ph6.5下的抗原结合活性低于在ph6.7-10.0下的抗原结合活性,除此之外可以具有任何性质,例如可以是激动抗原结合分子或拮抗抗原结合分子等。本发明的优选的抗原结合分子的例子有:拮抗抗原结合分子。拮抗抗原结合分子通常是抑制配体(激动剂)与受体的结合、抑制受体介导的向细胞内传递信号的抗原结合分子。本发明还提供:以下的至少1个位点的氨基酸被取代成组氨酸或非天然氨基酸的抗体。氨基酸位点以kabat编号(kabatea等人.1991.sequencesofproteinsofimmunologicalinterest.nih)表示。重链:h27、h31、h32、h33、h35、h50、h58、h59、h61、h62、h63、h64、h65、h99、h100b、h102轻链:l24、l27、l28、l32、l53、l54、l56、l90、l92、l94这些修饰位点中,认为h32、h61、l53、l90、l94是普遍性高的修饰位点。虽然没有特别限定,但在抗原为il-6受体(例如人il-6受体)的情况下,优选的修饰位点有以下位点。重链:h27、h31、h32、h35、h50、h58、h61、h62、h63、h64、h65、h100b、h102轻链:l24、l27、l28、l32、l53、l56、l90、l92、l94将多个位点组合起来取代成组氨酸或非天然氨基酸时,优选的组合的具体例子有:例如h27、h31、h35的组合,h27、h31、h32、h35、h58、h62、h102的组合,l32、l53的组合,l28、l32、l53的组合等。并且,重链与轻链的取代位点的优选组合的例子有:h27、h31、l32、l53的组合。虽然没有特别限定,但在抗原为il-6(例如人il-6)的情况下,优选的修饰位点有以下位点。重链:h32、h59、h61、h99轻链:l53、l54、l90、l94虽然没有特别限定,但在抗原为il-31受体(例如人il-31受体)的情况下,优选的修饰位点有h33。本发明的抗原结合分子所识别的抗原可以是任何抗原。本发明的抗体所识别的抗原的具体例子有:上述受体蛋白(膜结合型受体、可溶型受体)、细胞表面标志物等膜抗原或细胞因子等可溶型抗原、例如il-1、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-10、il-11、il-12、il-15、il-31、il-23、il-2受体、il-6受体、osm受体、gp130、il-5受体、cd40、cd4、fas、骨桥蛋白、crth2、cd26、pdgf-d、cd20、单细胞趋化活化因子、cd23、tnf-α、hmgb-1、α4整合素、icam-1、ccr2、cd11a、cd3、ifnγ、blys、hla-dr、tgf-β、cd52、il-31受体等。特别优选的抗原例如有:il-6受体。本发明的抗原结合分子如上所述。本发明中,作为抗原结合分子的优选的方案,可以列举抗体。具有抗原结合活性和fcrn结合区的抗体的例子有:igg抗体。使用igg抗体作为抗体时,对其种类没有限定,可以使用igg1、igg2、igg3、igg4等。对本发明的抗体的来源没有特别限定,可以是任何来源的抗体,可以使用例如小鼠抗体、人抗体、大鼠抗体、兔抗体、山羊抗体、骆驼抗体等。而且,可以是例如上述嵌合抗体、其中人源化抗体等取代了氨基酸序列的修饰抗体。还可以是上述的双重特异性抗体、结合有各种分子的抗体修饰物、包含抗体片段的多肽、糖链修饰抗体等。嵌合抗体的制作是公知的,例如,当为人-小鼠嵌合抗体时,连接编码抗体v区的dna和编码人抗体c区的dna,将其插入表达载体中并导入到宿主中产生抗体,从而可以得到嵌合抗体。“人源化抗体”也称作重构(reshaped)人抗体,是将来源于人以外的哺乳动物的抗体例如小鼠抗体的互补性决定区(cdr)移植到人抗体的cdr中而得到的抗体。用于鉴定cdr的方法是公知的(kabat等人,sequenceofproteinsofimmunologicalinterest(1987),nationalinstituteofhealth,bethesda,md.;chothia等人,nature(1989)342:877)。另外,其通常的基因重组方法也是公知的(参照欧洲专利申请公开号ep125023号公报、wo96/02576号公报)。人源化抗体可以利用公知的方法如下产生:例如,确定小鼠抗体的cdr,将该cdr和人抗体的构架区(fr)连接得到抗体,获取编码所得抗体的dna,之后通过使用通常的表达载体的系统产生人源化抗体。上述dna可以使用多个寡核苷酸作为引物,通过pcr法来合成,上述寡核苷酸制作成在cdr和fr的末端区均具有重叠部分(参照wo98/13388号公报中记载的方法)。选择经由cdr连接的人抗体的fr,使cdr形成良好的抗原结合位点。根据需要,可以修饰抗体可变区中fr的氨基酸,使重构人抗体的cdr形成合适的抗原结合位点(sato等人,cancerres.(1993)53:10.01-6)。在可修饰的fr中的氨基酸残基中,包括通过非共价键直接与抗原结合的部分(amit等人,science(1986)233:747-53)、影响或作用于cdr结构的部分(chothia等人,j.mol.biol.(1987)196:901-17)以及与vh-vl相互作用有关的部分(ep239400号专利公报)。当本发明中的抗体为嵌合抗体或人源化抗体时,上述抗体的c区优选使用来源于人抗体的c区。例如,在h链中可以使用cγ1、cγ2、cγ3、cγ4等;在l链中可以使用cκ、cλ等。为了增大或降低与fcγ受体或fcrn的结合、为了改善抗体的稳定性或抗体的产生,根据需要可以向人抗体c区中导入氨基酸突变。本发明中的嵌合抗体优选包含来源于人以外的哺乳动物的抗体的可变区和来源于人抗体的恒定区。而人源化抗体优选包含来源于人以外的哺乳动物的抗体的cdr和来源于人抗体的fr和c区。来源于人抗体的恒定区优选包含fcrn结合区,这样的抗体的例子有:igg(igg1、igg2、igg3、igg4)。本发明中的人源化抗体所使用的恒定区可以是属于任意同种型的抗体的恒定区。优选使用人igg的恒定区,但并不受限于此。对用于人源化抗体的来源于人抗体的fr也没有特别限定,可以是属于任意同种型的抗体的fr。本发明中的嵌合抗体和人源化抗体的可变区和恒定区,只要显示出原始抗体的结合特异性即可,可以通过缺失、取代、插入和/或添加等进行修饰。使用来源于人的序列得到的嵌合抗体和人源化抗体,由于其在人体内的免疫原性降低,所以当为了治疗目的等对人进行给药时有用。本发明的抗体可以利用任何方法而得到,例如,对于本来在ph5.8下的抗原结合活性高于在ph7.4下的抗原结合活性的抗体或抗原结合活性为相同程度的抗体,通过上述的组氨酸取代等,可以人为地使在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性,还可以从由以下所示的抗体文库或杂交瘤得到的多个抗体中筛选在ph5.8下的抗原结合活性低于在ph7.4下的抗原结合活性的抗体,从而选择本发明的抗体。将抗体中的氨基酸取代成组氨酸时,导入组氨酸突变之前的抗体的h链或l链的氨基酸序列还可以使用已知的序列,还可以使用按照本领域技术人员所公知的方法新获得的抗体的氨基酸序列。例如,抗体既可以由抗体文库获得,也可以由产生单克隆抗体的杂交瘤克隆编码抗体的基因而获得。关于抗体文库,已经有多个抗体文库为人所公知,另外抗体文库的制作方法也是公知的,所以本领域技术人员可以获取适当的抗体文库。例如,关于抗体噬菌体文库,可以参照clackson等人,nature1991,352:624-8;marks等人,j.mol.biol.1991,222:581-97;waterhouses等人,nucleicacidsres.1993,21:2265-6;griffiths等人,emboj.1994,13:324.0-60;vaughan等人,naturebiotechnology1996,14:309-14和日本特表平20-504970号公报等文献。此外,可以使用以真核细胞作为文库的方法(wo95/15393号小册子)或核糖体提示法等公知方法。而且,还已知使用人抗体文库,通过淘选获得人抗体的技术。例如,可以以人抗体的可变区作为单链抗体(scfv),通过噬菌体展示法使之在噬菌体表面表达,来选择与抗原结合的噬菌体。分析所选择的噬菌体的基因,可以确定编码与抗原结合的人抗体可变区的dna序列。一旦清楚了与抗原结合的scfv的dna序列,就可以根据该序列制作适当的表达载体,获得人抗体。上述方法已经众所周知,可以参考wo92/01047、wo92/20791、wo93/06213、wo93/11236、wo93/19172、wo95/01438、wo95/15388。由杂交瘤获得编码抗体的基因的方法,基本上使用公知技术,使用所需抗原或表达所需抗原的细胞作为致敏抗原,按照通常的免疫方法对其进行免疫,利用通常的细胞融合法使所得的免疫细胞与公知的亲本细胞融合,再利用通常的筛选法筛选产生单克隆抗体的细胞(杂交瘤),并使用逆转录酶由所得杂交瘤的mrna合成抗体可变区(v区)的cdna,将该cdna和编码所需抗体恒定区(c区)的dna连接而得到。更具体而言,并不特别限于以下例子,用于得到上述的编码h链和l链的抗体基因的致敏抗原包括:具免疫原性的完全抗原和不具免疫原性的不完全抗原两者,上述不完全抗原包括半抗原等。例如,可以使用目标蛋白的全长蛋白或部分肽等。此外,已知由多糖类、核酸、脂质等构成的物质能够成为抗原,对本发明抗体的抗原并没有特别限定。抗原的制备可以按照本领域技术人员所公知的方法进行,例如可以按照使用杆状病毒的方法(例如wo98/46777等)等进行制备。杂交瘤的制作,例如可以按照milstein等人的方法(g.kohler和c.milstein,methodsenzymol.1981,73:3-46)等来进行。当抗原的免疫原性低时,可以使之与白蛋白等具免疫原性的大分子结合来进行免疫。根据需要,还可以使抗原与其他分子结合,从而成为可溶性抗原。当使用膜抗原(例如受体等)这样的跨膜分子作为抗原时,可以使用膜抗原的胞外区部分作为片段,或者使用在细胞表面上表达跨膜分子的细胞作为免疫原。产生抗体的细胞可以通过使用上述适当的致敏抗原,对动物进行免疫而得到。或者,在体外对能够产生抗体的淋巴细胞进行免疫而成为产生抗体的细胞。进行免疫的动物可以使用各种哺乳动物,但通常使用啮齿类、兔形目、灵长类动物。可以例示:小鼠、大鼠、仓鼠等啮齿类;兔等兔形目;食蟹猴、恒河猴、狒狒、黑猩猩等的猴等的灵长类动物。此外,还已知具有人抗体基因的所有组成部分(repertories)的转基因动物,通过使用这样的动物,也可以得到人抗体(参照wo96/34096;mendez等人,nat.genet.1997,15:146-56)。不使用上述转基因动物,例如,通过在体外用所需抗原或表达所需抗原的细胞致敏人淋巴细胞,并使致敏淋巴细胞与人骨髓瘤细胞例如u266融合,也可以得到具抗原结合活性的所需人抗体(参照日本特公平1-59878号公报)。还可以通过用所需抗原对具有人抗体基因的全部的所有组成成分的转基因动物进行免疫得到所需的人抗体(参照wo93/12227、wo92/03918、wo94/02602、wo96/34096、wo96/33735)。动物的免疫可如下进行:将致敏抗原用磷酸盐缓冲液(pbs)或生理盐水等适当稀释并悬浮,根据需要混合佐剂并进行乳化,然后注射到动物的腹腔内或皮下来进行免疫。之后,优选每4-21天给予数次和弗式不完全佐剂混合的致敏抗原。确认抗体的产生可以通过常规方法测定动物血清中目标抗体的效价来进行。杂交瘤可如下制备:使用常用融合剂(例如聚乙二醇),使自利用所需抗原免疫的动物或淋巴细胞得到的产生抗体的细胞与骨髓瘤细胞融合来制备(golding,monoclonalantibodies(单克隆抗体):principlesandpractice,academicpress,1986,59-103)。根据需要培养、增殖杂交瘤细胞,通过免疫沉淀、放射免疫分析(ria)、酶联免疫吸附分析(elisa)等公知的分析方法,测定由该杂交瘤产生的抗体的结合特异性。之后,根据需要可以利用有限稀释法等方法亚克隆产生抗体的杂交瘤,上述抗体的目标特异性、亲和性或活性已测定。接下来,可以使用能够与抗体特异性结合的探针(例如,与编码抗体恒定区的序列互补的寡核苷酸等),由杂交瘤或产生抗体的细胞(致敏淋巴细胞等)克隆编码所选择的抗体的基因。还可以通过rt-pcr由mrna进行克隆。免疫球蛋白分为iga、igd、ige、igg和igm五个不同类别。这些类别又进一步分成几个亚类(同种型)(例如igg-1、igg-2、igg-3和igg-4;iga-1和iga-2等)。本发明中,对抗体制备中使用的h链和l链没有特别限定,可以来源于属于上述任一类别和亚类的抗体,特别优选为igg。这里,还可以通过基因工程技术修饰编码h链和l链的基因。例如,对于小鼠抗体、大鼠抗体、兔抗体、仓鼠抗体、山羊抗体、骆驼抗体等抗体,为了减弱其对人的异种抗原性等,可以适当制作人工修饰的基因重组型抗体,例如嵌合抗体、人源化抗体等。嵌合抗体是包含除人以外的哺乳动物例如小鼠抗体的h链、l链的可变区和人抗体的h链、l链的恒定区的抗体,可以通过将编码小鼠抗体可变区的dna和编码人抗体恒定区的dna连接,将其插入到表达载体中,并导入到宿主中产生抗体,从而得到嵌合抗体。人源化抗体也称作重构(reshaped)人抗体,可通过pcr法由多个寡核苷酸合成,上述寡核苷酸制作成在dna序列的末端具有重叠部分,上述dna序列设计成连接除人以外的哺乳动物例如小鼠抗体的互补性决定区(cdr)。将所得dna和编码人抗体恒定区的dna连接,然后插入到表达载体中,并将其导入到宿主中而产生人源化抗体(参照ep239400、wo96/02576)。经由cdr连接的人抗体的fr,选择互补性决定区形成良好的抗原结合位点的fr。根据需要,可以取代抗体可变区的构架区的氨基酸,使重构人抗体的互补性决定区形成适当的抗原结合位点(k.sato等人,cancerres.1993,53:10.01-10.06)。除上述的人源化以外,认为还为了改善例如与抗原的结合性等抗体的生物学特性而进行修饰。本发明中的修饰可以通过位点特异性突变(参照例如kunkel(1910.0)proc.natl.acad.sci.usa82:488)、pcr突变、盒式突变等方法来进行。通常,生物学特性得到改善的抗体突变体,其相对于原始抗体可变区的氨基酸序列,具有70%以上、更优选80%以上、进一步优选90%以上(例如95%以上、97%、98%、99%等)的氨基酸序列同源性和/或相似性。本说明书中,序列同源性和/或相似性定义为:根据需要进行序列排比和间隙导入,使序列同源性最大化之后,与原始抗体残基相同(相同残基)或相似(根据一般的氨基酸侧链的特性分类为同一组的氨基酸残基)的氨基酸残基的比例。通常,天然氨基酸残基根据其侧链的性质分为以下几组:(1)疏水性:丙氨酸、异亮氨酸、缬氨酸、蛋氨酸和亮氨酸;(2)中性亲水性:天冬酰胺、谷氨酰胺、半胱氨酸、苏氨酸和丝氨酸;(3)酸性:天冬氨酸和谷氨酸;(4)碱性:精氨酸、组氨酸和赖氨酸;(5)影响链取向的残基:甘氨酸和脯氨酸;以及(6)芳族性:酪氨酸、色氨酸和苯丙氨酸。通常,存在于h链和l链的可变区中的共6个互补性决定区(超可变区;cdr)相互作用,形成抗体的抗原结合位点。已知即使是其中一个可变区,尽管与包括所有结合位点时相比亲和性低,但也具有识别并结合抗原的能力。因此,本发明的编码h链和l链的抗体基因,可以编码包括h链和l链的各抗原结合位点的片段部分,只要由该基因所编码的多肽维持与所需抗原的结合性即可。如上所述,重链可变区通常由3个cdr区和4个fr区构成。在本发明的优选方案中,作为供于“修饰”的氨基酸残基,例如可以从位于cdr区或fr区的氨基酸残基中适当选择。通常,修饰cdr区的氨基酸残基,有时会降低与抗原的结合能力。因此,本发明中对供于“修饰”的氨基酸残基没有特别限定,优选从位于fr区的氨基酸残基中适当选择。即使是cdr,当确认通过修饰其结合能力并没有降低时,可以选择该位点。另外,本领域技术人员可以利用公共数据库等适当获得在人或小鼠等生物中可用作抗体可变区的fr的序列。本发明还提供编码本发明的抗体的基因。编码本发明的抗体的基因可以是任何基因,还可以是dna、rna、其他核酸类似体等。本发明还提供携带上述基因的宿主细胞。对该宿主细胞没有特别限定,例如有大肠杆菌或各种动物细胞等。宿主细胞可以用作例如用于制备或表达本发明的抗体的产生系统。用于制备多肽的产生系统包括:体外(invitro)和体内(invivo)产生系统。体外产生系统例如有:使用真核细胞的产生系统和使用原核细胞的产生系统。可用作宿主细胞的真核细胞例如有:动物细胞、植物细胞、真菌细胞。动物细胞包括:哺乳动物细胞、例如cho(j.exp.med.(1995)108:94.0)、cos、hek293、3t3、骨髓瘤细胞、bhk(幼仓鼠肾)、hela、vero等;两栖动物细胞、例如非洲爪蟾卵母细胞(xenopuslaevisoocytes)(valle等人,nature(1981)291:338-340);以及昆虫细胞、例如sf9、sf21、tn5。在本发明的抗体的表达中适合使用cho-dg44、cho-dx11b、cos7细胞、hek293细胞、bhk细胞。动物细胞中,以大量表达为目的时,特别优选cho细胞。关于向宿主细胞内导入载体,例如可以通过以下方法来进行:磷酸钙法、deae-葡聚糖法、使用正离子脂质体dotap(boehringermannheim制)的方法、电穿孔法、脂质转染法(lipofection法)等。作为植物细胞,例如有来源于烟草的细胞和浮萍(lemnaminor),它们作为蛋白生产系统而已知,利用愈伤组织培养该细胞的方法可以产生本发明的抗体。使用真菌细胞的蛋白表达系统是公知的,这些真菌细胞可以用作产生本发明的抗体的宿主,上述真菌细胞例如有:酵母,例如酵母菌(saccharomyces)属的细胞(啤酒酵母(saccharomycescerevisiae)、粟酒裂殖酵母(saccharmycespombe)等);以及丝状菌,例如曲霉属(aspergillus)的细胞(黑曲霉(aspergillusniger)等)。使用原核细胞时,有使用细菌细胞的产生系统。作为细菌细胞,除上述大肠杆菌(e.coli)外,已知还有使用枯草杆菌的产生系统,这些细菌细胞可用于产生本发明的抗体。<筛选方法>本发明提供筛选抗原结合分子在酸性ph下的抗原结合活性低于在中性ph下的抗原结合活性的抗原结合分子的方法。本发明还提供可以以1分子与多个抗原结合的抗原结合分子的筛选方法。本发明还提供血浆中滞留性优异的抗原结合分子的筛选方法。本发明还提供在细胞内解离在细胞外与抗原结合分子结合的抗原的抗原结合分子的筛选方法。本发明还提供以与抗原结合的状态摄入细胞内、以未与抗原结合的状态被释放到细胞外的抗原结合分子的筛选方法。本发明还提供血浆中抗原消除能力增加的抗原结合分子的筛选方法。本发明还提供用作药物组合物时特别有用的抗原结合分子的筛选方法。具体而言,本发明提供抗原结合分子的筛选方法,该筛选方法包括以下步骤。(a)获得ph6.7-ph10.0下的抗原结合分子的抗原结合活性的步骤、(b)获得ph4.0-ph6.5下的抗原结合分子的抗原结合活性的步骤、(c)选择ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤。在本发明的筛选方法中,只要ph6.7-ph10.0下的抗原结合分子的抗原结合活性是ph6.7-ph10.0之间的抗原结合活性即可,没有特别限定,作为优选的抗原结合活性,例如有ph7.0-ph8.0之间的抗原结合活性;作为更优选的抗原结合活性,例如有ph7.4下的抗原结合活性。另外,ph4.0-ph6.5下的抗原结合分子的抗原结合活性只要是ph4.0-ph6.5之间的抗原结合活性即可,没有特别限定,作为优选的抗原结合活性,例如有ph5.5-ph6.5之间的抗原结合活性;作为更优选的抗原结合活性,例如有ph5.8或ph5.5下的抗原结合活性。抗原结合分子的抗原结合活性可以利用本领域技术人员所公知的方法进行测定,有关除ph以外的条件,本领域技术人员可以适当确定。抗原结合分子的抗原结合活性可以以kd(dissociationconstant:解离常数)、表观kd(apparentdissociationconstant:表观解离常数)、解离速度kd(dissociationrate:解离速度)或表观kd(apparentdissociation:表观解离速度)等形式进行评价。这些参数可以按照本领域技术人员公知的方法进行测定,例如可以使用biacore(gehealthcare)、斯卡恰特作图法、facs等。本发明中,选择ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤,与选择ph4.0-ph6.5下的抗原结合活性低于ph6.7-ph10.0下的抗原结合活性的抗原结合分子的步骤意思相同。只要ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性,对ph6.7-ph10.0下的抗原结合活性与ph4.0-ph6.5下的抗原结合活性之差没有特别限定,优选ph6.7-ph10.0下的抗原结合活性为ph4.0-ph6.5下的抗原结合活性的2倍以上,进一步优选为10倍以上,更优选为40倍以上。本发明还提供抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)在ph6.7-ph10.0的条件下使抗原结合分子与抗原结合的步骤、(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤、(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤。本发明还提供抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)选择在ph4.0-ph6.5的条件下未与抗原结合的抗原结合分子的步骤、(b)在ph6.7-ph10.0的条件下使(a)中选择的抗原结合分子与抗原结合的步骤、(c)获得在ph6.7-ph10.0的条件下与抗原结合的抗原结合分子的步骤。本发明还提供抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)在ph6.7-ph10.0的条件下使抗原结合分子与抗原结合的步骤、(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤、(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤、(d)扩增编码解离的抗原结合分子的基因的步骤、(e)获得被洗脱的抗原结合分子的步骤。步骤(a)-(d)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(d)的步骤的方法。对步骤(a)-(d)的重复次数没有特别限定,通常为10次以内。本发明还提供抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)选择在ph4.0-ph6.5的条件下未与抗原结合的抗原结合分子的步骤、(b)在ph6.7-ph10.0的条件下使(a)中选择的抗原结合分子与抗原结合的步骤、(c)获得在ph6.7-ph10.0的条件下与抗原结合的抗原结合分子的步骤、(d)扩增编码解离的抗原结合分子的基因的步骤、(e)获得被洗脱的抗原结合分子的步骤。步骤(a)-(d)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(d)的步骤的方法。对步骤(a)-(d)的重复次数没有特别限定,通常为10次以内。在本发明的筛选方法中,当使用噬菌体文库等时,扩增编码抗原结合分子的基因的步骤还可以作为扩增噬菌体的步骤。在本发明的方法中,抗原与抗原结合分子的结合可以以任何状态进行,没有特别限定。例如,通过使抗原与固定化的抗原结合分子接触,可以使抗原结合分子与抗原结合;通过使抗原结合分子与固定化的抗原接触,可以使抗原结合分子与抗原结合。另外,通过使抗原结合分子与抗原在溶液中接触,可以使抗原结合分子与抗原结合。本发明还提供抗原结合分子在第一ph下的结合活性高于在第二ph下的结合活性的抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)在第一ph条件下使抗原结合分子与固定有抗原的柱结合的步骤、(b)在第二ph条件下从柱上洗脱在第一ph条件下与柱结合的抗原结合分子的步骤、(c)获得被洗脱的抗原结合分子的步骤。本发明还提供抗原结合分子在第一ph下的结合活性低于在第二ph下的结合活性的抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)在第一ph条件下使抗原结合分子通过固定有抗原的柱的步骤、(b)回收在步骤(a)中未与柱结合而洗脱的抗原结合分子的步骤、(c)在第二ph条件下使(b)中回收的抗原结合分子与柱结合的步骤、(d)获得在步骤(c)中与柱结合的抗原结合分子的步骤。本发明还提供第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的筛选方法,该筛选方法包括以下步骤。(a)在第一ph条件下使抗原结合分子文库与固定有抗原的柱结合的步骤、(b)在第二ph条件下从柱上洗脱抗原结合分子的步骤、(c)扩增编码被洗脱的抗原结合分子的基因的步骤、(d)获得被洗脱的抗原结合分子的步骤。步骤(a)-(c)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(c)的步骤的方法。对步骤(a)-(c)的重复次数没有特别限定,通常为10次以内。本发明中,只要第一ph和第二ph各自不是相同的ph即可,可以是任何ph。优选的第一ph与第二ph的组合的例子有:第一ph为ph6.7-10.0之间的ph、第二ph为ph4.0-ph6.5之间的ph的组合;更优选的组合的例子有:第一ph为ph7.0-ph8.0之间的ph、第二ph为ph5.5-ph6.5之间的ph的组合;进一步优选的组合的例子有:第一ph为ph7.4、第二ph为ph5.8或ph5.5的组合。其他优选的第一ph与第二ph的组合的例子有:第一ph为ph4.0-6.5之间的ph、第二ph为ph6.7-ph10.0之间的ph的组合;更优选的组合的例子有:第一ph为ph5.5-ph6.5之间的ph、第二ph为ph7.0-ph8.0之间的ph的组合;进一步优选的组合的例子有:第一ph为ph5.8或ph5.5、第二ph为ph7.4的组合。利用本发明的方法筛选的抗原结合分子可以是任何抗原结合分子,例如可以将上述的抗原结合分子用于本发明的筛选。例如,可以筛选具有天然序列的抗原结合分子,也可以筛选氨基酸序列被取代的抗原结合分子。本发明中筛选的抗原结合分子的优选例子有:例如抗原结合分子的至少1个氨基酸被组氨酸取代或插入至少1个组氨酸的抗原结合分子。对导入组氨酸取代或插入的位点没有特别限定,可以在任何位点导入。可以向1个位点导入组氨酸取代或插入,也可以向2个位点以上的多个位点中导入。本发明中筛选的抗原结合分子的优选例子有:例如包含被修饰的抗体恒定区的抗原结合分子。利用本发明的方法筛选的抗原结合分子,例如可以是通过组氨酸分区等方法向不同位点导入组氨酸取代或插入的多个不同的抗原结合分子。因此,本发明的筛选方法可以进一步包括:将抗原结合分子的至少1个氨基酸取代成组氨酸或插入至少1个组氨酸的步骤。本发明的筛选方法可以使用非天然氨基酸来代替组氨酸。因此,也可以将上述组氨酸与非天然氨基酸互换来理解本发明。本发明的筛选方法可以进一步包括修饰抗体恒定区的氨基酸的步骤。通过本发明的筛选方法筛选的抗原结合物质可以以任何方式进行制备,例如可以使用:事先存在的抗体、事先存在的文库(噬菌体文库等)、对动物进行免疫而得到的杂交瘤或由来自免疫动物的b细胞制作的抗体或文库、向这些抗体或文库中导入组氨酸或非天然氨基酸突变而得到的抗体或文库(提高组氨酸或非天然氨基酸的含有率的文库或在特定位点导入了组氨酸或非天然氨基酸突变的文库等)等。利用本发明的筛选方法,可以得到多次与抗原结合、血浆中滞留性优异的抗原结合分子。因此,本发明的筛选方法可以用作用于得到血浆中滞留性优异的抗原结合分子的筛选方法。利用本发明的筛选方法,对人、小鼠、猴等动物给药时,还可以得到能够与抗原进行两次以上结合的抗原结合分子。因此,本发明的筛选方法可以用作用于得到能够两次以上与抗原结合的抗原结合分子的筛选方法。利用本发明的筛选方法,对人、小鼠、猴等动物给药时,还可以得到能够与多于抗原结合分子的抗原结合位点数的数目的抗原结合的抗原结合分子。因此,本发明的筛选方法可以用作用于得到能够与多于抗原结合分子的抗原结合位点数的数目的抗原结合的抗原结合分子的筛选方法。例如,当抗体为中和抗体时,可以用作用于得到能够中和多于抗原结合分子的抗原结合位点数的数目的抗原的抗原结合分子的筛选方法。利用本发明的筛选方法,对人、小鼠、猴等动物给药时,还可以得到能够在细胞内解离在细胞外结合的抗原的抗原结合分子。因此,本发明的筛选方法可以用作用于得到在细胞内解离在细胞外结合的抗原的抗原结合分子的筛选方法。利用本发明的筛选方法,对人、小鼠、猴等动物给药时,还可以得到以与抗原结合的状态摄入细胞内、以未与抗原结合的状态被释放到细胞外的抗原结合分子。因此,本发明的筛选方法可以用作用于得到以与抗原结合的状态摄入细胞内、以未与抗原结合的状态被释放到细胞外的抗原结合分子的筛选方法。利用本发明的筛选方法,对人、小鼠、猴等动物给药时,还可以得到可以使抗原从血浆中快速消除的抗原结合分子。因此,本发明的筛选方法可以用作用于得到血浆中抗原消除能力增加(高)的抗原结合分子的筛选方法。这些抗原结合分子可以减少对患者的给药量或给药频率,结果,可以减少总给药量,因此认为其作为药品特别优异。因此,本发明的筛选方法可以用作作为药物组合物的抗原结合分子的筛选方法。本发明还提供与原始文库相比组氨酸的含有比例增加的文库。文库中所含的抗原结合分子所具有的组氨酸的比例增加的文库可用于上述的筛选方法或后述的制备方法。提高了组氨酸的含有比例的文库的制作方法可以利用本领域技术人员所公知的方法来制作,例如有以下方法。合成用于制作文库的核酸时,利用三核苷酸法(jmolbiol.2008feb29;376(4):1182-200),以相等的概率含有编码20种氨基酸的20种的3碱基密码子(三核苷酸),从而可以在文库化的位点以相等的概率含有20种氨基酸。此时,20种氨基酸中,通过使编码组氨酸的三核苷酸的比例高于其他氨基酸的比例,可以提高在文库化的位点出现组氨酸的可能性。<抗原结合分子制备方法>本发明提供抗原结合分子在核内体内的ph下的抗原结合活性低于在血浆中的ph下的抗原结合活性的抗原结合分子的制备方法。本发明还提供血浆中滞留性优异的抗原结合分子的制备方法。本发明还提供用作药物组合物时特别有用的抗原结合分子的制备方法。具体而言,本发明提供抗原结合分子的制备方法,该制备方法包括以下步骤:(a)获得在ph6.7-ph10.0下的抗原结合分子的抗原结合活性的步骤、(b)获得在ph4.0-ph6.5下的抗原结合分子的抗原结合活性的步骤、(c)选择在ph6.7-ph10.0下的抗原结合活性高于在ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤、(d)获得编码(c)中选择的抗原结合分子的基因的步骤、(e)使用(d)中得到的基因制备抗原结合分子的步骤。本发明还提供抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在ph6.7-ph10.0的条件下使抗原结合分子与抗原结合的步骤、(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤、(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤、(d)获得编码(c)中取得的抗原结合分子的基因的步骤、(e)使用(d)中得到的基因制备抗原结合分子的步骤。本发明还提供抗原结合分子的制备方法,该制备方法包括以下步骤:(a)选择在ph4.0-ph6.5的条件下不与抗原结合的抗原结合分子的步骤、(b)在ph6.7-ph10.0的条件下使(a)中选择的抗原结合分子与抗原结合的步骤、(c)获得在ph6.7-ph10.0的条件下与抗原结合的抗原结合分子的步骤、(d)获得编码(c)中取得的抗原结合分子的基因的步骤、(e)使用(d)中得到的基因制备抗原结合分子的步骤。本发明还提供抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在ph6.7-10.0的条件下使抗原结合分子与抗原结合的步骤、(b)将(a)的与抗原结合的抗原结合分子置于ph4.0-ph6.5的条件下的步骤、(c)获得在ph4.0-ph6.5的条件下解离的抗原结合分子的步骤、(d)扩增编码解离的抗原结合分子的基因的步骤、(e)获得被洗脱的抗原结合分子的步骤、(f)获得编码(e)中取得的抗原结合分子的基因的步骤、(g)使用(f)中得到的基因制备抗原结合分子的步骤。步骤(a)-(d)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(d)的步骤的方法。对步骤(a)-(d)的重复次数没有特别限定,通常为10次以内。本发明还提供抗原结合分子的筛选方法,该筛选方法包括以下步骤:(a)选择在ph4.0-ph6.5的条件下不与抗原结合的抗原结合分子的步骤、(b)在ph6.7-ph10.0的条件下使(a)中选择的抗原结合分子与抗原结合的步骤、(c)获得在ph6.7-ph10.0的条件下与抗原结合的抗原结合分子的步骤、(d)扩增编码解离的抗原结合分子的基因的步骤、(e)获得被洗脱的抗原结合分子的步骤、(f)获得编码(e)中取得的抗原结合分子的基因的步骤、(g)使用(f)中得到的基因制备抗原结合分子的步骤。步骤(a)-(d)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(d)的步骤的方法。对步骤(a)-(d)的重复次数没有特别限定,通常为10次以内。本发明还提供第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在第一ph条件下使抗原结合分子与固定有抗原的柱结合的步骤、(b)在第二ph条件下从柱上洗脱在第一ph条件下与柱结合的抗原结合分子的步骤、(c)获得被洗脱的抗原结合分子的步骤、(d)获得编码(c)中取得的抗原结合分子的基因的步骤、(e)使用(d)中得到的基因制备抗原结合分子的步骤。本发明还提供第一ph下的结合活性高于第二ph下的结合活性的抗原结合分子的制备方法,该制备方法包括以下步骤:(a)在第一ph条件下使抗原结合分子文库与固定有抗原的柱结合的步骤、(b)在第二ph条件下从柱上洗脱抗原结合分子的步骤、(c)扩增编码被洗脱的抗原结合分子的基因的步骤、(d)获得被洗脱的抗原结合分子的步骤、(e)获得编码(d)中取得的抗原结合分子的基因的步骤、(f)使用(e)中得到的基因制备抗原结合分子的步骤。步骤(a)-(c)可以重复两次以上。因此,本发明提供在上述方法中进一步包括重复两次以上步骤(a)-(c)的步骤的方法。对步骤(a)-(c)的重复次数没有特别限定,通常为10次以内。在本发明的制备方法中,使用噬菌体文库等时,扩增编码抗原结合分子的基因的步骤还可以作为扩增噬菌体的步骤。本发明的制备方法中使用的抗原结合物质可以按照任何方式来制备,例如可以使用:事先存在的抗体、事先存在的文库(噬菌体文库等)、由对动物进行免疫得到的杂交瘤或由来自免疫动物的b细胞制作的抗体或文库、向这些抗体或文库中导入组氨酸或非天然氨基酸突变而得到的抗体或文库(提高了组氨酸或非天然氨基酸的含量的文库或向特定位点导入了组氨酸或非天然氨基酸突变的文库等)等。上述制备方法中,ph6.7-ph10.0下的抗原结合分子的抗原结合活性只要是ph6.7-ph10.0之间的抗原结合活性即可,没有特别限定,作为优选的抗原结合活性,例如有ph7.0-ph8.0之间的抗原结合活性;作为进一步优选的抗原结合活性,例如有ph7.4下的抗原结合活性。另外,ph4.0-ph6.5下的抗原结合分子的抗原结合活性只要是ph4.0-ph6.5之间的抗原结合活性即可,没有特别限定,作为优选的抗原结合活性,例如有ph5.5-ph6.5之间的抗原结合活性;作为进一步优选的抗原结合活性,例如有ph5.8或ph5.5下的抗原结合活性。抗原结合分子的抗原结合活性可以利用本领域技术人员所公知的方法进行测定,关于除ph以外的条件,本领域技术人员可以适当确定。选择ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性的抗原结合分子的步骤,与选择ph4.0-ph6.5下的抗原结合活性低于ph6.7-ph10.0下的抗原结合活性的抗原结合分子的步骤意思相同。只要ph6.7-ph10.0下的抗原结合活性高于ph4.0-ph6.5下的抗原结合活性即可,对ph6.7-ph10.0下的抗原结合活性与ph4.0-ph6.5下的抗原结合活性之差没有特别限定,但优选ph6.7-ph10.0下的抗原结合活性为ph4.0-ph6.5下的抗原结合活性的2倍以上,进一步优选为10倍以上,更优选为40倍以上。在上述制备方法中,抗原与抗原结合分子的结合可以以任何状态进行,没有特别限定。例如,通过使抗原与固定化的抗原结合分子接触,可以使抗原结合分子与抗原结合;通过使抗原结合分子与固定化的抗原接触,也可以使抗原结合分子与抗原结合。另外,通过使抗原结合分子与抗原在溶液中接触,也可以使抗原结合分子与抗原结合。上述制备方法中,只要第一ph与第二ph各自是不同的ph即可,可以是任何的ph。优选的第一ph与第二ph的组合的例子有:第一ph为ph6.7-10.0之间的ph、第二ph为ph4.0-ph6.5之间的ph的组合;更优选的组合的例子有:第一ph为ph7.0-ph8.0之间的ph、第二ph为ph5.5-ph6.5之间的ph的组合;进一步优选的组合的例子有:第一ph为ph7.4、第二ph为ph5.8或ph5.5的组合。其他优选的第一ph与第二ph的组合的例子有:第一ph为ph4.0-ph6.5之间的ph、第二ph为ph6.7-ph10.0之间的ph的组合;更优选的组合的例子有:第一ph为ph5.5-ph6.5之间的ph、第二ph为ph7.0-ph8.0之间的ph的组合;进一步优选的组合的例子有:第一ph为ph5.8或ph5.5、第二ph为ph7.4的组合。利用上述制备方法制备的抗原结合分子可以是任何的抗原结合分子,例如优选的例子有:抗原结合分子的至少1个氨基酸被组氨酸取代或插入至少1个组氨酸的抗原结合分子。对导入这样的组氨酸突变的位点没有特别限定,可以在任何位点导入。可以向1个位点导入组氨酸突变,也可以向2个位点以上的多个位点导入组氨酸突变。因此,在本发明的制备方法中,可以进一步包括将抗原结合分子的至少1个氨基酸取代成组氨酸或插入组氨酸的步骤。在本发明的制备方法中,可以使用非天然氨基酸来代替组氨酸。因此,还可以将上述组氨酸与非天然氨基酸互换来理解本发明。作为利用上述制备方法制备的抗原结合分子的其他方案,例如有包含被修饰的抗体恒定区的抗原结合分子,因此,在本发明的制备方法中,可以进一步包括修饰抗体恒定区中的氨基酸的步骤。利用本发明的制备方法制备的抗原结合分子是血浆中滞留性优异的抗原结合分子。因此,本发明的制备方法可以用作血浆中滞留性优异的抗原结合分子的制备方法。利用本发明的制备方法制备的抗原结合分子,将其对人、小鼠、猴等动物给药时,认为其可以两次以上与抗原结合。因此,本发明的制备方法可以用作能够两次以上与抗原结合的抗原结合分子的制备方法。并且,利用本发明的制备方法制备的抗原结合分子,将其对人、小鼠、猴等动物给药时,认为其可以与多于抗原结合分子的抗原结合位点数的数目的抗原结合。因此,本发明的制备方法可以用作能够与多于抗原结合分子的抗原结合位点数的数目的抗原结合的抗原结合分子的制备方法。并且,利用本发明的制备方法制备的抗原结合分子,将其对人、小鼠、猴等动物给药时,认为其可以使在细胞外与抗原结合分子结合的抗原在细胞内自抗原结合分子上解离。因此,本发明的制备方法可以用作能够在细胞内解离在细胞外结合的抗原的抗原结合分子的制备方法。并且,利用本发明的制备方法制备的抗原结合分子,将其对人、小鼠、猴等动物给药时,认为其可以使以与抗原结合的状态摄入细胞内的抗原结合分子以未与抗原结合的状态被释放到细胞外。因此,本发明的制备方法可以用作以与抗原结合的状态摄入细胞内、以未与抗原结合的状态被释放到细胞外的抗原结合分子的制备方法。并且,利用本发明的制备方法制备的抗原结合分子,将其对人、小鼠、猴等动物给药时,认为其可以使抗原从血浆中快速消除。因此,本发明的制备方法可以用作血浆中抗原消除能力增加(高)的抗原结合分子的制备方法。这些抗原结合分子可以减少对患者的给药次数,认为其作为药品特别优异。因此,本发明的制备方法可以用作作为药物组合物的抗原结合分子的制备方法。本发明的制备方法中得到的基因,通常将其负载(插入)到适当的载体上,并导入宿主细胞中。对该载体没有特别限定,只要是稳定保持插入的核酸的载体即可,例如使用大肠杆菌作为宿主时,作为克隆用载体,优选pbluescript载体(stratagene公司制)等,但也可以使用各种市售载体。当为了生产本发明的抗原结合分子而使用载体时,表达载体特别实用。对表达载体没有特别限定,只要是在试管内、大肠杆菌内、培养细胞内、生物个体内表达抗原结合分子的载体即可,例如,在试管内表达抗原结合分子时,优选pbest载体(promega公司制);在大肠杆菌内表达抗原结合分子时,优选pet载体(invitrogen公司制);在培养细胞内表达抗原结合分子时,优选pme18s-fl3载体(genbank检索号ab009864);在生物个体内表达抗原结合分子时,优选pme18s载体(molcellbiol.8:466-472(1988))等。向载体中插入本发明的dna,可以利用常规方法、例如通过使用限制酶切位点的连接酶反应来进行(currentprotocolsinmolecularbiologyedit.ausubel等人(1987)出版.johnwiley&sons.section11.4-11.11)。对上述宿主细胞没有特别限定,根据目的使用各种宿主细胞。作为用于表达抗原结合分子的细胞,可以例示:细菌细胞(例如:链球菌(streptococcus)、葡萄球菌(staphylococcus)、大肠杆菌(e.coli)、链霉菌(streptomyces)、枯草杆菌(bacillussubtilis));真菌细胞(例如:酵母(yeast)、曲霉(aspergillus));昆虫细胞(例如:果蝇s2(drosophilas2)、草地夜蛾sf9(spodopterasf9));动物细胞(例如:cho、cos、hela、c127、3t3、bhk、hek293、bowes黑色素瘤细胞)和植物细胞。向宿主细胞中导入载体,例如可以通过磷酸钙沉淀法、电脉冲穿孔法(currentprotocolsinmolecularbiologyedit.ausubel等人.(1987)出版.johnwiley&sons.section9.1-9.9)、脂质转染法、显微注射法等公知方法来进行。宿主细胞的培养可以按照公知方法进行。例如,以动物细胞作为宿主时,培养液可以使用例如dmem、mem、rpmi1640、imdm。此时,可以结合使用fbs、胎牛血清(fcs)等血清补充液,也可以通过无血清培养来培养细胞。培养时的ph优选为约6-8。通常是在约30-40℃下培养约15-200小时,根据需要可以交换培养基或进行通气、搅拌。可以向目标多肽中插入适当的分泌信号,以使在宿主细胞中表达的抗原结合分子分泌到内质网的内腔、周质间隙或胞外环境中。相对于目标抗原结合分子而言,上述信号可以是内源性信号,也可以是异源信号。另一方面,作为在体内产生多肽的系统,例如有使用动物的产生系统或使用植物的产生系统。向这些动物或植物中导入目标多核苷酸,使在动物或植物体内产生多肽并回收。本发明中的“宿主”包括这些动物和植物。使用动物时,有使用哺乳动物和昆虫的产生系统。作为哺乳动物,可以使用山羊、猪、绵羊、小鼠、牛等(vickiglaser,spectrumbiotechnologyapplications(1993))。使用哺乳动物时,可以使用转基因动物。例如,制备编码本发明的抗原结合分子的多核苷酸,将其作为和山羊β-酪素等乳汁中固有产生的编码多肽的基因的融合基因。然后,将包括该融合基因的多核苷酸片段注入到山羊胚胎中,将该胚胎移植到雌山羊体内。接受了胚胎的山羊生出转基因山羊,从该转基因山羊或其后代产生的乳汁中可以得到目标抗原结合分子。为了使转基因山羊产生的含有抗原结合分子的乳汁量增加,可以给予转基因山羊适当的激素(ebert等人,bio/technology(1994)12:699-702)。作为产生本发明的抗原结合分子的昆虫,例如可以使用蚕。使用蚕时,通过使蚕感染插入有编码目标抗原结合分子的多核苷酸的杆状病毒,可以从该蚕的体液中得到目标抗原结合分子。并且,在本发明的抗原结合分子产生中使用植物时,例如可以使用烟草。使用烟草时,将编码目标抗原结合分子的多核苷酸插入到植物表达用载体例如pmon530中,将该载体导入根瘤土壤杆菌(agrobacteriumtumefaciens)这样的细菌中。用该细菌感染烟草例如烟草(nicotianatabacum),可以从该烟草的叶中得到所期望的抗原结合分子(ma等人,eur.j.immunol.(1994)24:131-8)。用同样的细菌感染浮萍(lemnaminor),克隆化后可以从浮萍的细胞中得到所期望的抗原结合分子(coxkm等人.nat.biotechnol.2006dec;24(12):1591-1597)。如此操作得到的抗原结合分子,可以将其从宿主细胞内或细胞外(培养基、乳汁等)分离,纯化成实质上纯粹且均匀的抗原结合分子。抗原结合分子的分离、纯化可以使用通常多肽的纯化中所使用的分离、纯化方法,但没有任何限制。例如,可以适当选择组合层析柱、滤器、超滤、盐析、溶剂沉淀、溶剂提取、蒸馏、免疫沉淀、sds-聚丙烯酰胺凝胶电泳、等电点电泳法、透析、重结晶等来分离、纯化抗原结合分子。层析例如有:亲合层析、离子交换层析、疏水层析、凝胶过滤、反相层析、吸附层析等(strategiesforproteinpurificationandcharacterization(蛋白质纯化与鉴定实验指南):alaboratorycoursemanual.eddanielr.marshark等人.(1996)coldspringharborlaboratorypress)。上述层析可以使用液相层析、例如hplc、fplc等液相层析来进行。用于亲合层析的柱例如有:蛋白a柱、蛋白g柱。蛋白a柱例如有:hyperd、poros、sepharosef.f.(pharmacia制)等。根据需要,可以在纯化抗原结合分子之前或之后,使适当的蛋白修饰酶与抗原结合分子作用,以任意地进行修饰或部分性地去除肽。作为蛋白修饰酶,例如可以使用胰蛋白酶、糜蛋白酶、赖氨酰内肽酶、蛋白激酶、糖苷酶等。<抗il-6受体抗体>本发明还提供以下(a)-(m)中任一项所述的抗il-6受体抗体。(a)抗体,该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第27位的tyr、第31位的asp、第32位的asp、第35位的trp、第51位的tyr、第59位的asn、第63位的ser、第106位的met、第108位的tyr中的至少1个被取代成his的氨基酸序列;(b)抗体(h3pi),该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第27位的tyr、第31位的asp和第35位的trp被取代成his的氨基酸序列;(c)抗体,该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第27位的tyr、第31位的asp、第32位的asp、第35位的trp、第59位的asn、第63位的ser、第108位的tyr被取代成his的氨基酸序列;(d)抗体(h170),该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第27位的tyr、第31位的asp、第32位的asp、第35位的trp、第59位的asn、第63位的ser、第108位的tyr被取代成his、且第99位的ser被取代成val、第103位的thr被取代成ile的氨基酸序列;(e)抗体,该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第31位的asp、第51位的tyr、第63位的ser、第106位的met和第108位的tyr被取代成his的氨基酸序列;(f)抗体(clh5),该抗体包含具有下述氨基酸序列的重链可变区:在seqidno:1(h53可变区)的氨基酸序列中第31位的asp、第51位的tyr、第63位的ser、第106位的met和第108位的tyr被取代成his、且第99位的ser被取代成phe、第103位的thr被取代成ile的氨基酸序列;(g)抗体,该抗体包含具有下述氨基酸序列的轻链可变区:在seqidno:2(pf1l可变区)的氨基酸序列中第28位的asp、第32位的tyr、第53位的glu、第56位的ser、第92位的asn中的至少一个被取代成his的氨基酸序列;(h)抗体(l73),该抗体包含具有下述氨基酸序列的轻链可变区:在seqidno:2(pf1l可变区)的氨基酸序列中第28位的asp、第32位的tyr和第53位的glu被取代成his的氨基酸序列;(i)抗体(l82),该抗体包含具有下述氨基酸序列的轻链可变区:在seqidno:1(h53可变区)的氨基酸序列中第32位的tyr和第53位的glu被取代成his的氨基酸序列;(j)抗体(cll5),该抗体包含具有下述氨基酸序列的轻链可变区:在seqidno:2(pf1l可变区)的氨基酸序列中第32位的tyr、第53位的glu、第56位的ser和第92位的asn被取代成his的氨基酸序列;(k)抗体,该抗体包含(b)的重链可变区和(h)的轻链可变区;(l)抗体,该抗体包含(d)的重链可变区和(i)的轻链可变区;(m)抗体,该抗体包含(f)的重链可变区和(h)的轻链可变区。作为具有在seqidno:1(h53可变区)的氨基酸序列中第27位的tyr、第31位的asp、第32位的asp、第35位的trp、第51位的tyr、第59位的asn、第63位的ser、第106位的met、第108位的tyr中的至少1个被取代成his的氨基酸序列的重链可变区的具体例子,例如有以下的重链可变区:重链可变区,其中具有seqidno:3(h3pi)的氨基酸序列重链可变区,其中具有seqidno:4(h170)的氨基酸序列重链可变区,其中具有seqidno:5(clh5)的氨基酸序列作为具有在seqidno:2(pf1l可变区)的氨基酸序列中第28位的asp、第32位的tyr、第53位的glu、第56位的ser、第92位的asn中的至少1个被取代成his的氨基酸序列的轻链可变区的具体例子,例如有以下的轻链可变区:轻链可变区,其中具有seqidno:6(l73)的氨基酸序列轻链可变区,其中具有seqidno:7(l82)的氨基酸序列轻链可变区,其中具有seqidno:8(cll5)的氨基酸序列上述h3pi、h170、clh5、l73、l82和cll5的各抗体中的氨基酸位点和氨基酸取代见下表1。氨基酸位点根据kabat编号来表示。[表1]位置2731323335505861626364659599100b102h3pihhhhh170hhhhhhhvihclh5hhhhfihh位置24272832535556909294l73hhhhl82hhhcll5hhhhh*h链的第33位和l链的第55位在wt中具有组氨酸的序列.本发明提供至少包含上述(a)-(j)中任一项所述的氨基酸取代的抗体以及该抗体的制备方法。因此,本发明的抗体还包含:除上述(a)-(j)中任一项所述的氨基酸取代外,还包含除上述(a)-(j)记载的氨基酸取代以外的氨基酸取代的抗体。作为除上述(a)-(j)记载的氨基酸取代以外的氨基酸取代,例如有cdr部分的氨基酸序列的取代、缺失、添加和/或插入等、或fr的氨基酸序列的取代、缺失、添加和/或插入等。本发明还提供以下(1)-(28)中任一项所述的抗il-6受体抗体。(1)抗体,该抗体包含具有seqidno:21(vh1-igg1)的第1位-第119位的氨基酸序列的重链可变区(vh1-igg1可变区);(2)抗体,该抗体包含具有seqidno:22(vh2-igg1)的第1位-第119位的氨基酸序列的重链可变区(vh2-igg1可变区);(3)抗体,该抗体包含具有seqidno:23(vh3-igg1)的第1位-第119位的氨基酸序列的重链可变区(vh3-igg1可变区);(4)抗体,该抗体包含具有seqidno:24(vh4-igg1)的第1位-第119位的氨基酸序列的重链可变区(vh4-igg1可变区);(5)抗体,该抗体包含具有seqidno:25(vl1-ck)的第1位-第107位的氨基酸序列的轻链可变区(vl1-ck可变区);(6)抗体,该抗体包含具有seqidno:26(vl2-ck)的第1位-第107位的氨基酸序列的轻链可变区(vl2-ck可变区);(7)抗体,该抗体包含具有seqidno:27(vl3-ck)的第1位-第107位的氨基酸序列的轻链可变区(vl3-ck可变区);(8)抗体(fv1-igg1),其包含(2)的重链可变区和(6)的轻链可变区;(9)抗体(fv2-igg1),其包含(1)的重链可变区和具有seqidno:7(l82)记载的氨基酸序列的轻链可变区;(10)抗体(fv3-igg1),其包含(4)的重链可变区和(5)的轻链可变区;(11)抗体(fv4-igg1),其包含(3)的重链可变区和(7)的轻链可变区;(12)抗体(vh3-igg2δgk),其包含具有seqidno:33记载的氨基酸序列的重链;(13)抗体(vh3-m58),其包含具有seqidno:34记载的氨基酸序列的重链;(14)抗体(vh3-m73),其包含具有seqidno:35记载的氨基酸序列的重链;(15)抗体(fv4-igg2δgk),其包含(12)的重链和具有seqidno:27(vl3-ck)的氨基酸序列的轻链;(16)抗体(fv4-m58),其包含(13)的重链和具有seqidno:27(vl3-ck)的氨基酸序列的轻链;(17)抗体(fv4-m73),其包含(14)的重链和具有seqidno:27(vl3-ck)的氨基酸序列的轻链;(18)抗体(vh2-m71),其包含具有seqidno:36(vh2-m71)的氨基酸序列的重链;(19)抗体(vh2-m73),其包含具有seqidno:37(vh2-m73)的氨基酸序列的重链;(20)抗体(vh4-m71),其包含具有seqidno:38(vh4-m71)的氨基酸序列的重链;(21)抗体(vh4-m73),其包含具有seqidno:39(vh4-m73)的氨基酸序列的重链;(22)抗体(fv1-m71),其包含(18)的重链和具有seqidno:26(vl2-ck)的氨基酸序列的轻链;(23)抗体(fv1-m73),其包含(19)的重链和具有seqidno:26(vl2-ck)的氨基酸序列的轻链;(24)抗体(fv3-m71),其包含(20)的重链和具有seqidno:25(vl1-ck)的氨基酸序列的轻链;(25)抗体(fv3-m73),其包含(21)的重链和具有seqidno:25(vl1-ck)的氨基酸序列的轻链;(26)抗体,其包含具有seqidno:25(vl1-ck)的氨基酸序列的轻链;(27)抗体,其包含具有seqidno:26(vl2-ck)的氨基酸序列的轻链;(28)抗体,其包含具有seqidno:27(vl3-ck)的氨基酸序列的轻链。本发明还提供以下(a)-(v)中任一项所述的fr或cdr。(a)seqidno:40记载的重链cdr1(vh1、2、3、4)、(b)seqidno:41记载的重链cdr2(vh1、2)、(c)seqidno:42记载的重链cdr2(vh3)、(d)seqidno:43记载的重链cdr2(vh4)、(e)seqidno:44记载的重链cdr3(vh1、2)、(f)seqidno:45记载的重链cdr3(vh3、4)、(g)seqidno:46记载的重链fr1(vh1、2)、(h)seqidno:47记载的重链fr1(vh3、4)、(i)seqidno:48记载的重链fr2(vh1、2、3、4)(j)seqidno:49记载的重链fr3(vh1)、(k)seqidno:50记载的重链fr3(vh2)、(l)seqidno:51记载的重链fr3(vh3、4)、(m)seqidno:52记载的重链fr4(vh1、2、3、4)(n)seqidno:53记载的轻链cdr1(vl1、2)、(o)seqidno:54记载的轻链cdr1(vl3)、(p)seqidno:55记载的轻链cdr2(vl1、vl3)、(q)seqidno:56记载的轻链cdr2(vl2)、(r)seqidno:57记载的轻链cdr3(vl1、2、3)、(s)seqidno:58记载的轻链fr1(vl1、2、3)、(t)seqidno:59记载的轻链fr2(vl1、2、3)、(u)seqidno:60记载的轻链fr3(vl1、2、3)、(v)seqidno:61记载的轻链fr4(vl1、2、3)。上述(a)-(v)的各序列汇总于图25中。本发明还提供包含上述(a)-(v)中任一项的fr或cdr的多肽。本发明的抗il-6受体抗体还包括:包含上述任一项所述的氨基酸取代的抗体的片段或其修饰物。作为抗体片段,例如有fab、f(ab’)2、fv或将h链和l链的fv经适当的接头连接而成的单链fv(scfv)、h链单结构域或l链单结构域(例如nat.biotechnol.2005sep;23(9):1126-36),unibody(wo2007059782a1)、smip(wo2007014278a2)。对抗体的来源没有特别限定,可以是人抗体、小鼠抗体、大鼠抗体、兔抗体等。本发明的抗体还可以是嵌合抗体、人源化抗体、完全人源化抗体等。具体而言,用酶、例如木瓜蛋白酶、胃蛋白酶处理抗体,生成抗体片段;或者,构建编码这些抗体片段的基因,将其导入表达载体中,之后使之在适当的宿主细胞中表达(例如参照co,m.s.等人,j.immunol.(1994)152,2968-2976;better,m.&horwitz,a.h.methodsinenzymology(1989)178,476-496;pluckthun,a.&skerra,a.methodsinenzymology(1989)178,497-515;lamoyi,e.,methodsinenzymology(1989)121,652-663;rousseaux,j.等人,methodsinenzymology(1989)121,663-66;bird,r.e.等人,tibtech(1991)9,132-137)。因此,本发明提供制备本发明的多肽或由编码本发明的多肽的基因编码的多肽的方法,该方法包括培养宿主细胞的步骤,所述宿主细胞包含导入有编码本发明的多肽的多核苷酸的载体。更具体而言,提供本发明的多肽的制备方法,该制备方法包括以下步骤:(a)培养宿主细胞的步骤,所述宿主细胞包含导入有编码本发明的多肽的基因的载体;(b)获得由该基因编码的多肽的步骤。scfv是通过连接抗体h链v区和l链v区而得到的。在该scfv中,h链v区和l链v区经由接头、优选肽接头进行连接(huston,j.s.等人,proc.natl.acad.sci.usa(1988)10.0,5879-5883)。scfv中的h链v区和l链v区可以来自作为上述抗体记载的任一种抗体。作为连接v区的肽接头,使用例如包含12-19个氨基酸残基的任意的单链肽。当本发明的抗il-6受体抗体包含恒定区时,恒定区可以是任何类型的恒定区,例如可以使用igg1、igg2、igg4等的恒定区。恒定区优选为人抗体的恒定区。还可以是对人igg1、人igg2、人igg4等的恒定区进行氨基酸序列的取代、缺失、添加和/或插入等得到的修饰体。本发明的抗il-6受体抗体所结合的il-6受体优选为人il-6受体。本发明的抗il-6受体抗体是血浆中滞留性优异的抗体,抗il-6受体抗体以可与作为抗原的可溶型il-6受体和膜型il-6受体结合的状态存在于血浆中的时间延长,在体内可溶型il-6受体和膜型il-6受体被抗il-6受体抗体结合的时间延长。另外,该抗il-6受体抗体可以与il-6受体结合两次以上,认为其可以中和3个以上的il-6受体。<药物组合物>本发明还涉及含有本发明的抗原结合分子、利用本发明的筛选方法分离的抗原结合分子、或利用本发明的制备方法制备的抗原结合分子的药物组合物。本发明的抗原结合分子或利用本发明的制备方法制备的抗原结合分子,其血浆中滞留性优异,被期待减少抗原结合分子的给药频率,所以作为药物组合物是有效的。本发明的药物组合物可以含有医药上可接受的载体。本发明中,药物组合物通常是指用于疾病的治疗或预防或者检查、诊断的药物。本发明的药物组合物可以按照本领域技术人员所公知的方法制成制剂。例如,可以制成和水或水以外的药学上可接受的液体的无菌溶液或悬浮液,以注射剂的形式进行胃肠外给药。例如,可以考虑和药理学上可接受的载体或介质,具体而言,和灭菌水或生理盐水、植物油、乳化剂、悬浮剂、表面活性剂、稳定剂、香味剂、赋形剂、媒介物(vehicle)、防腐剂、结合剂等适当组合,以通常认可的制药实施所要求的单位用量形态进行混合以制成制剂。上述制剂中的有效成分量设定为得到所指示的范围的适当容量。用于注射的无菌组合物,可以使用注射用蒸馏水这样的媒介物,按照通常的制剂流程进行配制。注射用水溶液例如有生理盐水和等渗溶液,该等渗溶液中包含葡萄糖或其他辅药(例如d-山梨醇、d-甘露糖、d-甘露糖醇、氯化钠)。可以结合使用适当的助溶剂,例如醇(乙醇等)、多元醇(丙二醇、聚乙二醇等)、非离子表面活性剂(polysorbate80(tm)、hco-50等)。油性液体有芝麻油、大豆油,可以结合使用苯甲酸苄酯和/或苄醇作为助溶剂。还可以和缓冲剂(例如磷酸盐缓冲液和乙酸钠缓冲液)、缓和剂(例如盐酸普鲁卡因)、稳定剂(例如苄醇和苯酚)、抗氧化剂混合。制备的注射液通常填充在适当的安瓿中。本发明的药物组合物,优选通过胃肠外给药途径进行给药。例如,可以制成注射剂型、经鼻给药剂型、经肺给药剂型、经皮给药型的组合物。例如,可以通过静脉注射、肌肉注射、腹腔内注射、皮下注射等进行全身或局部给药。给药方法可以根据患者的年龄、症状适当选择。含有抗原结合分子的药物组合物,其给药量例如可以设定为:每次每kg体重0.0001mg-1000mg的范围。或者,给药量还可以是例如每位患者0.001-100000mg,但本发明未必限于上述数值。给药量和给药方法根据患者的体重、年龄、症状等而变化,本领域技术人员可以考虑上述条件,设定适当的给药量和给药方法。本发明所记载的氨基酸序列中所含的氨基酸,有时在翻译后会接受修饰(例如,n末端的谷氨酰胺通过焦谷氨酰化成为焦谷氨酸的修饰是本领域技术人员所熟知的修饰),即使是这样氨基酸在翻译后被修饰的情形,当然也包含在本发明所记载的氨基酸序列中。本说明书中引用的所有的现有技术文献均作为参照而纳入本说明书中。实施例以下,通过实施例来具体说明本发明,但本发明并不限于这些实施例。[实施例1]修饰人源化pm1抗体的制作重组可溶型人il-6受体(sr344)的制备作为抗原的人il-6受体的重组人il-6受体如下制备。制备j.biochem.108,673-676(1990)中报道的、包含n末端侧第1位-第344位的氨基酸序列的可溶型人il-6受体(以下记作sr344)(yamasaki等人,science1988;241:825-828(genbank#x12830))的cho细胞恒定表达株。从由sr344表达cho细胞得到的培养上清中,通过bluesepharose6ff柱层析、固定有sr344特异性抗体的柱的亲和层析、凝胶过滤柱层析这3种柱层析纯化sr344。以主峰形式洗脱的组分作为最终纯品。重组可溶型食蟹猴il-6受体(cil-6r)的制备根据公开的恒河猴il-6受体基因序列(birney等人,ensembl2006,nucleicacidsres.2006jan1;34(databaseissue):d556-61)制作寡dna引物rhe6rf1(seqidno:16)、rhe6rr2(seqidno:17)。以由食蟹猴胰脏制备的cdna为模板,使用引物rhe6rf1和rhe6rr2,通过pcr法制备编码食蟹猴il-6受体全长基因的dna片段。以得到的dna片段为模板,使用寡dna引物cynoil6rn-ecori(seqidno:18)和cynoil6rc-noti-his(seqidno:19),通过pcr法扩增1131bp的dna片段(seqidno:20),该dna片段编码在包含食蟹猴il-6受体基因的信号区的可溶型区(met1-pro363)的c末端添加有6xhis的蛋白。得到的dna片段用ecori-noti进行消化,之后插入动物细胞表达载体中,使用该载体制作cho恒定表达株(产生cyno.sil-6r的cho细胞)。将产生cyno.sil-6r的cho细胞的培养液用histrap柱(gehealthcarebioscience)纯化,之后使用amiconultra-15ultracel-10k(millipore)进行浓缩,用superdex200pg16/60凝胶过滤柱(gehealthcarebioscience)进一步进行纯化,得到可溶型食蟹猴il-6受体(以下记作cil-6r)的最终纯品。重组食蟹猴il-6(cil-6)的制作食蟹猴il-6如下制备。制作编码在swissprotaccessionno.p79341中注册的212个氨基酸的核苷酸序列,将其克隆到动物细胞表达载体中,再将所得载体导入cho细胞中,从而制作恒定表达细胞株(产生cyno.il-6的cho细胞)。将产生cyno.il-6的cho细胞的培养液用sp-sepharose/ff柱(gehealthcarebioscience)纯化,之后使用amiconultra-15ultracel-5k(millipore)进行浓缩,再使用superdex75pg26/60凝胶过滤柱(gehealthcarebioscience)进一步进行纯化,并用amiconultra-15ultracel-5k(millipore)进行浓缩,作为食蟹猴il-6(以下记作cil-6)的最终纯品。人gp130表达baf3细胞株的建立为了得到显示il-6依赖性增殖的细胞株,如下建立表达人gp130的baf3细胞株。通过pcr法扩增全长人gp130cdna(hibi等人,cell1990;63:1149-1157(genbank#nm_002184)),之后将其克隆到表达载体pcos2zeo中,构建pcos2zeo/gp130,所述表达载体pcos2zeo是通过除去pchoi(hirata等人,febsletter1994;356:244-248)的dhfr基因表达位点、并插入zeocin耐性基因表达位点而得到的。通过pcr法扩增全长人il-6rcdna,之后将其克隆到pcdna3.1(+)(invitrogen)中,构建hil-6r/pcdna3.1(+)。将10μg的pcos2zeo/gp130混合在悬浮于pbs中的baf3细胞(0.8×107细胞)中,之后使用genepulser(bio-rad)以0.33kv、950μfd的容量施加脉冲。将通过电穿孔处理导入有基因的baf3细胞在含有0.2ng/ml小鼠白介素-3(peprotech)、10%胎牛血清(以下记作fbs;hyclone)的rpmi1640培养基(invitrogen)中培养一昼夜,加入含有100ng/ml人白介素-6(r&dsystems)、100ng/ml人白介素-6可溶型受体(r&dsystems)和10%fbs的rpmi1640培养基进行筛选,建立人gp130表达baf3细胞株(以下记作baf3/gp130)。由于该baf/gp130在人白介素-6(r&dsystems)和sr344的存在下增殖,所以可以将其用于评价抗il-6受体抗体的增殖抑制活性(即il-6受体中和活性)。人源化抗il-6受体抗体的制作在cancerres.1993feb15;53(4):851-6中,向人源化的小鼠pm1抗体(以后,将野生型简记为wt、将h链wt简记为h(wt)(氨基酸序列seqidno:9),将l链wt记作l(wt)(氨基酸序列seqidno:10))的构架序列和cdr序列中导入突变,制作作为修饰h链的h53(氨基酸序列seqidno:1)、pf1h(氨基酸序列seqidno:11)、作为修饰l链的l28(氨基酸序列seqidno:12)、pf1l(氨基酸序列seqidno:2)。具体而言,使用quikchange位点定向诱变试剂盒(stratagene),按照附录说明书所述的方法制作突变体,将得到的质粒片段插入动物细胞表达载体中,制作目标h链表达载体和l链表达载体。所得的表达载体的核苷酸序列按照本领域技术人员公知的方法来确定。人源化抗il-6受体抗体的表达和纯化抗体的表达采用下述方法进行。将来自人胚肾癌细胞的hek293h株(invitrogen)悬浮于含有10%胎牛血清(invitrogen)的dmem培养基(invitrogen)中,以5-6×105个细胞/ml的细胞密度在粘附细胞用培养皿(直径10cm;corning)的各皿中分别接种10ml,在37℃、5%co2的培养箱内培养一昼夜,之后吸除培养基,添加6.9mlcho-s-sfm-ii培养基(invitrogen)。通过脂质转染法将制备的质粒导入细胞中。回收所得的培养上清,之后以约2000g的离心力在室温下离心5分钟以除去细胞,再通过0.22μm的滤器millex(r)-gv(millipore)进行灭菌,得到培养上清。对于所得的培养上清,使用rproteinasepharosetmfastflow(amershambiosciences),按照本领域技术人员公知的方法进行纯化。关于纯化抗体浓度,使用分光亮度计测定280nm下的吸亮度。利用pace法由所得的值算出吸光系数,使用该吸光系数算出抗体浓度(proteinscience1995;4:2411-2423)。[实施例2]ph依赖性结合抗体h3pi/l73的制作可以多次中和抗原的抗体的制备方法igg分子是二价的,所以当其在2个位点与抗原结合时,以1分子的igg分子最多可以中和2分子的抗原,但无法中和3分子以上的抗原。因此,当为中和抗体时,为了使其中和效果持续一定期间,必需给予在该一定期间内产生的抗原量以上的抗体量,仅凭提高抗体的药物动力学或提高亲和力的技术,在减少必需的抗体给药量方面存在界限。因此,只要能够以1分子的igg分子中和2分子以上的抗原,就可以以相同的给药量提高中和效果的持续性,并且减少用于达到相同的持续性所必需的给药量。当为中和抗体时,作为靶的抗原的种类,存在以下两种情形:抗原存在于血浆中的可溶型抗原和抗原在细胞表面表达的膜型抗原。当抗原为膜型抗原时,给予的抗体与细胞表面上的膜抗原结合,之后,抗体保持着与膜抗原结合的状态,与抗原一起通过内化作用摄入细胞内的核内体中,之后抗体保持着与抗原结合的状态向溶酶体移动,抗体和抗原一起被溶酶体分解。膜抗原的由内化作用介导的从血浆中消除被称作抗原依赖性消除,这在多数抗体分子中都有报道(drugdiscovtoday.2006jan;11(1-2):81-8)。当1分子的igg抗体以二价与抗原结合时,其与2分子的抗原结合并被内化,直接被溶酶体分解,所以当为普通抗体时,1分子的igg抗体无法中和2分子以上的抗原(图1)。igg分子的血浆中滞留性长(消除慢),这是由于作为igg分子的补救受体而已知的fcrn在起作用(nat.rev.immunol.2007sep;7(9):715-25)。通过胞饮作用摄入核内体中的igg分子,其在核内体内的酸性条件下与在核内体内表达的fcrn结合。无法与fcrn结合的igg分子摄入溶酶体内,被溶酶体分解,但与fcrn结合的igg分子向细胞表面移动,在血浆中的中性条件下自fcrn上解离,又返回到血浆中(图2)。与膜抗原结合的igg分子通过内化作用摄入细胞内的核内体中,保持着与抗原结合的状态移动到溶酶体中被分解,当igg抗体以二价与抗原结合时,其中和2分子的抗原,与抗原一同被分解。认为通过内化作用摄入细胞内的核内体中时,只要igg抗体在核内体内的酸性条件下能够自抗原上解离,解离的抗体就可以与在核内体内表达的fcrn结合。自抗原上解离并与fcrn结合的igg分子向细胞表面移动,在血浆中的中性条件下自fcrn上解离,从而再次返回到血浆中,返回血浆中的igg分子可以再次与新的膜抗原结合。通过重复上述过程,1分子的igg分子可以反复与膜型抗原结合,因此1分子的igg分子可以中和多个抗原(图3)。当抗原为可溶型抗原时,给予的抗体在血浆中与抗原结合,以抗原与抗体的复合体的形式滞留在血浆中。如上所述,通常抗体的血浆中滞留性由于fcrn的作用而非常长(消除速度非常慢),相对于此,抗原的血浆中滞留性短(消除速度快),因此与抗体结合的抗原与抗体具有相同程度的血浆中滞留性(消除非常慢)。抗原在体内常常是以一定的速度产生,不存在抗体时,抗原以抗原的产生速度和抗原的消除速度平衡的状态的浓度存在于血浆中。在抗体的存在下,大部分的抗原与抗体结合,抗原的消除变得非常慢,因此血浆中的抗原浓度与不存在抗体时相比有所上升(kidneyint.2003,64,697-703、j.nationalcancerinstitute2002,94(19),1484-1493、j.allergyandclinicalimmunology1997,100(1),110-121、eur.j.immunol.1993,23;2026-2029)。假使抗体与抗原的亲和力无限大,抗原的浓度上升,抗体自血浆中慢慢消除,在抗体与抗原的浓度一致的时刻以后,抗体的抗原中和效果被中断。关于对可溶型抗原的中和效果,解离常数(kd)越大,就可以以越少的抗体浓度进行中和,但无论亲和力多强,都无法以所存在的抗原浓度的1/2以下的抗体浓度来中和抗原(biochembiophysrescommun.2005sep9;334(4):1004-13)。与未结合有抗原的igg分子一样,抗原所结合的igg分子在血浆中也通过胞饮作用摄入核内体,在核内体内的酸性条件下与在核内体内表达的fcrn结合。与fcrn结合的igg分子保持着与抗原结合的状态向细胞表面移动,在血浆中的中性条件下自fcrn上解离,从而,igg分子保持着与抗原结合的状态再次返回到血浆中,因此其在血浆中无法与新的抗原结合。认为此时若igg分子在核内体内的酸性条件下能够自抗原上解离,则解离的抗原无法与fcrn结合,因此该抗原被溶酶体分解。另一方面,igg分子通过与fcrn结合,可以再次返回到血浆中。返回血浆中的igg分子已经在核内体内解离抗原,所以其在血浆中能够再次与新的抗原结合。通过重复该过程,1分子的igg分子可以反复与可溶型抗原结合,因此1分子的igg分子可以中和多个抗原(图4)。这样,认为不管抗原是膜型抗原还是可溶型抗原,只要igg抗体在核内体内的酸性条件下能够自抗原上解离,1分子的igg分子就可以反复中和抗原。为了使igg抗体在核内体内的酸性条件下自抗原上解离,必需使酸性条件下抗原与抗体的结合较在中性条件下大幅减弱。由于必需在细胞表面中和膜抗原,所以在细胞表面的ph即ph7.4下必需与抗原强力结合。据报道,核内体内的ph通常为ph5.5-ph6.0(natrevmolcellbiol.2004feb;5(2):121-32),所以如果是在ph5.5-ph6.0下与抗原微弱结合的抗体,认为在核内体内的酸性条件下抗体会自抗原上解离。即,如果是在细胞表面的ph即ph7.4下与抗原强力结合、在核内体内的ph即ph5.5-ph6.0下与抗原微弱结合的抗体,认为1分子的igg分子可以中和多个抗原,可以提高药物动力学。通常,蛋白-蛋白相互作用包括疏水相互作用、静电相互作用和氢键,其结合强度通常以结合常数(affinity,亲和力)或表观结合常数(avidity,亲合力)表示。在中性条件下(ph7.4)和酸性条件下(ph5.5-ph6.0)结合强度发生变化的ph依赖性结合存在于天然存在的蛋白-蛋白相互作用中。例如,上述的igg分子与作为igg分子的补救受体而已知的fcrn的结合,在酸性条件下(ph5.5-ph6.0)强力结合,在中性条件下(ph7.4)极微弱地结合。在这些众多的ph依赖性结合发生变化的蛋白-蛋白相互作用中,组氨酸残基与该相互作用有关。组氨酸残基的pka存在于6.0-6.5附近,因此在中性条件下(ph7.4)与酸性条件下(ph5.5-ph6.0)之间组氨酸残基的质子的解离状态发生变化。即,在中性条件下(ph7.4)组氨酸残基不带电荷,以中性发挥氢原子接纳体的作用;在酸性条件下(ph5.5-ph6.0)组氨酸残基带有正电荷,发挥氢原子供体的作用。据报道,即使在上述的igg-fcrn相互作用中,存在于igg侧的组氨酸残基也与ph依赖性结合有关(molcell.2001apr;7(4):867-77)。因此,通过将与蛋白-蛋白相互作用有关的氨基酸残基取代成组氨酸残基、或者向进行相互作用的位点导入组氨酸,可以对蛋白-蛋白相互作用赋予ph依赖性。在抗体-抗原间的蛋白-蛋白相互作用中也进行这样的尝试,通过向抗卵白溶菌酶抗体的cdr序列中导入组氨酸,成功地获得了在酸性条件下与抗原的结合性降低的抗体突变体(febsletter(309卷,no.1,85-88,1992))。还有人报道了:通过向cdr序列中导入组氨酸,在癌组织的低ph下与抗原特异性结合、在中性条件下微弱结合的抗体(wo2003105757)。有人报道了按照这种方式向抗原抗体反应中导入ph依赖性的方法,但迄今为止,通过在体液中的ph即ph7.4下与抗原强力结合、在核内体内的ph即ph5.5-ph6.0下与抗原微弱结合,1分子的igg分子中和多个抗原的抗体未见报道。即,没有关于通过导入在维持中性条件下的结合的同时只大幅降低在酸性条件下的结合的修饰,与修饰前的抗体相比,修饰后的抗体在体内与抗原多次结合,从而药物动力学提高、在相同的给药量下中和效果的持续性提高的抗体的修饰的报道。il-6受体在体内以可溶型il-6受体和膜型il-6受体两种形式存在(natclinpractrheumatol.2006nov;2(11):619-26)。抗il-6受体抗体与可溶型il-6受体和膜型il-6受体两者结合,中和它们的生物学作用。认为抗il-6受体抗体与膜型il-6受体结合后,保持着与膜型il-6受体结合的状态通过内化作用摄入细胞内的核内体中,之后抗il-6受体抗体保持着与膜型il-6受体结合的状态向溶酶体移动,一起被溶酶体分解。有报道称,实际上人源化抗il-6受体抗体显示出非线性清除率,抗原依赖性消除大大有助于人源化抗il-6受体抗体的消除(thejournalofrheumatology,2003,30;71426-1435)。即,1分子的人源化抗il-6受体抗体以(一价或二价)与1分子或2分子的膜型il-6受体结合,内化后被溶酶体分解。因此,认为如果能够制作在维持天然型的人源化抗il-6受体抗体在中性条件下的结合的同时只大幅降低在酸性条件下的结合的修饰抗体(ph依赖性结合抗il-6受体抗体),那么以1分子的人源化抗il-6受体抗体可以中和多个分子的il-6受体,由此,与天然型的人源化抗il-6受体抗体相比,认为ph依赖性结合抗il-6受体抗体在体内以相同的给药量可以提高中和效果的持续性。ph依赖性结合人源化il-6受体抗体h3pi/l73的制作作为向抗原抗体反应中导入ph依赖性结合的方法,有人报道了向cdr中导入组氨酸的方法(febsletter(309卷,no.1,85-88,1992))。为了确认暴露在实施例1中制作的h53/pf1l的可变区表面的氨基酸残基和可能与抗原发生相互作用的残基,使用moe软件(chemicalcomputinggroupinc.),利用同源模建制作h53/pf1l的fv区模型。由根据h53/pf1l的序列信息制作的立体结构模型,选定h27、h31、h35、l28、l32、l53(kabat编号、kabatea等人.1991.sequencesofproteinsofimmunologicalinterest.nih)作为突变位点,认为通过向上述突变位点中导入组氨酸,可以导入与抗原的ph依赖性结合。向实施例1中制作的h53中导入将h27、h31、h35的残基取代成组氨酸的突变,以所得的链作为h3pi(氨基酸序列seqidno:3);向实施例1中制作的pf1l中导入将l28、l32、l53的残基取代成组氨酸的突变,以所得的链作为l73(氨基酸序列seqidno:6)。h3pi/l73的表达载体的制作、表达和纯化对选定的位点进行用于制作修饰抗体的氨基酸修饰。向实施例1中制作的h53(核苷酸序列seqidno:13)和pf1l(核苷酸序列seqidno:14)中导入突变,制作h3pi(氨基酸序列seqidno:3)和l73(氨基酸序列seqidno:6)。具体而言,使用quikchange位点定向诱变试剂盒(stratagene),按照附录说明书所述的方法进行制作,将得到的质粒片段插入动物细胞表达载体中,制作目标h链表达载体和l链表达载体。所得的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。h链使用h3pi、l链使用l73的h3pi/l73的表达、纯化按照实施例1中记载的方法进行。[实施例3]利用使用了噬菌体展示技术的cdrhis修饰赋予ph依赖性抗原结合能力人源化pm1抗体的scfv分子的制作进行作为人源化抗il-6r抗体的人源化pm1抗体(cancerres.1993feb15;53(4):851-6)的scfv化。通过pcr扩增vh、vl区,制作在vh、vl之间具有接头序列ggggsggggsggggs(seqidno:15)的人源化pm1hlscfv。通过组氨酸分区选定可以导入组氨酸的位点利用以制作的人源化pm1hlscfvdna为模板的pcr,制作各cdr氨基酸中的1个氨基酸成为组氨酸的组氨酸文库。使用以欲进行文库化的氨基酸的密码子作为相当于组氨酸的密码子即cat的引物,通过pcr反应构筑文库部分,除此以外的部分通过普通pcr来制作,利用装配pcr法进行连接进行构筑。构筑的文库用sfii消化,之后插入到同样用sfii消化的噬菌粒载体pelbglaci载体中,然后转化xl1-blue(stratagene)。使用得到的菌落,利用噬菌体elisa进行抗原结合性评价和hlscfv序列分析。按照j.mol.biol1992;227:381-388,使用用1μg/mlsr344包被的板进行噬菌体-elisa。对于确认到与sr344的结合性的克隆,使用特异性引物进行序列分析。通过利用了抗etag抗体(gehealthcare)和抗m13抗体(gehealthcare)的elisa法求出噬菌体效价。使用该值,根据sr344的噬菌体elisa的结果,选定与人源化pm1hlscfv相比,即使将cdr的残基取代成组氨酸,结合能力也没有大的变化的位点。这些位点见表2。各残基的编号遵从kabat编号(kabatea等人.1991.sequencesofproteinsofimmunologicalinterest.nih)。[表2]对结合能力没有重大影响的组氨酸取代位点h31,h50,h54,h56,h57,h58,h59,h60,h61,h62,h63,h64,h65,h100a.h100b,h102l24,l26,l27,l28,l30,l31,l32,l52,l53,l54,l56,l90,l92,l93,l94cdr组氨酸修饰文库的构建对表2所示的、即使取代成组氨酸结合能力也没有大的变化的cdr残基(可导入组氨酸的位点)的氨基酸进行成为原始序列(天然型序列)或组氨酸的文库的设计。根据实施例1中制作的h链pf1h、l链pf1l的序列构建文库,使其在文库位点中成为原始序列或组氨酸(原始序列或组氨酸中的任一个)。使用设计成使欲进行文库化的位点成为原始氨基酸的密码子或组氨酸的密码子的引物,通过pcr反应构建文库部分,除此以外的位置则通过普通pcr进行制作、或与文库部分一样使用合成引物通过pcr反应进行制作,通过装配pcr法进行连接以构建文库(j.mol.biol1996;256:77-88)。使用该文库,按照j.immunologicalmethods1999;231:119-135,构建核糖体展示用文库。为了进行大肠杆菌无细胞系体外翻译,在5’侧添加sda序列(核糖体结合位点)、t7启动子,将gene3部分序列作为核糖体展示用的接头,使用sfii与3’侧进行连接。通过念珠淘选由文库获得ph依赖性结合scfv为了浓缩只与sr344具有结合能力的scfv,按照naturebiotechnology2000dec;18:1287-1292,利用核糖体展示法进行两次淘选。使用nhs-peo4-生物素(pierce)将制备的sr344生物素化,作为抗原。使用40nm的生物素化抗原进行淘选。以得到的dna池(dnapool)为模板,使用特异性引物进行pcr,从而复原hlscfv。用sfii进行消化,之后插入到同样用sfii消化的噬菌粒载体pelbglaci载体中,然后转化xl1-blue(stratagene)。使具有目标质粒的大肠杆菌在2yt/100μg/ml氨苄青霉素/2%葡萄糖培养基中增殖至0.4-0.6o.d./ml。向其中加入辅助噬菌体(helperphage)(m13ko7、4.5×1011pfu),在37℃下静置培养30分钟,再于37℃下振荡培养30分钟,之后移入50mlfalcon管中,以3000rpm的转速离心10分钟,再悬浮于2yt/100μg/ml氨苄青霉素/25μg/ml卡那霉素/0.5mmiptg中,然后使之在30℃下增殖一夜。关于噬菌体液,利用2.5mnacl/10%peg使培养了一夜的培养液沉淀,之后用pbs稀释,作为噬菌体文库液。向噬菌体文库液中加入10%m-pbs(含有10%脱脂乳的pbs)、1mtris-hcl,使达到终浓度2.5%m-pbs、ph7.4。淘选采用一般的方法、即使用固定在磁珠上的抗原的淘选方法(jimmunolmethods.2008mar20;332(1-2):2-9、jimmunolmethods.2001jan1;247(1-2):191-203、biotechnolprog.2002mar-apr;18(2):212-20)。具体而言,向制备的噬菌体文库中加入40pmol的生物素标记sr344,使之在37℃下与抗原接触60分钟。加入用5%m-pbs(含有5%脱脂乳的pbs)清洗的链霉亲和素包被的念珠(dynalm-280),使之在37℃下结合15分钟。将念珠用0.5ml的pbst(含有0.1%tween-20的pbs、ph7.4)和pbs(ph7.4)各清洗5次。将念珠在37℃下悬浮于1mlpbs(ph5.5)中,立即回收噬菌体。将回收的噬菌体溶液添加到10ml对数增殖期(od6000.4-0.5)xl1-blue中,在37℃下静置30分钟使之感染。将感染的大肠杆菌接种在2yt/100μg/ml氨苄青霉素/2%葡萄糖的225mm×225mm的板上。再次从该大肠杆菌开始培养,与上述同样进行噬菌体的培养,反复淘选8次。利用噬菌体elisa进行的评价将上述单一菌落接种在100μl2yt/100μg/ml氨苄青霉素/2%葡萄糖/12.5μg/ml四环素中,在30℃下培养一夜。将其中的2μl接种在300μl2yt/100μg/ml氨苄青霉素/2%葡萄糖中,在37℃下培养4小时,之后加入辅助噬菌体(m13ko7)9×108pfu,在37℃下静置培养30分钟,在37℃下进行30分钟的搅拌培养,使之感染。之后,将培养基更换为含有300μl2yt/100μg/ml氨苄青霉素/25μg/ml卡那霉素/0.5mmiptg的培养基。接着,在30℃下培养一夜,回收离心上清。向40μl离心上清中加入360μl50mm的pbs(ph7.4),供给elisa。将streptawell96微量滴定板(roche)用100μl含有62.5ng/ml生物素标记sr344的pbs包被一夜。用pbst清洗,除去抗原,之后用250μl2%bsa-pbs封闭1小时以上。除去2%bsa-pbs,向其中加入制备的培养上清,在37℃下静置1小时,使抗体结合。清洗后,加入50mmpbs(ph7.4)或50mmpbs(ph5.5),在37℃下静置30分钟,进行培养。清洗后,利用经2%bsa-pbs稀释的hrp结合抗m13抗体(amershamparmaciabiotech)和tmb单一溶液(zymed)进行检测,添加硫酸使反应停止,之后测定450nm的吸亮度。但是,在此次使用固定在磁珠上的抗原的淘选中,没有得到具有强ph依赖性结合能力的克隆。对于虽然弱但确认到了ph依赖性结合能的克隆,使用特异性引物进行序列分析。这些克隆中,以高概率成为组氨酸的位点见表3。[表3]通过噬菌体文库(磁珠淘选)发现的组氨酸取代位点h50,h58,h61,h62,h63,h64,h65,h102l24,l27,l28,l32,l53,l56,l90,l92,l94通过柱淘选从文库中获得ph依赖性结合scfv在通常的使用固定在磁珠上的抗原的淘选中,没有得到具有强ph依赖性结合能的克隆。认为原因在于:在使用固定在磁珠上的抗原的淘选或使用固定在板上的抗原的淘选中,由于是回收所有在酸性条件下从磁珠或板上解离的噬菌体,所以即使是ph依赖性弱的克隆的噬菌体也被回收,最终被浓缩的克隆中含有具强ph依赖性的克隆的可能性小。于是,作为更严格的条件下的淘选方法,研究了使用固定有抗原的柱的淘选(图5)。迄今为止,尚无利用使用了固定有抗原的柱的淘选获得具有ph依赖性结合能力的克隆的报道。认为在使用固定有抗原的柱的淘选中,使在中性条件下结合的噬菌体在酸性条件下洗脱时,ph依赖性弱的克隆在柱内与抗原再次结合而不易被洗脱,而ph依赖性强、在柱内不易发生再次结合的克隆选择性地从柱上被洗脱。在使用固定在磁珠上的抗原的淘选或使用固定在板上的抗原的淘选中,虽然是回收“所有”在酸性条件下解离的噬菌体,但在使用固定有抗原的柱的淘选中,通过使酸性条件的缓冲液流经柱而开始洗脱,只回收适当的组分,从而认为可以选择性地回收具强ph依赖性结合能力的噬菌体。首先,制作固定有作为抗原的sr344的柱。用1mlpbs清洗200μl链霉亲和素sepharose(gehealthcare),之后将其悬浮于500μlpbs中,使其与400pmol生物素标记sr344在室温下接触1小时。之后,向空柱(amershampharmciabiotech)中充填上述sepharose,用约3ml的pbs清洗柱。用0.5%bsa-pbs(ph7.4)将上述进行了peg沉淀的文库噬菌体稀释至1/25,通过0.45nm滤器后添加到柱中。用约6ml的pbs(ph7.4)清洗后,使50mmmes-nacl(ph5.5)流过,达到低ph则洗脱解离的抗体。回收适当的洗脱组分,将回收的噬菌体溶液添加到10ml对数增殖期(od6000.4-0.5)xl1-blue中,在37℃下静置30分钟使之感染。将感染的大肠杆菌接种在2yt/100μg/ml氨苄青霉素/2%葡萄糖的225mm×225mm的板上。再次从该大肠杆菌开始培养,与上述同样进行噬菌体的培养,重复进行6次淘选。利用噬菌体elisa进行评价利用噬菌体elisa进行所得噬菌体的评价。对于明显确认到ph依赖性的克隆,使用特异性引物进行序列分析。其结果,得到多个与wt相比明显看出ph依赖性结合的克隆。如图6所示,与wt相比,克隆cl5(h链clh5、l链cll5)(clh5:氨基酸序列seqidno:5、cll5:氨基酸序列seqidno:8)确认到特别强的ph依赖性结合。由此可知:在通常的使用固定在磁珠上的抗原的淘选中未获得的、显示出强ph依赖性结合的抗体,通过使用固定有抗原的柱的淘选可以获得,作为从文库中获得ph依赖性结合抗体的方法,使用固定有抗原的柱的淘选非常有效。发现ph依赖性结合的多个克隆的氨基酸序列分析的结果,在浓缩的克隆中以高概率成为组氨酸的位点见表4。[表4]利用噬菌体文库(柱淘选)得到的组氨酸取代位点h31,h50,h58,h62,h63,h65,h100b,h102l24,l27,l28,l32,l53,l56,l90,l92,l94[实施例4]人源化il-6受体抗体的组氨酸修饰体的表达和纯化人源化il-6受体抗体的组氨酸修饰抗体的表达载体的制作、表达和纯化为了使通过噬菌体elisa明显确认到ph依赖性的克隆进行igg化,通过pcr分别扩增vh和vl,通过xhoi/nhei消化和ecori消化插入到动物细胞表达用载体中。各dna片段的核苷酸序列按照本领域技术人员公知的方法进行确定。将h链使用clh5、l链使用实施例2中得到的l73的clh5/l73以igg的形式进行表达、纯化。表达、纯化按照实施例1中记载的方法进行。利用突变位点的组合,制作具有更高ph依赖性的抗体。根据噬菌体文库中his被浓缩的位点、结构信息等,将实施例2中作为h链得到的h3pi中的h32、h58、h62、h102取代成组氨酸、并将h95取代成缬氨酸、将h99取代成异亮氨酸,制作h170(seqidno:4)。修饰体的制作按照实施例1中记载的方法进行。另外,将实施例2作为l链制作的l73的第28位的组氨酸取代成天冬氨酸,制作l82(seqidno:7)。修饰体的制作按照实施例1记载的方法进行。按照实施例1记载的方法,将h链使用h170、l链使用l82的h170/l82以igg的形式进行表达、纯化。[实施例5]评价ph依赖性结合抗体的il-6r中和活性评价igg化克隆的人il-6受体中和活性对于人源化pm1抗体(野生型:wt)和实施例2、4中制作的h3pi/l73、clh5/l73、h170/l82这4种,评价il-6受体中和活性。具体而言,使用显示出il-6/il-6受体依赖性增殖的baf3/gp130,评价il-6受体中和活性。将baf3/gp130用含有10%fbs的rpmi1640培养基清洗3次,之后悬浮于含有60ng/ml人白介素-6(toray)、60ng/ml重组可溶型人il-6受体(sr344)和10%fbs的rpmi1640培养基中,使达到5×104细胞/ml的密度,再向96孔平板(corning)的各孔中分别注入50μl。接下来,用含有10%fbs的rpmi1640培养基稀释已纯化的抗体,向各孔中分别混合50μl。在37℃、5%co2的条件下培养3天,以20μl/孔加入用pbs稀释至两倍的wst-8试剂(细胞计数试剂盒8;株式会社同仁化学研究所),之后立即使用sunriseclassic(tecan)测定450nm的吸亮度(参比波长为620nm)。培养2小时后,再次测定450nm的吸亮度(参比波长为620nm),以2小时的吸亮度变化为指标,评价il-6受体中和活性。其结果,如图7所示,与人源化pm1抗体(野生型:wt)相比,h3pi/l73、clh5/l73、h170/l82具有同等的生物学中和活性。[实施例6]ph依赖性结合抗体的biacore分析分析ph依赖性结合克隆与可溶型il-6受体的结合对于人源化pm1抗体(野生型:wt)和实施例2、4中制作的h3pi/l73、clh5/l73、h170/l82这4种,使用biacoret100(gehealthcare)进行ph5.8和ph7.4下的抗原抗体反应的速度论的分析(缓冲液为10mmmes、ph7.4或ph5.8、150mmnacl、0.05%tween20)。利用胺偶联法使各种抗体结合在固定有重组proteina/g(pierce)的传感器芯片上,向其中注入以分析物形式制备成9.8-400nm浓度的sr344。实时观测ph依赖性结合克隆与sr344的结合和解离(图8和图9)。所有测定均在37℃下进行。使用biacoret100评估软件(gehealthcare)计算结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表5)。再算出各自的在ph5.8和ph7.4下的亲和力比,评价ph依赖性结合。所有测定均在37℃下进行。计算各自的在ph5.8和ph7.4下的亲和力比,结果,h3pi/l73、h170/l82、clh5/l73与sr344的ph依赖性结合(亲和力)分别为41倍、394倍、66倍,与wt相比,所有克隆均显示出15倍以上的高的ph依赖性结合。迄今为止,尚无在血浆中的ph即ph7.4下与抗原强力结合、在核内体内的ph即ph5.5-ph6.0下与抗原微弱结合的抗il-6受体抗体的报道。在本次研究中,得到了维持与wt的人源化il-6受体抗体同等的生物学中和活性和ph7.4下的亲和力不变、只特异性地使ph5.8下的亲和力下降10倍以上的抗体。[表5]对于sr344的ph依赖性结合克隆与可溶型il-6受体的结合比较分析ph依赖性结合克隆与膜型il-6受体的结合对于制作的上述ph依赖性结合克隆,使用biacoret100(gehealthcare),观测其在ph5.8、ph7.4下与膜型il-6受体的抗原抗体反应。通过评价其与固定在传感器芯片上的il-6受体的结合,评价其与膜型il-6受体的结合。按照本领域技术人员公知的方法将sr344生物素化,利用链霉亲和素与生物素的亲和性,经由链霉亲和素将生物素化sr344固定在传感器芯片上。所有测定均在37℃下进行,流动相的缓冲液为10mmmesph5.8、150mmnacl、0.05%tween20,在ph7.4的条件下向其中注入ph依赖性结合克隆,使其与sr344结合(注入样品的缓冲液为10mmmesph7.4、150mmnacl、0.05%tween20),之后在流动相的ph5.8下观测各克隆的ph依赖性解离(图10)。使样品浓度为0.5μg/ml,使用biacoret100评估软件(gehealthcare),只拟合在10mmmesph7.4、150mmnacl、0.05%tween20中结合、在10mmmesph5.8、150mmnacl、0.05%tween20中解离时的ph5.8下的解离相,由此算出在ph5.8下的解离速度(kd(1/s))。同样,使样品浓度为0.5μg/ml,使用biacoret100评估软件(gehealthcare),只拟合在10mmmesph7.4、150mmnacl、0.05%tween20中结合、在10mmmesph7.4、150mmnacl、0.05%tween20中解离时的ph7.4下的解离相,由此算出ph7.4下的解离速度常数(kd(1/s))。各克隆的ph依赖性解离速度常数见表6。[表6]比较对于sr344的ph依赖性结合克隆自膜型il-6受体上解离的解离速度常数解离度的ph依赖性的大小依次为h3pi/l73、clh5/l73、h170/l82,所有克隆均显示出高于wt的ph依赖性的从膜型il-6受体上的解离。但ph依赖性结合、解离的顺序在可溶型il-6受体和膜型il-6受体中有所不同。由结果可知:在与可溶型il-6受体的结合分析中显示出最高的ph依赖性结合的h170/l82,在与膜型il-6受体的结合分析中显示出最低的ph依赖性结合。已知通常igg分子以一价(亲和力)与可溶型抗原结合,相对于此,igg分子以二价(亲合力)与膜型抗原结合。在这样的可溶型抗原和膜型抗原中,认为结合方式的不同对h170/l82的ph依赖性结合有影响。[实施例7]确认ph依赖性结合抗体与抗原的多次结合如实施例2所述,认为ph依赖性结合抗体可以与抗原多次结合。即,抗原所结合的ph依赖性结合抗体非特异性地进入核内体内,在核内体内的酸性条件下自可溶型抗原上解离。抗体通过与fcrn结合而再次返回到血浆中,由于返回到血浆中的抗体上未结合有抗原,所以该抗体可以再次与新的抗原结合。通过反复进行该过程,ph依赖性结合抗体可以多次与抗原结合。但是,关于不具有ph依赖性结合的igg抗体,不会出现在核内体的酸性条件下所有抗原都自抗体上解离的情况,所以通过fcrn返回到血浆中的抗体保持着结合抗原的状态,其无法再次与新的抗原结合。因此,在大部分情况下,1分子的igg抗体只能中和2个抗原(以二价进行结合的情况)。于是,与人源化pm1抗体(野生型:wt)相比,评价实施例2、4中制作的h3pi/l73、clh5/l73、h170/l82这3种ph依赖性结合抗体能否与作为抗原的sr344多次结合。通过biacore(gehealthcare)评价:通过在ph7.4下结合、在ph5.8下解离,从而可以与抗原多次结合的情况。利用胺偶联法使将要评价的抗体与固定有重组proteina/g(pierce)的传感器芯片结合,使ph7.4的流动相流过(步骤1)。使调整至ph7.4的sr344溶液以分析物的形式流过,在ph7.4下使sr344与抗体结合(步骤2)。该ph7.4下的结合是在模仿在血浆中与抗原的结合。之后,仅使调整至ph5.8的缓冲液(不含sr344的溶液)以分析物的形式流过,使与抗体结合的抗原暴露在酸性条件下(步骤3)。该ph5.8下的解离是在模仿在核内体内的抗体抗原复合体的结合状态。之后,再次进行步骤2。这是在模仿通过fcrn返回到血浆中的抗体再次与新的抗原结合。之后,再次进行步骤2,使抗体抗原复合体暴露于酸性条件下。通过如此地在37℃下多次重复“步骤2→步骤3”,可以模仿体内的状态:重复进行抗体自血浆中通过胞饮作用进入核内体内、再通过fcrn返回到血浆中的过程(natrevimmunol.2007sep;7(9):715-25)。对于制作的上述ph依赖性结合克隆,使用biacoret100(gehealthcare)分析在ph5.8、ph7.4下与抗原sr344的多次结合能力。具体如下进行。所有测定均在37℃下进行,首先,利用胺偶联法使上述的样品抗体结合在固定有重组proteina/g(pierce)的传感器芯片上,流动相的缓冲液为10mmmesph5.8、150mmnacl、0.05%tween20(步骤1)。在ph7.4的条件下,用3分钟向其中注入以分析物的形式制备成约40nm浓度的sr344(注入sr344的缓冲液为10mmmesph7.4、150mmnacl、0.05%tween20),使之结合(步骤2)。之后,停止注入sr344,使ph5.8的流动相流经约70秒,从而将抗体/sr344复合体暴露在酸性条件下(步骤3)。以该结合(步骤2)和酸性暴露(步骤3)作为1个组合(set),连续重复进行10个组合,实时观测该传感图,见图11。wt由于在步骤3的酸性暴露时sr344的解离少,所以在接下来的步骤2中能够与新的抗原结合的抗体的比例非常少。相对于此,可知ph依赖性结合克隆、其中特别是h170/l82和clh5/l73在步骤3的酸性暴露时的解离非常大,所结合的sr344基本上都发生解离,所以在接下来的步骤2中抗体基本上都能与新的抗原结合。由结果可知:即使重复进行10个组合的结合(步骤2)和酸性暴露(步骤3),h170/l82和clh5/l73在每个组合中基本上都能与新的抗原结合。使用所得的传感图,利用biacoret100评估软件(biacore)计算各样品在每一组合中的sr344结合量,10个组合的经时性累积值见图12。第10个组合得到的累积ru值相当于10次循环中结合的总抗原量。结果显示:与wt相比,ph依赖性结合克隆、其中特别是h170/l82和clh5/l73结合的总抗原量最多,与wt相比,能够与4倍量左右的抗原反复结合。由此可知:通过对wt赋予ph依赖性结合,可以反复与抗原结合,中和多个抗原。[实施例8]利用人il-6受体转基因小鼠进行的ph依赖性结合抗体的pk/pd试验il-6受体在体内以可溶型il-6受体和膜型il-6受体两种形式存在(natclinpractrheumatol.2006nov;2(11):619-26)。抗il-6受体抗体与可溶型il-6受体和膜型il-6受体结合,中和它们的生物学作用。认为抗il-6受体抗体与膜型il-6受体结合后,保持着与膜型il-6受体结合的状态,通过内化作用摄入细胞内的核内体中,之后抗il-6受体抗体保持着与膜型il-6受体结合的状态向溶酶体移动,一起被溶酶体分解。实施例6中评价的ph依赖性结合抗il-6受体抗体即h3pi/l73、clh5/l73、h170/l82只要能够在核内体内的酸性条件下解离、从而经由fcrn返回到血浆中,则返回到血浆中的抗体可以再次与抗原结合,可以以1分子抗体中和多个膜型il-6受体。已制作的ph依赖性结合抗il-6受体抗体能否实现在核内体内的酸性条件下解离、从而经由fcrn返回到血浆中,而这可以评价这些抗体的药物动力学与wt相比是否有所改善。于是,评价人源化pm1抗体(野生型:wt)和实施例2、4中制作的h3pi/l73、clh5/l73、h170/l82这4种在人il-6受体转基因小鼠(hil-6rtg小鼠、procnatlacadsciusa.1995may23;92(11):4862-6)体内的药物动力学。将wt和h3pi/l73、clh5/l73、h170/l82以25mg/kg对hil-6rtg小鼠进行静脉内单剂量给药,给药前和给药后经时性采血。采集的血液立即在4℃下以15,000rpm的转速离心15分钟,得到血浆。分离的血浆在实施测定前一直保存在被设定为-20℃以下的冷库中。小鼠血浆中浓度测定按照elisa法进行。制备血浆中浓度为6.4、3.2、1.6、0.8、0.4、0.2、0.1μg/ml的校准曲线试样。将校准曲线试样和小鼠血浆测定试样分别注入用抗人igg(γ-链特异性)f(ab’)2(sigma社制)固定的多孔板(immunoplate)(nunc-immunoplate,maxisorp(nalgenuncinternational社制))中,在室温下静置1小时,之后使山羊抗人igg-biot(southernbiotechnologyassociates社制)和链霉亲和素碱性磷酸酶缀合物(rochediagnostics社制)依次反应,使用bluephosmicrowell磷酸酶底物系统(kirkegaard&perrylaboratories社制)作为底物进行显色反应,使用酶标仪(microplatereader)测定650nm的吸亮度。使用分析软件softmaxpro(moleculardevices社制),由校准曲线的吸亮度算出小鼠血浆中浓度。wt和h3pi/l73、clh5/l73、h170/l82的血浆中浓度变化见图13。与wt相比,h3pi/l73、clh5/l73、h170/l82的药物动力学均有所改善。其中,h3pi/l73和clh5/l73的药物动力学大幅改善。与膜型il-6受体结合的天然型抗il-6受体抗体(wt)通过内化作用摄入细胞内的核内体中,保持着与抗原结合的状态移动到溶酶体中被分解,因此血浆中滞留性短。相对于此,ph依赖性结合抗il-6受体抗体的药物动力学大幅改善,由此认为:ph依赖性结合抗il-6受体抗体在核内体内的酸性条件下自作为抗原的膜型il-6受体上解离,从而经由fcrn再次返回到血浆中。与wt相比,h3pi/l73、clh5/l73、h170/l82的药物动力学均有所改善,但h170/l82的血浆中滞留性延长效果较h3pi/l73、clh5/l73的小。认为igg分子通常以二价与膜型抗原结合,由此认为抗il-6受体抗体也以二价(亲合力)与膜型il-6受体结合,之后被内化。如实施例6所示,在利用biacore进行的分析中可知:170/l82在与可溶型il-6受体结合时,在ph5.8下快速地自il-6受体上解离(图9),但其与膜型il-6受体结合时,在ph5.8下自il-6受体上解离的速度非常慢(图10)。由此认为:h170/l82的血浆中滞留性延长效果小的原因在于,其与膜型il-6受体结合时在ph5.8下的解离慢,所以被内化后在核内体内无法充分解离。即,相对于膜型抗原,1个igg分子中和多个膜型抗原,由此可知:与以一价进行结合(亲和力)时的ph依赖性相比,以二价进行结合(亲合力)后的解离的ph依赖性更重要。[实施例9]利用食蟹猴进行的ph依赖性结合抗体的pk/pd试验在实施例8中,ph依赖性结合抗il-6受体抗体的药物动力学大幅改善,由此认为:ph依赖性结合抗il-6受体抗体在核内体内的酸性条件下自作为抗原的膜型il-6受体上解离,从而经由fcrn再次返回到血浆中。如果再次返回到血浆中的抗体能够再次与膜型il-6受体结合,则与天然型抗il-6受体抗体相比,认为ph依赖性结合抗il-6受体抗体在相同给药量下更长时间地持续中和作为抗原的膜型il-6受体。另外,il-6受体中还存在可溶型il-6受体,由此认为:有关可溶型il-6受体的中和,在相同给药量下也更长时间地持续中和。评价wt和h3pi/l73在食蟹猴体内的药物动力学。将wt和h3pi/l73以1mg/kg对食蟹猴进行静脉内单剂量给药,投与前和给药后经时性进行采血。采集的血液立即在4℃下以15,000rpm的转速离心15分钟,得到血浆。分离的血浆在实施测定前一直保存在被设定为-20℃以下的冷库中。食蟹猴血浆中浓度测定按照elisa法进行。首先,将抗人igg(γ-链特异性)f(ab’)2抗体片段(sigma社制)分别注入nunc-immuno板、maxisorp(nalgenuncinternational社制)中,在4℃下静置一夜,制作抗人igg固相化板。制备血浆中浓度为3.2、1.6、0.8、0.4、0.2、0.1、0.05μg/ml的校准曲线试样和稀释了100倍以上的食蟹猴血浆测定试样。向100μl这些校准曲线试样和血浆测定试样中加入200μl20ng/ml的食蟹猴il-6r,在室温下静置1小时。之后,分别注入到抗人igg固相化板上,进一步在室温下静置1小时。之后,使生物素化抗人il-6r抗体(r&d社制)在室温下反应1小时,再使链霉亲和素-polyhrp80(stereospecificdetectiontechnologies社制)在室温下反应1小时,使用tmbonecomponenthrpmicrowellsubstrate(biofxlaboratories社制)作为底物进行显色反应,利用1n-硫酸(showachemical社制)使反应停止,之后用酶标仪测定450nm的吸亮度。使用分析软件softmaxpro(moleculardevices社制),由校准曲线的吸亮度算出食蟹猴血浆中浓度。wt和h3pi/l73的静脉内给药后的血浆中浓度变化见图14。其结果,即使在食蟹猴体内,也与人il-6受体转基因小鼠一样,与wt相比,h3pi/l73的药物动力学大幅改善。作为ph依赖性结合抗il-6受体抗体的h3pi/l73的药物动力学大幅改善,由此认为:h3pi/l73在核内体内的酸性条件下自作为抗原的膜型il-6受体上解离,从而经由fcrn再次返回到血浆中。为了评价通过静脉内给予wt和h3pi/l73时食蟹猴膜型il-6受体被中和的程度,研究检体对由食蟹猴il-6诱导的血浆c反应性蛋白(crp)的影响。一旦il-6与膜型il-6受体结合则分泌crp,所以crp成为膜型il-6受体的中和指标。在给予wt和h3pi/l73后第3天-第10天,以5μg/kg在腰背部连日皮下给予含有1%灭活食蟹猴血浆的食蟹猴il-6(实施例1中制作的食蟹猴il-6)。从即将开始给予食蟹猴il-6之前(第3天)起到给药后24小时间隔(第4天-第11天)均从隐静脉采血,分离血浆。利用ciasrcrp(关东化学株式会社),使用自动分析装置(tba-120fr、东芝medicalsystems株式会社)测定各个体的crp浓度。wt和h3pi/l73的由食蟹猴il-6诱导时的crp浓度变化见图15。其结果,与wt相比,h3pi/l73的crp抑制期间大幅延长。由此认为:作为ph依赖性结合抗il-6受体抗体的h3pi/l73在核内体内的酸性条件下自作为抗原的膜型il-6受体上解离,从而经由fcrn再次返回到血浆中,通过再次与膜型il-6受体结合以进行中和,与wt相比长时间地抑制crp的产生。即,结果显示:h3pi/l73可以以1分子的抗体多次与膜型il-6受体结合以进行中和。与wt相比,h3pi/l73抑制crp的产生的时间延长,由此表明:与wt相比,h3pi/l73延长了作为抗原的膜型il-6受体被抗体结合的时间。为了评价通过静脉内给予wt和h3pi/l73时食蟹猴可溶型il-6受体被中和的程度,测定食蟹猴血浆中的非结合型食蟹猴可溶型il-6受体浓度。将30μl食蟹猴的血浆添加到在0.22μm的滤杯(millipore)中已干燥的适量的rproteinasepharosefastflow(gehealthcare)树脂中,使血浆中存在的所有igg型抗体(食蟹猴igg、抗人il-6受体抗体和抗人il-6受体抗体-食蟹猴可溶型il-6受体复合体)均吸附在蛋白a上。之后,用高速离心机进行旋转下降(spinned-down),回收通过的溶液(以下记作“通过溶液(passsolution)”)。由于通过溶液中不含与蛋白a结合的抗人il-6受体抗体-食蟹猴可溶型il-6受体复合体,所以通过测定蛋白a通过溶液中的食蟹猴可溶型il-6受体浓度,可以测定非结合型的可溶型il-6受体浓度。向制备成4000、2000、1000、500、250、125、62.5pg/ml的食蟹猴il-6受体校准曲线试样和上述的进行了蛋白a处理的血浆样品中混合用sulfo-tagnhsester(mesoscalediscovery社制)进行了钌化的单克隆抗人il-6r抗体(r&d社制)和生物素化的抗人il-6r抗体(r&d社制),在室温下反应1小时。之后,将其分别注入到sa包被的校准ma240096孔板(mesoscalediscovery社制)上。再于室温下反应1小时,清洗后分别注入readbuffert(×4)(mesoscalediscovery社制),立即用sectorimager2400(mesoscalediscovery社制)进行测定。使用分析软件softmaxpro(moleculardevices社制),由校准曲线的响应值算出食蟹猴il-6受体浓度。wt和h3pi/l73的非结合型的食蟹猴可溶型il-6受体浓度变化见图16。其结果,与wt相比,h3pi/l73的食蟹猴可溶型il-6受体的中和期间大幅延长。由此认为:作为ph依赖性结合抗il-6受体抗体的h3pi/l73在核内体内的酸性条件下自作为抗原的可溶型il-6受体上解离,经由fcrn再次返回到血浆中,再次与可溶型il-6受体结合以进行中和。与wt相比,h3pi/l73的抑制非结合型的食蟹猴可溶型il-6受体的时间延长,由此显示:与wt相比,h3pi/l73延长了作为抗原的可溶型il-6受体被抗体结合的时间。由此发现:相对于野生型抗il-6受体抗体,在血浆中的ph即ph7.4下与抗原强力结合、在核内体内的ph即ph5.8下减弱与抗原的结合的ph依赖性结合抗il-6受体抗体,其直至抗体从血浆中消除的时间、以及体内的可溶型il-6受体和膜型il-6受体被抗体结合的时间大幅延长。由此,可以减少对患者的给药量或给药频率,其结果,可以减少总给药量,因此认为ph依赖性结合抗il-6受体抗体作为il-6拮抗剂的药品特别优异。[实施例10]通过可变区的最优化提高与膜型il-6受体的ph依赖性结合可变区h3pi/l73和clh5/l82的最优化实施例9中显示:具有ph依赖性结合能力的抗体发挥优异的效果,所以为了进一步提高ph依赖性结合能力,向实施例3中得到的clh5的cdr序列中导入突变,制作vh1-igg1(seqidno:21)、vh2-igg1(seqidno:22)。另外,向h3pi的构架序列和cdr序列中导入突变,制作作为修饰h链的vh3-igg1(seqidno:23)、vh4-igg1(seqidno:24)。向l73、l82的cdr序列中导入突变,制作作为修饰l链的vl1-ck(seqidno:25)、vl2-ck(seqidno:26)、vl3-ck(seqidno:27)。具体而言,使用quikchange位点定向诱变试剂盒(stratagene),按照附录说明书记载的方法制作突变体,将得到的质粒片段插入哺乳动物细胞表达载体中,制作目标h链表达载体和l链表达载体。所得的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。以h链使用vh2-igg1(seqidno:22)、l链使用vl2-ck(seqidno:26)的抗体作为fv1-igg1,以h链使用vh1-igg1(seqidno:21)、l链使用l82的抗体作为fv2-igg1,以h链使用vh4-igg1(seqidno:24)、l链使用vl1-ck(seqidno:25)的抗体作为fv3-igg1,以h链使用vh3-igg1(seqidno:23)、l链使用vl3-ck(seqidno:27)的抗体作为fv4-igg1。这些抗体中,进行fv2-igg1和fv4-igg1的表达、纯化。表达、纯化按照实施例1中记载的方法进行。分析ph依赖性结合克隆与可溶型il-6受体的结合针对人源化pm1抗体(野生型:wt)、以及实施例2和10中制作的wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1这4种抗体,使用biacoret100(gehealthcare)进行ph7.4下的抗原抗体反应的速度论的分析(缓冲液为10mmmesph7.4、150mmnacl、0.05%tween20)。利用胺偶联法使各种抗体结合在固定有抗iggγ链特异性f(ab)2(pierce)的传感器芯片上,向其中注入以分析物形式制备成9.8-40nm浓度的sr344。实时观测ph依赖性结合克隆与sr344的结合和解离。所有测定均在37℃下进行。使用biacoret100评估软件(gehealthcare),计算结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表7)。[表7]对于sr344的ph依赖性结合克隆自可溶型il-6受体上的解离速度常数比较算出各自的ph7.4的亲和力,结果,对于sr344的wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1的解离常数(亲和力、kd值)分别为2.7nm、1.4nm、2.0nm、1.4nm,几乎是同等的值。结果显示:fv2-igg1、fv4-igg1与可溶型il-6受体的结合能力与wt为同等以上。分析ph依赖性结合克隆与膜型il-6受体的结合针对制作的wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1这4种抗体,使用biacoret100(gehealthcare)观测在ph5.8、ph7.4下与膜型il-6受体的抗原抗体反应。通过评价上述抗体与固定在传感器芯片上的il-6受体的结合,来评价它们与膜型il-6受体的结合。按照本领域技术人员公知的方法将sr344生物素化,利用链霉亲和素与生物素的亲和性,经由链霉亲和素将生物素化sr344固定在传感器芯片上。所有测定均在37℃下进行,流动相的缓冲液为10mmmesph5.8、150mmnacl、0.05%tween20,在ph7.4的条件下向其中注入ph依赖性结合克隆,使之与sr344结合后(注入样品的缓冲液为10mmmesph7.4、150mmnacl、0.05%tween20),观测在流动相的ph5.8下各克隆的ph依赖性解离(图17)。使样品浓度为0.25μg/ml,通过使用biacoret100评估软件(gehealthcare)只拟合使之在10mmmesph7.4、150mmnacl、0.05%tween20中结合、在10mmmesph5.8、150mmnacl、0.05%tween20中解离时的ph5.8下的解离相,算出ph5.8下的解离速度常数(kd(1/s))。同样,使样品浓度为0.25μg/ml,通过使用biacoret100评估软件(gehealthcare)只拟合使之在10mmmesph7.4、150mmnacl、0.05%tween20中结合、在10mmmesph7.4、150mmnacl、0.05%tween20中解离时的ph7.4下的解离相,算出ph7.4下的解离速度常数(kd(1/s))。各克隆的ph依赖性解离速度常数见表8。[表8]对于sr344的ph依赖性结合克隆自膜型il-6受体上的解离速度常数比较算出各自的ph依赖性,结果,对于sr344的wt、h3pi/l73-igg1、fv2-igg1、fv4-igg1这4种抗体与膜型il-6受体的结合的ph依赖性分别为1.0倍、2.59倍、7.18倍、5.56倍,fv2-igg1、fv4-igg1显示出高于h3pi/l73-igg1的ph依赖性的自膜型il-6受体上的解离。由以上可知:fv2-igg1、fv4-igg1维持着与wt同等以上的对可溶型il-6受体的亲和力,同时显示出强于h3pi/l73-igg1的与膜型il-6受体的ph依赖性结合。[实施例11]利用人il-6受体转基因小鼠进行的可变区最优化的ph依赖性结合抗体的pk/pd试验利用在实施例8中使用的人il-6受体转基因小鼠,评价实施例10中制作、评价的fv2-igg1与fv4-igg1以及wt与h3pi/l73-igg1的药物动力学。将wt和h3pi/l73-igg1、fv2-igg1、fv4-igg1以25mg/kg对hil-6rtg小鼠进行静脉内单剂量给药,与实施例8同样地进行血浆中各抗体的浓度的测定。wt和h3pi/l73-igg1、fv2-igg1、fv4-igg1的血浆中浓度变化见图18。与实施例8一样,与wt相比,h3pi/l73-igg1的药物动力学提高,并且fv2-igg1和fv4-igg1的药物动力学较h3pi/l73-igg1进一步提高。有关在实施例9中在食蟹猴体内测定的非结合型il-6受体浓度,按照同样的方法在本试验的hil-6rtg小鼠中进行测定时,也确认到:fv2-igg1和fv4-igg1的可溶型il-6受体的中和期间较h3pi/l73-igg1有所延长(数据未显示)。如实施例10所示,与h3pi/l73-igg1相比,fv2-igg1和fv4-igg1与膜型il-6受体的ph依赖性结合有所提高,由此显示:通过提高与膜型il-6受体的ph依赖性结合,可以使药物动力学和可溶型il-6受体的中和期间较h3pi/l73-igg1进一步提高。[实施例12]通过恒定区的最优化提高与膜型il-6受体的ph依赖性结合fv4-igg1的恒定区的最优化有报道称:通常抗体与膜型抗原的结合根据抗体的恒定区而发生变化(jimmunolmethods.1997jun23;205(1):67-72)。迄今为止制作的ph依赖性结合抗体的恒定区为igg1同种型。于是,为了提高与膜型il-6受体的ph依赖性结合,研究恒定区的最优化。向作为天然型恒定区的恒定区igg2(seqidno:28)中导入突变,制作恒定区igg2δgk(seqidno:29)。进一步向恒定区igg2δgk中导入突变,制作恒定区m58(seqidno:30)。向恒定区igg2和恒定区m58中进一步导入突变,制作恒定区m71(seqidno:31)和m73(seqidno:32)。制作将实施例10中制作的vh3-igg1的恒定区取代成igg2δgk的vh3-igg2δgk(seqidno:33)、将恒定区取代成m58的vh3-m58(seqidno:34)、将恒定区取代成m73的vh3-m73(seqidno:35)。具体而言,将实施例10中使用的vh3的恒定区部分通过nhei/noti消化和连接取代成目标恒定区,来构建表达载体。所得的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。进行下述抗体的表达、纯化:h链使用vh3-igg2δgk(seqidno:33)、l链使用vl3-ck(seqidno:27)的fv4-igg2;h链使用vh3-m58(seqidno:34)、l链使用vl3-ck(seqidno:27)的fv4-m58;以及h链使用vh3-m73(seqidno:35)、l链使用vl3-ck(seqidno:27)的fv4-m73。表达、纯化按照实施例1记载的方法进行。分析恒定区最优化的fv4与可溶型il-6受体的结合关于制作的fv4-igg1、fv4-igg2、fv4-m58、fv4-m73和wt,按照与实施例10相同的方法实时观测其与sr344的结合和解离。同样地进行分析,算出结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表9)。[表9]对于sr344的ph依赖性结合克隆自可溶型il-6受体上的解离速度常数比较算出各自的在ph7.4下的亲和力,结果,对于sr344的fv4-igg1、fv4-igg2、fv4-m58、fv4-m73的解离常数(亲和力、kd值)分别为1.4nm、1.3nm、1.4nm、1.4nm,几乎是同等的值,显示:即使修饰恒定区,对于sr344的ph依赖性结合克隆与可溶型il-6受体的结合能力也未发生变化。由此认为:关于fv1、fv2、fv3,即使同样地修饰恒定区,其与可溶型il-6受体的结合能力也未发生变化。分析恒定区最优化的fv4与膜型il-6受体的结合关于制作的fv4-igg1、fv4-igg2、fv4-m58、fv4-m73和wt,按照与实施例10相同的方法,使用biacoret100(gehealthcare)观测在ph5.8、ph7.4下与膜型il-6受体的抗原抗体反应。在ph7.4的条件下注入ph依赖性结合克隆,使之与sr344结合后,观测在ph5.8的流动相中各克隆的ph依赖性解离,结果见图19。进一步按照与实施例10相同的方法进行分析,各克隆的ph依赖性解离速度见表10。[表10]对于sr344的ph依赖性结合克隆自膜型il-6受体上的解离速度常数比较计算各自的ph依赖性,结果,对于sr344的fv4-igg1、fv4-igg2、fv4-m58、fv4-m73的ph依赖性分别为5.6倍、17.0倍、17.6倍、10.1倍,fv4-igg2、fv4-m58、fv4-m73均显示出高于fv4-igg1的ph依赖性的自膜型il-6受体上的解离。根据与使用了fv4的可变区的可溶型il-6受体的结合分析结果和与膜型il-6受体的结合分析结果,可以发现:通过将恒定区由igg1取代成igg2、m58和m73,可以在不改变与可溶型il-6受体的亲和力的情况下,只改善与膜型il-6受体的ph依赖性结合。此外,认为fv1、fv2、fv3也是同样的情形。[实施例13]利用人il-6受体转基因小鼠进行的恒定区最优化的ph依赖性结合抗体的pk/pd试验利用实施例8中使用的人il-6受体转基因小鼠(hil-6rtg小鼠),评价实施例13中制作的fv4-igg1、fv4-igg2、fv4-m58的药物动力学,研究恒定区对药物动力学的影响。将wt和fv4-igg1、fv4-igg2、fv4-m58以25mg/kg对hil-6rtg小鼠进行静脉内单剂量给药,与实施例8同样地进行血浆中各抗体的浓度的测定。wt和fv4-igg1、fv4-igg2、fv4-m58的血浆中浓度变化见图20。与实施例11一样,与wt相比,fv4-igg1的药物动力学提高,并且fv4-igg2、fv4-m58的药物动力学较fv4-igg1也有所提高。关于实施例9中在食蟹猴体内测定的非结合型il-6受体浓度,按照同样的方法在本试验的hil-6rtg小鼠中测定时,也确认到:与fv4-igg1相比,fv4-igg2、fv4-m58的可溶型il-6受体的中和期间延长(数据未显示)。如实施例10所示,与fv4-igg1相比,fv4-igg2、fv4-m58与膜型il-6受体的ph依赖性结合提高,由此显示:通过将恒定区由igg1取代成igg2或m58,可以提高与膜型il-6受体的ph依赖性结合,并提高药物动力学和可溶型il-6受体的中和期间。由此认为:不仅是在fv4中,在fv1、fv2、fv3中通过将恒定区由igg1取代成igg2或m58,与igg1相比,药物动力学和可溶型il-6受体的中和期间也有所提高。[实施例14]可变区和恒定区最优化的ph依赖性结合抗体的制作使用与现有方法相同的方法,制作vh2-igg1的恒定区为m71、m73的vh2-m71(seqidno:36)、vh2-m73(seqidno:37)、以及vh4-igg1的恒定区为m71、m73的vh4-m71(seqidno:38)、vh4-m73(seqidno:39)。进行下述抗体的表达、纯化:h链使用vh2-m71、l链使用vl2-ck的fv1-m71;h链使用vh2-m73、l链使用vl2-ck的fv1-m73;h链使用vh4-m71、l链使用vl1-ck的fv3-m71;以及h链使用vh4-m73、l链使用vl1-ck的fv3-m73。表达、纯化按照实施例1所述的方法进行。分析可变区和恒定区最优化的ph依赖性结合抗体与可溶型il-6受体的结合对于人源化pm1抗体(野生型:wt)和目前为止制作的h3pi/l73-igg1、fv1-m71、fv1-m73、fv2-igg1、fv3-m71、fv3-m73、fv4-igg1、fv4-igg2、fv4-m58、fv4-m73这11种抗体,按照与实施例10相同的方法实时观测其与sr344的结合和解离。同样地进行分析,算出结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表11)。[表11]对于sr344的ph依赖性结合克隆自可溶型il-6受体上的解离速度常数比较由结果可以发现:与wt相比,得到的10种ph依赖性结合克隆均对可溶型il-6受体具有同等以上的解离常数(亲和力、kd值)。分析可变区和恒定区最优化的ph依赖性结合抗体与膜型il-6受体的结合对于人源化pm1抗体(野生型:wt)和目前为止制作的h3pi/l73-igg1、fv1-m71、fv1-m73、fv2-igg1、fv3-m71、fv3-m73、fv4-igg1、fv4-igg2、fv4-m58、fv4-m73这11种抗体,按照与实施例10相同的方法,使用biacoret100(gehealthcare)观测在ph5.8、ph7.4下与膜型il-6受体的抗原抗体反应。在ph7.4的条件下注入ph依赖性结合克隆,使之与sr344结合后,观测在流动相的ph5.8下各克隆的ph依赖性解离,结果见图21(fv1-m71、fv1-m73、fv3-m71、fv3-m73的结果见图21、其他的见图17和图19)。进一步按照与实施例10相同的方法进行分析,所有11种克隆的解离速度常数的ph依赖性见表12。[表12]对于sr344的ph依赖性结合克隆自膜型il-6受体上的解离速度常数的ph依赖性得到的10种ph依赖性结合克隆对膜型il-6受体显示出ph依赖性结合能力。进一步发现:与h3pi/l73-igg1相比,fv1-m71、fv1-m73、fv2-igg1、fv3-m71、fv3-m73、fv4-igg1、fv4-igg2、fv4-m58、fv4-m73的任意一种与膜型il-6受体的ph依赖性结合均有所提高,而在实施例9中发现,与wt相比,所述的h3pi/l73-igg1在食蟹猴体内直至抗体从血浆中消除的时间、以及体内的可溶型il-6受体和膜型il-6受体被抗体结合的时间大幅延长。[实施例15]利用食蟹猴进行的可变区和恒定区最优化的ph依赖性结合抗体的pk/pd试验公知的高亲和性抗il-6受体抗体的制作为了表达公知的高亲和性抗il-6受体抗体、即us2007/0280945a1中记载的高亲和性抗il-6受体抗体vq8f11-21higg1(us2007/0280945a1,氨基酸序列19和27),构建动物细胞表达用载体。利用组合有合成寡dna的pcr法(装配pcr)制作抗体可变区。恒定区则利用pcr法由实施例1中使用的表达载体进行扩增。利用装配pcr法使抗体可变区与恒定区结合,之后插入到哺乳动物表达用载体中。将得到的h链和l链dna片段插入哺乳动物细胞表达载体中,制作目标h链表达载体和l链表达载体。所得的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。使用制作的表达载体进行表达、纯化。表达、纯化按照实施例1所述的方法进行,得到高亲和性抗il-6受体抗体(高亲和性ab)。利用食蟹猴进行的pk/pd试验评价作为ph依赖性结合抗体的h3pi/l73-igg1和fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73以及公知的高亲和性抗il-6受体抗体(高亲和性ab)在食蟹猴体内的药物动力学和药效。将h3pi/l73-igg1和fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73以0.5mg/kg对食蟹猴进行静脉内单剂量给药,再将高亲和性ab以1.0mg/kg进行静脉内单剂量给药,给药前和给药后经时性进行采血。与实施例9同样地进行血浆中各抗体的浓度的测定。h3pi/l73-igg1和fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73、高亲和性ab的血浆中浓度变化见图21。为了评价食蟹猴膜型il-6受体被中和的程度的药效,与实施例9同样,在给予抗体后第3天-第10天(当为高亲和性ab时则从第6天-第10天),将食蟹猴il-6以5μg/kg对腰背部连日进行皮下给药,测定24小时后各个体的crp浓度。给予各抗体时的crp浓度变化见图22。为了评价食蟹猴可溶型il-6受体被中和的程度的药效,与实施例9同样,测定食蟹猴血浆中的非结合型的食蟹猴可溶型il-6受体浓度。给予各抗体时的非结合型食蟹猴可溶型il-6受体浓度变化见图23。由此发现:与h3pi/l73-igg1相比,fv1-m71、fv1-m73、fv2-igg1、fv3-m73、fv4-m73的血浆中抗体浓度均维持在高水平,而crp浓度和非结合型食蟹猴可溶型il-6受体浓度维持在低水平。即,与h3pi/l73-igg1相比,这些抗体使膜型il-6受体和可溶型il-6受体被抗体结合的时间(换言之,被中和的时间)延长。与以1.0mg/kg进行给药的公知的高亲和性抗il-6受体抗体(高亲和性ab)相比,这些ph依赖性结合抗il-6受体抗体以一半的给药量即0.5mg/kg即可确认到同等以上的中和效果和持续性,由此可知:与公知的高亲和性抗il-6受体抗体相比,ph依赖性结合抗体具有优异的中和效果和持续性。表12记载的抗体中,即使是本试验中未利用食蟹猴进行pk/pd试验的抗体,与h3pi/l73-igg1相比,也确认到其与膜型il-6受体的ph依赖性结合提高,由此认为:与h3pi/l73-igg1相比,这些抗体使膜型il-6受体和可溶型il-6受体被抗体结合的时间(换言之,被中和的时间、中和效果的持续性)延长。在实施例9中发现:与wt相比,h3pi/l73-igg1的直至抗体从血浆中消除的时间、以及体内的可溶型il-6受体和膜型il-6受体被抗体结合的时间(中和效果的持续性)大幅延长。中和效果的持续性较h3pi/l73-igg1优异的fv1-m71、fv1-m73、fv2-igg1、fv3-m71、fv3-m73、fv4-igg1、fv4-igg2、fv4-m58、fv4-m73与wt进行比较时,认为它们的中和效果的持续性明显改善。根据上述结果认为:相对于抗il-6受体抗体,在血浆中的ph即ph7.4下与抗原强力结合、在核内体内的ph即ph5.8下减弱与抗原的结合的ph依赖性结合抗il-6受体抗体,其可以减少对患者的抗il-6受体抗体的给药量或给药频率,结果,可以大幅减少总给药量,认为其作为il-6拮抗剂的药品极其优异。[实施例16]ph依赖性结合的抗il-6抗体的制作抗il-6抗体的表达和纯化在实施例1-15的人源化抗il-6受体抗体中,对于人源化抗il-6受体抗体的可变区,以其cdr序列为中心导入组氨酸等取代,从而成功地研制了多个对人源化抗il-6受体抗体与il-6受体的结合赋予ph依赖性的抗体,发现这些抗体均与il-6受体反复结合,pk/pd大幅改善。于是,在不同于抗il-6受体抗体的、与抗原结合的抗体中,利用同样的方法,研究能否对抗原与抗体的结合赋予ph依赖性。选择人il-6作为不同的抗原,制作与wo2004/039826中记载的人il-6结合的、包含h链(wt)(氨基酸序列seqidno:62)和l链(wt)(氨基酸序列seqidno:63)的抗il-6抗体(抗il6野生型)。按照本领域技术人员公知的方法,将编码目标抗体氨基酸序列的基因片段插入动物细胞表达载体中,制作目标h链表达载体和l链表达载体。得到的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。抗il6野生型的表达和纯化按照实施例1所述的方法进行。ph依赖性抗il-6抗体的制作对于包含h链(wt)(氨基酸序列seqidno:62)和l链(wt)(氨基酸序列seqidno:63)的抗il-6抗体(抗il6野生型),通过向cdr的氨基酸中导入组氨酸取代,进行对抗体与il-6的结合赋予ph依赖性的研究。对于cdr的氨基酸,研究组氨酸取代并进行筛选,结果,与ph7.4下的结合相比,ph5.5下的结合大幅降低,得到了若干个显示出ph依赖性结合的克隆。ph依赖性克隆中的组氨酸取代位点见表13。其中,例如有包含h链(c1)(氨基酸序列seqidno:64)和l链(c1)(氨基酸序列seqidno:65)的抗il6克隆1、以及包含h链(c1)(氨基酸序列seqidno:64)和l链(c2)(氨基酸序列seqidno:66)的抗il6克隆2。抗il6克隆1和抗il6克隆2的表达和纯化按照实施例1中记载的方法进行。[表13]ph依赖性克隆中的组氨酸取代位点h32、h59、h61、h99l53、l54、l90、l94分析ph依赖性结合克隆与人il-6的结合对于上述制作的抗il6野生型、抗il6克隆1和抗il6克隆2这3种,使用biacoret100(gehealthcare)进行ph5.5和ph7.4下的抗原抗体反应的速度论的分析(缓冲液为dpbs(-)ph7.4或ph5.5,150mmnacl)。利用胺偶联法使各种抗体结合在固定有重组蛋白a/g(pierce)的传感器芯片上,向其中注入以分析物形式制备成适当浓度的人il-6(toray)。所有测定均在37℃下进行。使用biacoret100评估软件(gehealthcare)算出结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表14)。再算出各自的在ph5.5和ph7.4下的亲和力比,评价ph依赖性结合。[表14]对于il-6的ph依赖性结合克隆与il-6的结合比较计算各自的在ph5.5和ph7.4下的亲和力比(kd(ph5.5)/kd(ph7.4)),结果,对于人il-6的抗il6野生型、抗il6克隆1、抗il6克隆2的ph依赖性结合分别为0.8倍、10.3倍、13.5倍,与wt相比,所有克隆均显示出10倍以上的高的ph依赖性结合。抗il6克隆2在ph7.4和ph5.5下的传感图见图26。由此显示:不仅是抗il-6受体抗体,即使在抗il-6抗体中,通过以cdr序列为中心导入组氨酸等氨基酸取代,可以制作具有在血浆中的中性条件下与抗原强力结合、在核内体中的酸性条件下与抗原的结合降低的ph依赖性结合的抗体。如实施例1-15所示,具有ph依赖性结合的抗il-6受体抗体与il-6受体反复结合,pk/pd大幅改善,由此认为:与抗il6野生型相比,具有ph依赖性结合的抗il6克隆1、抗il6克隆2与更多的抗原反复结合,pk/pd大幅改善。[实施例17]ph依赖性结合的抗il-31受体抗体的制作抗il-31受体抗体的表达和纯化实施例1-15中,在人源化抗il-6受体抗体中,对于人源化抗il-6受体抗体的可变区,通过以其cdr序列为中心导入组氨酸等取代,成功地制作了多个对人源化抗il-6受体抗体与il-6受体的结合赋予ph依赖性的抗体,发现这些抗体均与il-6受体反复结合,pk/pd大幅改善。于是,在不同于抗il-6受体抗体的、与抗原结合的抗体中,利用同样的方法,研究能否对抗原与抗体的结合赋予ph依赖性。选择小鼠il-31受体作为不同的抗原,制作与wo2007/142325中记载的小鼠il-31受体结合的、包含h链(wt)(氨基酸序列seqidno:67)和l链(wt)(氨基酸序列seqidno:68)的抗il-31受体抗体(抗il31r野生型)。按照本领域技术人员公知的方法将编码目标抗体氨基酸序列的基因片段插入动物细胞表达载体中,制作目标h链表达载体和l链表达载体。得到的表达载体的核苷酸序列按照本领域技术人员公知的方法进行确定。抗il31r野生型的表达和纯化按照实施例1所述的方法进行。ph依赖性抗il-31受体抗体的制作对于包含h链(wt)(氨基酸序列seqidno:67)和l链(wt)(氨基酸序列seqidno:68)的抗il-31受体抗体(抗il31r野生型),通过向其cdr的氨基酸中导入组氨酸取代,进行对抗体与il-31受体的结合赋予ph依赖性的研究。对于cdr的氨基酸研究组氨酸取代并进行筛选,结果,与ph7.4下的结合相比,ph5.5下的结合大幅降低,得到了若干个显示出ph依赖性结合的克隆。ph依赖性克隆中的组氨酸取代位点见表15。其中的一个位点为:包含h链(c1)(氨基酸序列seqidno:69)和l链(wt)的抗il31r克隆1。抗il31r克隆1的表达和纯化按照实施例1所述的方法进行。[表15]ph依赖性克隆中的组氨酸取代位点h33分析ph依赖性结合克隆与可溶型il-31受体的结合对于上述制作的抗il31r野生型、抗il31r克隆1这2种,使用biacoret100(gehealthcare)进行ph5.5和ph7.4下的抗原抗体反应的速度论的分析(缓冲液为dpbs(-)ph7.4或ph5.5、150mmnacl、0.01%tween20、0.02%nan3)。利用胺偶联法使各种抗体结合在固定有重组蛋白a/g(pierce)的传感器芯片上,向其中注入以分析物形式制备成适当浓度的可溶型小鼠il-31受体(按照wo2007/142325中记载的方法制备)。所有测定均在25℃下进行。使用biacoret100评估软件(gehealthcare)算出结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表16)。再算出各自的在ph5.5和ph7.4下的亲和力比,评价ph依赖性结合。[表16]对于小鼠il-31受体的ph依赖性结合克隆与小鼠il-31受体的结合比较计算各自的在ph5.5和ph7.4下的亲和力比(kd(ph5.5)/kd(ph7.4)),结果,对于小鼠il-31受体的抗il31r野生型、抗il31r克隆1的ph依赖性结合分别为3.2倍、1000倍,与wt相比,克隆1显示出约300倍的高的ph依赖性结合。抗il31r克隆在ph7.4和ph5.5下的传感图见图27。由此显示:不仅是抗il-6受体抗体和抗il-6抗体,即使在抗il-31受体抗体中,通过以cdr序列为中心导入组氨酸等氨基酸取代,可以制作具有在血浆中的中性条件下与抗原强力结合、在核内体中的酸性条件下与抗原的结合降低的ph依赖性结合的抗体。如实施例1-15所示,具有ph依赖性结合的抗il-6受体抗体与il-6受体反复结合,pk/pd大幅改善,由此认为:与抗il31r野生型相比,具有ph依赖性结合的抗il31r克隆1与更多的抗原反复结合,pk/pd大幅改善。[实施例18]ph依赖性结合抗体与抗原的反复结合小鼠给药抗体的表达和纯化作为人源化il-6受体抗体,制作以下4种抗体:对il-6受体未显示出ph依赖性结合的普通抗体、即包含h(wt)(氨基酸序列seqidno:9)和l(wt)(氨基酸序列seqidno:10)的wt-igg1以及包含h54(氨基酸序列seqidno:70)和l28(氨基酸序列seqidno:12)的h54/l28-igg1、以及对il-6受体显示出ph依赖性结合的抗体、即包含实施例3中制作的h170(氨基酸序列seqidno:4)和l82(氨基酸序列seqidno:7)的h170/l82-igg1以及包含实施例10中制作的vh3-igg1(seqidno:23)和vl3-ck(seqidno:27)的fv4-igg1。按照实施例1所示的方法,进行这4种抗体的表达和纯化。分析各种抗体与可溶型il-6受体的结合对于制备的wt-igg1、h54/l28-igg1、h170/l82-igg1和fv4-igg1这4种抗体,使用biacoret100(gehealthcare)进行ph7.4和ph5.8下的抗原抗体反应的速度论的分析(缓冲液为10mmmesph7.4或ph5.8,150mmnacl,0.05%表面活性剂-p20)。利用胺偶联法使各种抗体结合在固定有重组蛋白a/g(pierce)的传感器芯片上,向其中注入以分析物形式制备成适当浓度的sr344。实时观测各种抗体与sr344的结合和解离。所有测定均在37℃下进行。使用biacoret100评估软件(gehealthcare)算出结合速度常数ka(1/ms)和解离速度常数kd(1/s),根据该值算出解离常数kd(m)(表17)。[表17]对于sr344的各种抗体对可溶型il-6受体的结合速度(ka)、解离速度(kd)、解离常数(kd)的比较计算各自的在ph5.8和ph7.4下的亲和力(kd值)比,结果,对于sr344的wt-igg1、h54/l28-igg1、h170/l82-igg1以及fv4-igg1的ph依赖性结合(kd值之比)分别为1.6倍、0.7倍、61.9倍和27.3倍。另外,计算各自的在ph5.8和ph7.4下的解离速度(kd值)比,结果,对于sr344的wt-igg1、h54/l28-igg1、h170/l82-igg1和fv4-igg1的ph依赖性解离速度(kd值之比)分别为2.9倍、2.0倍、11.4倍和38.8倍。由此确认:作为普通抗体的wt-igg1和h54/l28-igg1几乎没有显示出ph依赖性结合,而h170/l82-igg1和fv4-igg1显示出ph依赖性结合。另外,由于这些抗体在ph7.4下的亲和力(kd值)几乎同等,所以认为它们与血浆中的sr344的结合为同等程度。使用小鼠的体内动力学试验对不表达人i1-6受体的小鼠(c57bl/6j;这些抗人il-6受体抗体不与小鼠的il-6受体结合)单独给予sr344(人il-6受体:实施例1中制作)、或同时给予sr344和抗人il-6受体抗体后,评价sr344和抗人il-6受体抗体的体内动力学。以10ml/kg对尾静脉单剂量给予sr344溶液(5μg/ml)或sr344和抗人il-6受体抗体的混合溶液(分别为5μg/ml、0.1mg/ml)。此时,相对于sr344过剩存在足够量的抗人il-6受体抗体,由此认为:几乎所有的sr344均与抗体结合。在给药后15分钟、2小时、8小时、1天、2天、3天、4天、7天、14天、21天、28天进行采血。采集的血液立即在4℃下以15,000rpm的转速离心15分钟,得到血浆。分离的血浆在实施测定前一直保存在被设定为-20℃以下的冷库中。作为抗人il-6受体抗体,使用上述的wt-igg1、h54/l28-igg1、h170/l82-igg1和fv4-igg1。利用elisa法测定血浆中抗人il-6受体抗体浓度小鼠血浆中的抗人il-6受体抗体浓度利用elisa法进行测定。首先,将抗人igg(γ-链特异性)f(ab’)2抗体片段(sigma)分别注入nunc-immuno板、maxisorp(nalgenuncinternational)中,在4℃下静置一夜,制作抗人igg固相化板。制备血浆中浓度为0.8、0.4、0.2、0.1、0.05、0.025、0.0125μg/ml的校准曲线试样和稀释了100倍以上的小鼠血浆测定试样。向100μl这些校准曲线试样和血浆测定试样中加入200μl20ng/ml的sr344,在室温下静置1小时。之后,分别注入到抗人igg固相化板中,进一步在室温下静置1小时。之后,使生物素化抗人il-6r抗体(r&d)在室温下反应1小时,再使链霉亲和素-polyhrp80(stereospecificdetectiontechnologies)在室温下反应1小时,使用tmbonecomponenthrpmicrowellsubstrate(biofxlaboratories)作为底物进行显色反应,利用1n-硫酸(showachemical)使反应停止,之后使用酶标仪测定450nm的吸亮度。使用分析软件softmaxpro(moleculardevices),由校准曲线的吸亮度算出小鼠血浆中浓度。通过该方法测定的、静脉内给药后的血浆中抗体浓度变化见图28。利用电化学发光法测定血浆中sr344浓度小鼠的血浆中sr344浓度利用电化学发光法来测定。制备调整成2000、1000、500、250、125、62.5、31.25pg/ml的sr344校准曲线试样和稀释了50倍以上的小鼠血浆测定试样,混合用sulfo-tagnhsester(mesoscalediscovery)进行了钌化的单克隆抗人il-6r抗体(r&d)、生物素化的抗人il-6r抗体(r&d)和wt-igg1溶液,在37℃下反应一夜。此时,wt-igg1的终浓度为较样品中所含的抗人il-6受体抗体浓度过剩的333μg/ml,目的在于形成样品中的几乎所有的sr344均与wt-igg1结合的状态。之后,分别注入到ma400pr链霉亲和素板(mesoscalediscovery)中。再于室温下反应1小时,清洗后分别注入readbuffert(×4)(mesoscalediscovery),立即使用sectorpr400reader(mesoscalediscovery)进行测定。使用分析软件softmaxpro(moleculardevices),由校准曲线的响应值算出sr344浓度。通过该方法测定的、静脉内给药后的血浆中sr344浓度变化见图29。ph依赖性结合的效果关于未显示出ph依赖性结合的抗体即wt-igg1和h54/l28-igg1、以及显示出ph依赖性结合的抗体即h170/l82-igg1和fv4-igg1的抗体浓度变化,wt-igg1、h54/l28-igg1和fv4-igg1几乎同等,而h170/l82-igg1消除稍快。使用药物动力学分析软件winnonlin(pharsight)分析血浆中浓度变化的数据,结果,wt-igg1、h54/l28-igg1、fv4-igg1、h170/l82-igg1的血浆中半衰期分别为21.0天、28.8天、26.2天、7.5天。如实施例2所述,当抗原为可溶型抗原时,给予的抗体在血浆中与抗原结合,以抗原与抗体的复合体的形式滞留在血浆中。通常,抗体的血浆中滞留性由于fcrn的功能而非常长(消除速度非常慢),相对于此,抗原的血浆中滞留性短(消除速度快),所以与抗体结合的抗原与抗体具有同等程度的长的血浆中滞留性(消除非常慢)。单独给予人源化il-6受体抗体的抗原即sr344(可溶型人il-6受体)时,sr344也同样显示出极快的消除(血浆中半衰期为0.2天)。同时给予sr344和作为未显示出ph依赖性结合的普通抗体的wt-igg1或h54/l28-igg1时,sr344的消除速度明显下降,显示出长的血浆中滞留性(血浆中半衰期:wt-igg15.3天、h54/l28-igg16.3天)。这是由于sr344几乎全部与同时给药的抗体结合,如上所述,与抗体结合的sr344由于fcrn的功能而与抗体具有相同程度的长的血浆中滞留性的缘故。同时给予sr344和显示出ph依赖性结合的抗体即h170/l82-igg1或fv4-igg1时,与同时给予wt-igg1或h54/l28-igg1时相比,sr344的消除明显变快(血浆中半衰期:h170/l82-igg11.3天、fv4-igg10.6天)。该趋势在fv4-igg1中特别显着。fv4-igg1在ph7.4下的亲和力与wt-igg1和h54/l28-igg1为同等以上,由此认为:sr344几乎全部与fv4-igg1结合。尽管与wt-igg1和h54/l28-igg1相比,fv4-igg1显示出同等或稍长的血浆中滞留性、消除慢,但与fv4-igg1结合的sr344的消除明显变快。这可以通过图4所示的本技术的观念(concept)来说明。关于未显示出ph依赖性结合的普通抗体,抗体-可溶型抗原复合体在血浆中通过胞饮作用摄入核内体中,在核内体内的酸性条件下与在核内体内表达的fcrn结合,与fcrn结合的抗体-可溶型抗原复合体直接向细胞表面移动,再次返回到血浆中,所以与抗体结合的抗原与抗体具有相同程度的长的血浆中滞留性(消除非常慢)。另一方面,由于显示出ph依赖性结合的抗体在核内体内的酸性条件下解离抗原,所以只有抗体与fcrn结合,再次返回到血浆中,自抗体上解离的抗原没有返回到血浆中,而是被溶酶体分解,所以与未显示出ph依赖性结合的抗体的情形相比,抗原的消除明显变快。即,将sr344和未显示出ph依赖性结合的抗体即wt-igg1或h54/l28-igg1同时给药时,在血浆中和核内体内sr344与wt-igg1或h54/l28-igg1结合,所以sr344的消除与抗体同等程度地变慢,但将sr344和显示出ph依赖性结合的抗体即h170/l82-igg1或fv4-igg1同时给药时,在核内体内的低ph环境下sr344自抗体上解离,所以sr344的消除变得极快。即,关于作为显示出ph依赖性结合的抗体的h170/l82-igg1或fv4-igg1,在核内体内的低ph环境下sr344发生解离,由此认为:通过fcrn再次返回到血浆中的h170/l82-igg1或fv4-igg1中的大多数未与sr344结合。由此表明:如图4所示,显示出ph依赖性结合的抗体在核内体内的低ph环境下解离抗原,以未与抗原结合的状态通过fcrn返回到血浆中,从而在血浆中可以再次与新的抗原结合,通过重复进行此过程,显示出ph依赖性结合的抗体可以多次与抗原反复结合。如实施例7所示,这反映出在biacore中ph依赖性结合克隆可以与抗原反复结合,增强抗体与抗原的ph依赖性结合,从而可以增加抗体与抗原反复结合的次数。当抗原为可溶型抗原时,如果在血浆中的中性条件下与抗体结合的抗原在核内体内解离,抗体通过fcrn返回到血浆中,则抗体在血浆中的中性条件下可以再次与抗原结合,所以具有在核内体内的酸性条件下解离抗原的性质的抗体可以多次与抗原结合。和与抗体结合的抗原在核内体内未发生解离时(抗原保持着与抗体结合的状态返回到血浆中)相比,与抗体结合的抗原在核内体内解离时,抗原被运送到溶酶体中被分解,所以抗原从血浆中消除的速度增加。即,以抗原从血浆中消除的速度为指标,还可以判断抗体能否与抗原多次结合。关于抗原从血浆中消除的速度的测定,例如,如本实施例所示,通过对体内给予抗原和抗体,并测定给药后的血浆中的抗原浓度来进行。与未显示出ph依赖性结合的普通抗体相比,显示出ph依赖性结合的抗体的1个抗体可以多次与抗原反复结合,因此认为其可以大幅减少抗体的给药量和大幅延长给药间隔。基于本机理的、与抗原的多次反复结合立足于ph依赖性抗原抗体反应,所以无论是何种抗原,如果能够制作在血浆中的ph7.4下结合、在核内体内的酸性ph下自抗原上解离的显示出ph依赖性结合的抗体,则1个抗体可以多次与抗原反复结合。即,本技术不仅对il-6受体、il-6、il-31受体有用,对于不依赖于抗原的种类、抗任何抗原的抗体,本技术作为可适用的技术通常也是有用的。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1