蛋白质编码方法及蛋白质翻译后修饰位点预测方法及系统与流程
本发明涉及生物信息学领域,更具体地,涉及蛋白质编码方法及蛋白质翻译后修饰位点预测方法及系统。
背景技术:
蛋白质翻译后修饰是真核和原核生物最重要的机制之一,它涉及化学基团与蛋白质氨基酸侧链的连接。各种蛋白质翻译后修饰ptm在多种细胞过程中发挥着至关重要的作用,这些过程调节蛋白质的功能、物理化学性质、构象、稳定性和响应发育信号或环境刺激的分子相互作用。例如,蛋白质磷酸化是最普遍存在的蛋白质翻译后修饰ptm,可诱导信号转导和细胞凋亡;赖氨酸琥珀酰化在代谢途径中起着至关重要的作用;蛋白质乙酰化和甲基化参与染色质重编程和转录调控;赖氨酸泛素化介导蛋白质降解。并且,越来越多的研究发现蛋白质翻译后修饰ptm的失调与多种疾病(包括癌症)的发展和进展有关。由于各种限制,通过诸如高通量液相色谱/质谱(lc-ms)技术的传统实验技术鉴定蛋白质翻译后修饰位点仍然是低效、昂贵且耗时的。因此,开发能够识别蛋白质翻译后修饰位点的计算方法已变得越来越有必要。尽管有多种蛋白质翻译后修饰位点预测方法,例如,xue等利用基于肽段相似度打分的方法构建了多个蛋白质修饰位点的预测器。qiu等利用支持向量机构建多个蛋白质修饰位点的预测器。但当这些方法仍然存在一些缺陷,如:利用一种算法构建一个简化模型无法充分挖掘多类型多特征数据的信息。现有的预测方法仅考虑蛋白质的一个或几个特征,而修饰的发生往往跟蛋白质序列、结构、氨基酸理化性质等多个因素有关。此外,现有预测方法往往忽略了物种之间存在的差异,修饰位点周围的序列或结构可能在不同物种中显著变化。因而发展新的高精度预测方法非常关键。
技术实现要素:
本发明解决了现有技术中蛋白质翻译后修饰位点预测方法无法实现多特征数据的预测,且无法高精度预测不同物种中的蛋白质翻译后修饰。
按照本发明的第一方面,提供了一种蛋白质编码方法,所述蛋白质编码方法用于表示待编码肽段与阳性数据集肽段的相似度,含有以下步骤:
(1)收集修饰位点信息:首先收集蛋白质翻译后目标类型的修饰位点信息;将所述目标类型的修饰位点在蛋白质上的对应位点作为阳性位点,将该蛋白质上与所述阳性位点相同的其它氨基酸位点作为阴性位点;将蛋白质的一级序列切割成以所述阳性位点或阴性位点为中心,该中心上游为n个氨基酸,该中心下游为n个氨基酸,总长度为2n+1个氨基酸序列;所述n大于等于1;所有含有所述阳性位点的所述氨基酸序列构成阳性数据集,所有含有所述阴性位点的所述氨基酸序列构成阴性数据集;
(2)位置权重训练:步骤(1)所述阳性数据集和阴性数据集中的每个肽段与阳性数据集基于位置权重和氨基酸替换得分的相似度打分的公式为:
其中:l为所述阳性数据集中每个肽段的长度2n+1;n为所述阳性数据集中肽段的数量;tij是阳性数据集中肽段ti在位置j上的氨基酸,i的取值范围为1≤i≤n;pj为肽段在位置j上的氨基酸;m[pj,tij]为氨基酸pj和tij在blosum62氨基酸替换矩阵中的分值;wj为该肽段中位置j上的权重;
所述阳性数据集和阴性数据集中的每条肽段分别与阳性数据集中的每条肽段依次打分,其中肽段不与其自身打分,初始位置权重wj为1,获得肽段中除中心位置以外的其它2n个位置的得分;然后将该2n个位置的得分使用惩罚逻辑回归执行交叉验证,使auc值最大的权重向量由肽段中各个位置上的权重wj组成;
(3)待编码肽段的编码:待编码肽段与阳性数据集间的氨基酸对的平均相似度s为:
其中:l是待编码肽段的长度,j为氨基酸所在位置,cj为待编码肽段与阳性数据集间的任意一个氨基酸对在位置j上出现的次数,m为所述氨基酸对在blosum62氨基酸替换矩阵中的分值,wj为步骤(2)训练得到的待编码肽段位置j上的权重;待编码肽段与阳性数据集间的所有的氨基酸对的相似度得分构成该待编码肽段的数字向量特征。
按照本发明的另一方面,提供了多特征算法模型的蛋白质翻译后修饰位点预测方法,含有以下步骤:
(1)收集修饰位点信息:收集蛋白质翻译后目标类型的修饰位点信息;将所述目标类型的修饰位点在蛋白质上的对应位点作为阳性位点,将该蛋白质上与所述阳性位点相同的其它氨基酸位点作为阴性位点;将所述阳性位点和阴性位点按照蛋白质所属物种进行分类;将蛋白质的一级序列切割成以所述阳性位点或阴性位点为中心,该中心上游为n个氨基酸,该中心下游为n个氨基酸,总长度为2n+1个氨基酸的序列;所述n大于等于1;
(2)特征编码:将权利要求1所述的蛋白质编码方法以及其它的编码方案逐个对步骤(1)所述总长度为2n+1个氨基酸的序列进行特征编码,得到数字向量特征,将所述数字向量特征分别利用惩罚逻辑回归、支持向量机和随机森林验证每种编码方案的auc性能,将auc性能大于0.5的编码方案作为备用编码方案;挑选所述备用编码方案对步骤(1)所述总长度为2n+1个氨基酸的序列进行特征编码得到的数字向量特征;
(3)模型训练:利用深度神经网络和惩罚逻辑回归分别对步骤(2)所述不同类别的阳性位点和阴性位点的数字向量特征构建预测模型,得到多个预测模型;将每个预测模型的预测结果作为新的特征并利用惩罚逻辑回归构建最终模型;
(4)蛋白质翻译后修饰位点预测:通过步骤(3)所述最终模型预测蛋白质翻译后修饰位点;所述预测能得到步骤(2)所述备用编码方案中的特征以及步骤(1)所述蛋白质所属物种信息。
优选地,步骤(1)所述目标类型的修饰位点信息为赖氨酸琥珀酰化位点信息、磷酸化位点信息、泛素化位点信息、甲基化位点信息或乙酰化位点信息。
优选地,步骤(1)所述n小于等于30。
优选地,步骤(2)所述其它的编码方案为pseaac编码方案、cksaap编码方案、正交二进制编码方案、aaindex编码方案、自相关特征集编码方案、pssm编码方案、asa编码方案,ss编码方案和bta编码方案;
所述pseaac编码方案用于表示肽段中每种氨基酸出现的频率的数字向量特征;
所述cksaap编码方案用于表示肽段中被k个氨基酸间隔的任意两种或者两种相同氨基酸出现的次数的数字向量特征,所述k大于等于0小于等于(2n-1);
所述正交二进制编码方案用于表示肽段中每个氨基酸二进制向量的数字向量特征;
所述aaindex编码方案用于表示肽段中每个氨基酸在aaindex数据库中理化性质下编码的数字向量特征;
所述自相关特征编码方案用于表示肽段中被k个氨基酸间隔的任意两种或者两种相同氨基酸的aaindex数据库中理化性质相关性的数字向量特征;
所述pssm编码方案用于表示肽段中每个氨基酸位置分别出现特定氨基酸的概率的数字向量特征;
所述asa编码方案用于表示肽段中每个氨基酸的可及表面积的数字向量特征;
所述ss编码方案用于表示肽段中每个氨基酸发生α-螺旋、β-折叠和转角的概率的数字向量特征;
所述bta编码方案用于表示肽段中每个氨基酸发生二级结构的角度的数字向量特征。
按照本发明的另一方面,提供了一种蛋白质编码系统,包括:
收集修饰位点信息模板:所述收集修饰位点信息模板用于收集蛋白质翻译后目标类型的修饰位点信息;将所述目标类型的修饰位点在蛋白质上的对应位点作为阳性位点,将该蛋白质上与所述阳性位点相同的其它氨基酸位点作为阴性位点;将蛋白质的一级序列切割成以所述阳性位点或阴性位点为中心,该中心上游为n个氨基酸,该中心下游为n个氨基酸,总长度为2n+1个氨基酸序列;所述n大于等于1;所有含有所述阳性位点的所述氨基酸序列构成阳性数据集,所有含有所述阴性位点的所述氨基酸序列构成阴性数据集;
位置权重训练模块:所述位置权重训练模块用于训练位置权重;所述阳性数据集和阴性数据集中的每个肽段与阳性数据集基于位置权重和氨基酸替换得分的相似度打分的公式为:
其中:l为所述阳性数据集中每个肽段的长度2n+1;n为所述阳性数据集中肽段的数量;tij是阳性数据集中肽段ti在位置j上的氨基酸,i的取值范围为1≤i≤n;pj为肽段在位置j上的氨基酸;m[pj,tij]为氨基酸pj和tij在blosum62氨基酸替换矩阵中的分值;wj为该肽段中位置j上的权重;
所述阳性数据集和阴性数据集中的每条肽段分别与阳性数据集中的每条肽段依次打分,其中肽段不与其自身打分,初始位置权重wj为1,获得肽段中除中心位置以外的其它2n个位置的得分;然后将该2n个位置的得分使用惩罚逻辑回归执行交叉验证,使auc值最大的权重向量由肽段中各个位置上的权重wj组成;
待编码肽段的编码模块:所述待编码肽段的编码模块用于编码待编码肽段;待编码肽段与阳性数据集间的氨基酸对的平均相似度s为:
其中:l是待编码肽段的长度,j为氨基酸所在位置,cj为待编码肽段与阳性数据集间的任意一个氨基酸对在位置j上出现的次数,m为所述氨基酸对在blosum62氨基酸替换矩阵中的分值,wj为训练得到的待编码肽段位置j上的权重;待编码肽段与阳性数据集间的所有的氨基酸对的相似度得分构成该待编码肽段的数字向量特征。
按照本发明的另一方面,提供了多特征算法模型的蛋白质翻译后修饰位点预测系统,包括:
收集修饰位点信息模块:所述收集修饰位点信息模块用于收集蛋白质翻译后目标类型的修饰位点信息;将所述目标类型的修饰位点在蛋白质上的对应位点作为阳性位点,将该蛋白质上与所述阳性位点相同的其它氨基酸位点作为阴性位点;将所述阳性位点和阴性位点按照蛋白质所属物种进行分类;将蛋白质的一级序列切割成以所述阳性位点或阴性位点为中心,该中心上游为n个氨基酸,该中心下游为n个氨基酸,总长度为2n+1个氨基酸的序列;所述n大于等于1;
特征编码模块:所述特征编码模块用于对氨基酸序列进行特征编码;将权利要求1所述的蛋白质编码方法以及其它的编码方案逐个对所述总长度为2n+1个氨基酸的序列进行特征编码,得到数字向量特征,将所述数字向量特征分别利用惩罚逻辑回归、支持向量机和随机森林验证每种编码方案的auc性能,将auc性能大于0.5的编码方案作为备用编码方案;挑选所述备用编码方案对所述总长度为2n+1个氨基酸的序列进行特征编码得到的数字向量特征;
模型训练模块:所述模型训练模块用于利用深度神经网络和惩罚逻辑回归分别对所述不同类别的阳性位点和阴性位点的数字向量特征构建预测模型,得到多个预测模型;将每个预测模型的预测结果作为新的特征并利用惩罚逻辑回归构建最终模型;
蛋白质翻译后修饰位点预测模块:所述蛋白质翻译后修饰位点预测模块用于通过所述最终模型预测蛋白质翻译后修饰位点;所述预测能得到所述备用编码方案中的特征以及所述蛋白质所属物种信息。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,主要具备以下的技术优点:
(1)本发明先收集修饰位点数据,进行数据预处理后得到阳性和阴性数据集,将序列分别按不同的编码方法进行编码,优选地按pseaac编码方案、cksaap编码方案、正交二进制编码方案、aaindex编码方案、自相关特征集编码方案、pssm编码方案、基于组的预测系统(gpssimilarity)编码方案、asa编码方案,ss编码方案和bta编码方案进行特征编码;,并对特征进行评估。利用深度神经网络(dnn)和惩罚逻辑回归(plr)的混合学习框架分别对每种特征构建预测模型,将每个模型的预测结果打分作为新的特征并利用惩罚逻辑回归(plr)对其构建具有多特征算法的最终模型,用评价指标对模型进行性能评估。最后,构建蛋白质修饰位点的预测平台,用于在线预测。
(2)本发明公开了一种基于多特征混合算法模型的蛋白质修饰位点预测方法,通过交叉验证和独立测试比对表明,本方法构建的预测模型具有鲁棒性好、准确度高等优点。本发明的预测方法可以对蛋白质位点预测提供新思路,预测结果可以为验证蛋白质翻译后修饰位点的研究提供很好的借鉴作用,对研究蛋白质翻译后修饰的机理和生物功能有重大意义。
(3)本发明的目的在于提供一种蛋白质翻译后修饰位点的预测方法,由于整合深度学习与传统机器学习的混合框架以及多种特征的使用,因此该方法可以捕获更多蛋白信息从而有助于提高预测的准确度,可以快速的大规模鉴定蛋白质修饰位点。
附图说明
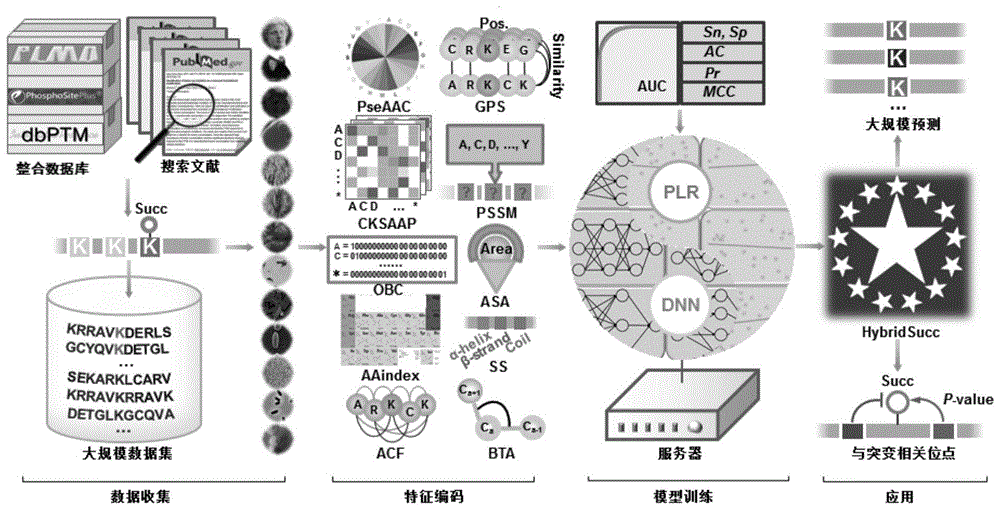
图1是按照本发明方法的流程图。
图2a是开发集成深度神经网络(dnn)和惩罚逻辑回归(plr)混合算法框架图;图2b为10倍交叉验证结果图;图2c为本发明hybridsucc的auc结果。
图3是利用本发明hybridsucc预测的琥珀酰化位点与已知的琥珀酰化位点周边序列在序列和结构上的比较:其中图3a反映了序列上各个位置的氨基酸偏好性的比较;图3b为显著出现在中心附近模体的比较;图3c为琥珀酰化修饰位点出现在蛋白质位置上的比较;图3d为琥珀酰化修饰赖氨酸位点的a-helix,β-strand和coil的比较;图3e为琥珀酰化修饰赖氨酸暴露面积上的比较;图3f为琥珀酰化修饰赖氨酸出现在无序或有序区域的偏好性的比较。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
本发明所述多特征混合算法模型的蛋白质翻译后修饰位点预测方法含有以下步骤:
(1)收集修饰位点数据:
从相关文献以及数据库中收集蛋白质翻译后修饰位点,删除重复的冗余位点。从uniprot数据库下载蛋白质的一级序列。将实验鉴定的修饰位点视为阳性数据,而将相同蛋白质中剩余的位点视为阴性数据,并按照物种或酶特异性分类。将蛋白质序列切割成以位点为中心,上游为n个氨基酸,下游为n个氨基酸,总长度为2n+1个氨基酸的序列;所述n大于等于1;
(2)特征编码:
将步骤(1)所述蛋白质序列对以下十种编码方案逐个进行特征编码,并且利用支持向量机评估利用10倍交叉验证每种编码的auc性能,将auc性能大于0.5作为备用编码方案。总共十种特征编码方案:pseaac编码方案(可参考文献xuy,dingyx,dingj,leiyh,wulyanddengny.isuc-pseaac:predictinglysinesuccinylationinproteinsbyincorporatingpeptideposition-specificpropensity.scirep2015;5:10184.)、cksaap编码方案(可参考文献xuhd,shisp,wenppandqiujd.succfind:anovelsuccinylationsitesonlinepredictiontoolviaenhancedcharacteristicstrategy.bioinformatics2015;31:3748-3750.)、正交二进制编码方案(可参考文献chenh,xuey,huangn,yaoxandsunz.memo:awebtoolforpredictionofproteinmethylationmodifications.nucleicacidsres2006;34:w249-w253.)、aaindex编码方案(可参考文献xuhd,shisp,wenppandqiujd.succfind:anovelsuccinylationsitesonlinepredictiontoolviaenhancedcharacteristicstrategy.bioinformatics2015;31:3748-3750.)、自相关特征集编码方案(可参考文献zhaox,ningq,chaihandmaz.accurateinsilicoidentificationofproteinsuccinylationsitesusinganiterativesemi-supervisedlearningtechnique.jtheorbiol2015;374:60-65.)、pssm编码方案(可参考文献hasanmm,khatunms,mollahmnh,yongcandguod.asystematicidentificationofspecies-specificproteinsuccinylationsitesusingjointelementfeaturesinformation.intjnanomedicine2017;12:6303-6315.)、基于组的预测系统(gpssimilarity)编码方案(本发明要求保护的编码方案)、asa编码方案(可参考文献lopezy,sharmaa,dehzangia,lalsp,taherzadehg,sattaraandtsunodat.success:evolutionaryandstructuralpropertiesofaminoacidsproveeffectiveforsuccinylationsiteprediction.bmcgenomics2018;19:923.),ss编码方案(可参考文献lopezy,sharmaa,dehzangia,lalsp,taherzadehg,sattaraandtsunodat.success:evolutionaryandstructuralpropertiesofaminoacidsproveeffectiveforsuccinylationsiteprediction.bmcgenomics2018;19:923.)和bta编码方案(可参考文献lopezy,sharmaa,dehzangia,lalsp,taherzadehg,sattaraandtsunodat.success:evolutionaryandstructuralpropertiesofaminoacidsproveeffectiveforsuccinylationsiteprediction.bmcgenomics2018;19:923.).
第一种编码方案,所述pseaac编码方案,用于计算包含阳性位点或阴性位点,总长度为2n+1个氨基酸的序列中,每种氨基酸的频率;由于有些修饰位点出现在蛋白质的两端,因此“*”被添加去补齐成2n+1肽段。将“*”视为第21种氨基酸,计算包括“*”在内的21种氨基酸的频率,每个肽段被编码为21维数字向量:vi=(fa,fc,fd,…,fy,f*)21;其中,fa,fc,fd,…,fy,f*分别表示每个肽段中21种氨基酸的的频率。
第二种编码方案,所述cksaap编码,其反映了蛋白质序列的k-间隔氨基酸对的组成(由k个其他氨基酸间隔的氨基酸对),如果氨基酸对aa在肽段上出现l次,则caa=l。所有的k=0,1,…,kmax被评估,选择试auc性能最好的k。每个肽段被编码为21*21*(kmax+1)维数字向量:
第三种编码方案,所述正交二进制编码方案,其中每个氨基酸由20维二进制向量表示。21种氨基酸按照简写的字母顺序排序,对于第j个氨基酸,第j位置设为1,其他位置为0,例如简写为a的氨基酸a被编码为[1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0],简写为c的氨基酸c被编码为[0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0],阳性位点或者阴性位点不编码,即中心位置不编码,因此,每个肽段被编码为2n*21维的向量。
第四种编码方案,所述aaindex编码方案,在aaindex数据库中有566种理化性质;对于理化性质h,每个肽段被编码为:vi=h1,h2,h3,…,h2n;
选择auc性能最好的10种理化性质,每个肽段被编码为2n*10维数字向量:vi=v1,v2,v3,…,v10。
第五种编码方案,自相关特征编码方案(acf),给定理化性质h,中心位置不编码,每个肽段编码为数字向量:vi=h1,h2,h3,…,h2n;
自相关函数定义为:
其中l为肽段长度2n+1,m如果等于0,则表示相邻2个氨基酸;rk表示被k个其他氨基酸间隔的2个氨基酸的相关性;因此,肽段被编码为vi=[r0,r1,r2,r3,…,r2n]1,[r0,r1,r2,r3,…,r2n]2,…,[r0,r1,r2,r3,…,r2n]i,…,[r0,r1,r2,r3,…,r2n]10;i表示第i个理化性质;其中r0,r1,r2,r3,…,r2n分别是在特定理化性质下,被0,1,2,……2n个氨基酸间隔的2个氨基酸的相关性。
第六种类型是pssm编码方案,其从psi-blast生成的位置特异性评分矩阵中提取特征。利用psi-blast程序包比对肽段到swiss-prot蛋白质序列得到pssm矩阵。每个肽段被编码为20*(2n+1)维数字向量:
第七种编码方案,基于组的预测系统(gps),评分策略的基本假设是类似的短肽表现出相似功能的生化特性。所述蛋白质编码方法用于表示待编码肽段与阳性数据集肽段的相似度,基于的假设为相似的肽段往往具有相似的功能;含有以下步骤:
(1)收集修饰位点信息:首先收集蛋白质翻译后目标类型的修饰位点信息;将所述目标类型的修饰位点在蛋白质上的对应位点作为阳性位点,将该蛋白质上与所述阳性位点相同的其它氨基酸位点作为阴性位点;将蛋白质的一级序列切割成以所述阳性位点或阴性位点为中心,该中心上游为n个氨基酸,该中心下游为n个氨基酸,总长度为2n+1个氨基酸序列;所述n大于等于1;所有含有所述阳性位点的所述氨基酸序列构成阳性数据集,所有含有所述阴性位点的所述氨基酸序列构成阴性数据集;
(2)位置权重训练:步骤(1)所述阳性数据集和阴性数据集中的每个肽段与阳性数据集基于位置权重和氨基酸替换得分的相似度打分的公式为:
其中:l为所述阳性数据集中每个肽段的长度2n+1;n为所述阳性数据集中肽段的数量;tij是阳性数据集中肽段ti在位置j上的氨基酸,i的取值范围为1≤i≤n;pj为肽段在位置j上的氨基酸;m[pj,tij]为氨基酸pj和tij在blosum62氨基酸替换矩阵中的分值;wj为该肽段中位置j上的权重;
所述阳性数据集和阴性数据集中的每条肽段分别与阳性数据集中的每条肽段依次打分,其中肽段不与其自身打分,初始位置权重wj为1,获得20维数字向量;然后将该20维数字向量使用惩罚逻辑回归执行交叉验证,使auc值最大的权重向量由肽段中各个位置上的权重wj组成;
(3)待编码肽段的编码:待编码肽段与阳性数据集间的氨基酸对的平均相似度s为:
其中:l是待编码肽段的长度,j为氨基酸所在位置,cj为待编码肽段与阳性数据集间的任意一个氨基酸对在位置j上出现的次数,m为所述氨基酸对在blosum62氨基酸替换矩阵中的分值,wj为步骤(2)训练得到的待编码肽段位置j上的权重;待编码肽段与阳性数据集间的所有的氨基酸对的相似度得分构成该待编码肽段的数字向量特征。
对于21种氨基酸,一共有[21*(21+1)]/2=231个sab分数(sab=sba);因此,每个肽段被编码为231维数字向量:v=(saa,sac,sad,…,s**)231;其中saa,sac,sad,…,s**分别表示待编码肽段与阳性数据集任意两种或两个相同氨基酸相似度的数字向量特征。
第八种类型是asa编码方案,其源自预测的氨基酸可及表面积的信息。利用spider2工具预测肽段上每个氨基酸asa值,每个肽段被编码为:vi=(a1,a2,…,a2n+1);其中a1,a2,…,a2n+1分别表示肽段上每个氨基酸的可及表面积asa值。
第九种类型是ss编码,其中每个氨基酸由α-helix,β-strand和coil的发生率表示,其利用spider2工具预测得到,每个肽段被编码为:vi=(s1,s2,…,s2n+1)α-helix(s1,s2,…,s2n+1)β-strand(s1,s2,…,s2n+1)coil,其中s1,s2,…,s2n+1分别表示肽段中每个每个氨基酸发生α-螺旋、β-折叠或转角的概率的数字向量特征。
第十种类型是bta编码,4个角度包括φ,ψ,cαi-1-cαi-cαi+1(θ),cαi-cαi+1(τ)被spider2工具预测得到,每个肽段被编码为:
(3)模型训练:
利用深度神经网络(dnn)和惩罚逻辑回归(plr)的混合学习框架分别对每种特征构建预测模型,得到多个预测模型,将每个模型的预测结果打分作为新的特征并利用惩罚逻辑回归(plr)对其构建最终模型,并用评价指标对模型进行性能评估,确保模型的可靠性,准确性。
(4)预测平台的构建以及应用
采用构建的预测模型开发在线网络预测平台,只需在预测平台的指定区域输入蛋白质序列,即可预测出该序列上修饰位点信息。通过预测平台,我们可以进行大规模预测,修饰与癌症的分析,突变对修饰发生影响分析等。
实施例1
以蛋白质赖氨酸琥珀酰化为例,我们使用本发明方法构建名为hybridsucc的预测模型,其流程图如图1所示。具体步骤为:
1、我们收集并整合了来自科学文献的7,415种蛋白质的21,770个琥珀酰化位点,从uniprot数据库下载蛋白质的一级序列。将已鉴定的赖氨酸琥珀酰化位点视为阳性数据,而将相同蛋白质中剩余的赖氨酸位点视为阴性数据,并按照物种特异性分类,按照这些琥珀酰化位点所述的蛋白质将位点分类为包括人,小鼠,酵母,水稻,大鼠,大肠杆菌在内的13个物种。将蛋白质序列切割成以位点为中心,上游为10个氨基酸,下游为10个氨基酸,长度为21的序列。
2、将蛋白质序列进行特征编码,基于数据集,将阳性和阴性数据集用10种编码方案分别编码,通过惩罚逻辑回归,支持向量机,随机森林执行10倍交叉验证每种编码的auc性能。最终auc都大于0.5,因此,所有编码都有效。
3、开发集成深度神经网络(dnn)和惩罚逻辑回归(plr)的混合学习框架训练模型,其细节结构如图2a所示,为解决物种特异性琥珀酰化位点预测中的训练数据小的问题,深度神经网络(dnn)的训练分为预训练与迁移学习两个阶段,即对每个特征的所有数据构建预训练模型后,特异性数据在该预训练模型上进行迁移学习从而构建每个特征的物种特异性模型。惩罚逻辑回归(plr)的训练分为三个阶段:首先利用lasso算法对每个特征的物种特异性数据训练出权重,然后通过随机权重突变以及随机权重置零1000次,若获取更好的性能则更新权重,从而构建每个特征的物种特异性模型。最后将每个模型的预测结果打分作为新的特征并利用惩罚逻辑回归(plr)对其构建最终模型。如图2b所示,10倍交叉验证结果显示,对于不同物种,hybridsucc的auc在0.840到0.961之间。明显高于单独使用一个特征得到的性能。如图2c所示,相较于单个dnn或plr算法,hybridsucc的auc实现了2.05-17.98%的相对提高。
4、我们仔细评估了我们方法的准确性和稳健性。结果显示,hybridsucc优于所有其他已提出的其他琥珀酰化位点预测模型,比如跟目前性能最高的模型相比,在通用物种上琥珀酰化位点预测的auc值从0.742提高到0.885的,相对改善超过19.27%,并且与其他已知模型相比,通过更广泛和有效的物种特异性琥珀酰化预测获得更可靠的结果,比如:根据结果,对于人类,4,6,8和10倍交叉验证的auc值分别为0.947,0.950,0.95和0.952,以及其他哺乳动物如小鼠和大鼠,hybridsucc通过多重交叉验证也表现良好,实现了高的平均auc值0.916和0.959。此外,对于酿酒酵母,4,6,8和10倍交叉验证的auc值分别显着达到0.955,0.958,0.960和0.960。除了在动物和真菌中具有出色的预测性能外,我们还发现hybridsucc可以在水稻中获得高预测性能,其4,6,8和10交叉验证auc值分别为0.938,0.939,0.942和0.944。此外,如图3所示,图3a反映了序列上各个位置的氨基酸偏好性的比较;图3b为显著出现在中心附近模体的比较;图3c为琥珀酰化修饰位点出现在蛋白质位置上的比较;图3d为琥珀酰化修饰赖氨酸位点的a-helix,β-strand和coil的比较;图3e为琥珀酰化修饰赖氨酸暴露面积上的比较;图3f为琥珀酰化修饰赖氨酸出现在无序或有序区域的偏好性的比较。已知琥珀酰化位点与预测的琥珀酰化位点的序列和结构的高度相似说明了琥珀酰化位点预测的可靠性,表明我们对实验结果的预测有用。
5、为方便广大使用者,利用php和javascript开发了基于hybridsucc模型的琥珀酰化位点预测平台(http://hybridsucc.biocuckoo.org/)。用户只需输入fasta格式的蛋白质序列,点击提交即可,预测结果如表1所示,id指用户输入的蛋白质名称或id。position指预测的琥珀酰化位点在蛋白质上的位置。peptide指以预测的琥珀酰化位点为中心的肽段情况。score为该位点的得分,得分越高越可能是琥珀酰化位点。cutoff指阈值,在阈值之上的为被预测的琥珀酰化位点。probability为该位点为琥珀酰化位点的概率。
表1是人类的pkm2蛋白质的琥珀酰化位点预测结果
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!