制备具有预定手性的单壁碳纳米管的方法与流程
一种制备SWCNT的方法为通过在催化剂表面上进行碳源分子(如乙醇或乙烯)的气相反应(也称为化学气相沉积CVD)。为了引发碳源分子的自发反应且因此在催化剂表面上生长SWCNT,催化剂粒子需要具有或多或少对应于要制备的SWCNT的直径的直径。此外,这些CVD方法典型地在相当高的温度下进行。通过气相反应制备SWCNT需要存在纳米大小的催化剂粒子,如例如Y.Homma等人在Nano Letters,第6卷,No.12,2006,第2642-2645页以及T.Maruyama等人在Mater.Express,第1卷,No.4,2011,第267-272页中所描述的。
如由R.Herges等人在Fragments of Fullerenes and Carbon Nanotubes,2012,John Wiley&Sons的第10章(“Aromatic Belts as Sections of Nanotubes”)中所论述,已开发了若干种基于模板的CNT合成方法,如由纳米管帽或纳米管带开始。对于CNT生长策略,提及了不同方法,尤其包括金属催化气相反应(CVD)。据描述,此类金属催化气相反应中所用的催化剂粒子具有的直径约为要制备的碳纳米管的直径。
US 2012/0177561 A1描述了通过将多环芳族化合物转化成SWCNT端帽来制备SWCNT,所述端帽充当SWCNT前体组件,其接着在狄尔斯-阿尔德(Diels-Alder)反应中生长为最终SWCNT,该反应在溶剂中进行,优选在不存在金属催化剂下进行。
K.Amsharov和A.Müller,Eur.J.Org.Chem.,2012,6155-6164描述了可适用于制备SWCNT前体组件的多环芳族化合物的制备。然而,实际上既未描述前体组件的制备也未描述前体组件生长为SWCNT。
本发明的一个目的是以高异构纯度提供具有预定手性的单壁碳纳米管(SWCNT)。
根据本发明的第一方面,该目的通过一种制备具有直径dSWCNT的单壁碳纳米管(SWCNT)的方法解决,该方法包括:
(i)提供前体组件,其包含单壁碳纳米管的片段SSWCNT,
片段SSWCNT由至少一个由邻位稠合苯环形成的环构成,且具有开放的第一端E1和与第一端E1相反的第二端E2,
所述前体组件任选进一步包含连接于片段SSWCNT的第二端E2的帽,
(ii)通过与碳源化合物在含金属的催化剂的表面上的气相反应使所述前体组件生长,其中前体组件与含金属的催化剂的表面经由片段SSWCNT的开放端E1接触,且含金属的催化剂为具有满足以下关系的平均直径dcat的粒子形式:dcat>2×dSWCNT,或为连续膜形式。
在本发明中已出人意料地实现了,若所述前体组件包含由至少一个由邻位稠合苯环形成的环构成的纳米管片段SSWCNT且该纳米管片段SSWCNT与金属催化剂经由其开放端E1接触,而纳米管片段SSWCNT的另一端可开放或通过SWCNT帽闭合,则前体组件通过气相反应选择性生长为预定手性的SWCNT不需要在直径与SWCNT直径相当的催化剂粒子存在下进行,而是可在较大催化剂粒子(其不需要具有较窄粒度分布)或甚至连续催化剂膜存在下有效实现。也如下文所论述,通过使用此类前体组件用于气相生长,不仅可使用较大尺寸CVD催化剂粒子或甚至连续催化剂膜,还可在低生长温度下提高产率。此外,步骤(ii)中使用较大催化剂粒子使风险最小化或甚至排除了碳源化合物在催化剂表面上自发地产生具有非限定结构的CNT且确保了这些碳源化合物仅与步骤(i)中已提供的SWCNT前体组件反应。
如技术人员所知,纳米管由手性向量C=na1+ma2规定,其代表了石墨烯片的滚动方向且因此代表了CNT手性。因此,碳纳米管的结构可由一对整数(n,m)代表。若m=0,则碳纳米管为所谓的锯齿状碳纳米管。若n=m,则碳纳米管为所谓的扶手椅型碳纳米管。另外的情形下,碳纳米管归类为手性的。
通过本发明的方法,可获得这些单壁碳纳米管结构中的任一种。换言之,本发明的单壁碳纳米管(SWCNT)可为扶手椅型单壁碳纳米管((n,n)-SWCNT)、锯齿状单壁碳纳米管((n,0)-SWCNT)或手性单壁碳纳米管((n,m)-SWCNT,其中n≠m)。
若SWCNT为扶手椅型单壁碳纳米管(即(n,n)-SWCNT),则整数n可在宽范围内变化,例如2至30,优选3至20或5至15。
若SWCNT为锯齿状单壁碳纳米管(即(n,0)-SWCNT),则整数n可在宽范围内变化,例如3至50,优选4至35或5至15。
若SWCNT为手性单壁碳纳米管(即(n,m)-SWCNT,其中n≠m),则整数n和m各自可在宽范围内变化,例如5≤n+m≤60或6≤n+m≤20。
如上文所指出,在本发明方法的步骤(i)中,提供单壁碳纳米管的前体组件,其中前体组件包含单壁碳纳米管的片段SSWCNT,该片段SSWCNT由至少一个由邻位稠合苯环形成的环构成且具有开放的第一端E1和与第一端E1相反的第二端E2。
该片段SSWCNT也可称为纳米管带。若许多这些片段SSWCNT经由它们的末端彼此连接,则它们形成预定的单壁碳纳米管。
片段SSWCNT的第二端E2也可为开放的。或者,前体组件进一步包含单壁碳纳米管的帽,该帽连接于片段SSWCNT的第二端E2(即,从而导致具有开放端E1和闭合端E2的纳米管片段SSWCNT)。
构成碳纳米管片段SSWCNT的所述一或多个环由邻位稠合苯环形成。其中任意两个相邻芳环具有两个且仅两个共同相邻原子的系统称为“邻位稠合”。若邻位稠合苯环序列中的第一苯环和最终苯环也彼此稠合,则形成碳纳米管片段SSWCNT的片段环。形成片段环的稠合苯环可全部为线性序列(此类环也称为环并苯(cyclacene))或角序列(此类环也称为环并苯(cyclophenacene)),或形成片段环的一些稠合苯环为线性序列而相同片段环中的其他稠合苯环为角序列。片段SSWCNT可具有另外的片段环(各片段环由邻位稠合苯环形成)。若存在两个或更多个片段环且相邻片段环彼此稠合,则这意味着第一环的“上边缘”及相邻环的“下边缘”由相同碳原子形成。然而,也可能的是片段SSWCNT仅由单个片段环构成。此外,也可能的是,片段SSWCNT在其端E1或E2处或在两端处并未由完整片段环封端,当然前提是存在至少一个由邻位稠合苯环形成的片段环。
优选地,片段SSWCNT由1至10个片段环构成(即,组成),各片段环由邻位稠合苯环形成。
优选地,前体组件由多环芳族化合物制备。
如技术人员所知,多环芳族化合物为含有稠合芳环作为结构组件的化合物。
在一个优选的实施方案中,前体组件由所述片段SSWCNT和要制备的SWCNT的帽构成,该帽连接于片段SSWCNT的第二端E2(从而导致具有闭合端E2和开放端E1的片段SSWCNT)。
技术人员已知,SWCNT的端帽(或其部分)可由多环芳族化合物制备。可参考US 2012/0177561 A1和K.Y.Amsharov,A.Müller,Eur.J.Org.Chem.,2012,第6155-6164页。在本发明中,若进行分子内环化,则优选使用多环芳族化合物,其不仅提供预定SWCNT的端帽自身而且提供另外“延伸的端帽”,该“延伸的端帽”含有连接于帽的碳纳米管片段SSWCNT(即由邻位稠合苯环形成的至少一个环)。
若片段SSWCNT具有通过SWCNT帽闭合的端E2,则片段环数目可为例如1至10。然而,由于多环芳族化合物的有机合成的复杂性可随分子大小增加而提高,因此片段环数目优选保持较低,例如1至3个片段环或1至2个片段环或甚至仅一个片段环。
图1示出了可由其制备(6,6)单壁碳纳米管的一种例示性前体组件的结构。在此例示性结构中,前体组件不仅由SWCNT片段(灰色阴影)构成而且另外在SWCNT片段的一端含有帽。SWCNT片段(灰色阴影区域)由片段环构成,这些片段环中的每一个由邻位稠合苯环形成。
用于制备前体组件(其优选由片段SSWCNT和连接于该片段SSWCNT的第二端E2的SWCNT帽构成)的多环芳族化合物可为例如具有中心苯环与基团R1至R6的式(1)化合物:
其中可相同或不同的基团R1、R3和R5各自具有下式(2):
其中
基团R7至R13中的至少一个为经取代或未经取代的苯基或多环芳族基;或
基团R7至R13中的彼此相邻的至少两个一起形成单环或多环芳族基;
可相同或不同的基团R2、R4和R6为氢;或具有下式(3):
其中
R7至R13具有与式(2)中相同的含义;或
表示连接中心苯环与式(2)中的基团R7或R13的化学键。
如上文已提及,基团R1、R3和R5可彼此相同或不同。同样情况适用于基团R2、R4和R6。此外,若存在的话,式(3)的基团可与式(2)的基团相同或不同。
任选地,若基团R2、R4和R6中的一个或多个具有式(3),则式(2)基团R1、R3和R5之一中的R7和相邻的式(3)基团R2、R4和R6之一中的R13,和/或式(2)基团R1、R3和R5之一中的R13和相邻的式(3)基团R2、R4和R6之一中的R7可一起表示/形成化学键。
若式(2)中的基团R7至R13之一为经取代的苯基,则一或多个取代基可为例如苯基。同样情况适用于式(3)中的基团R7至R13。
若式(2)中的基团R7至R13之一为多环芳族基,则该多环芳族基中的稠合苯环数目可在例如2至8,优选2至6或2至4内变化。同样情况适用于式(3)中的基团R7至R13。
若式(2)的基团R7至R13中的彼此相邻的至少两个一起形成单环芳族基,则其优选为六员芳环。同样情况适用于式(3)中的基团R7至R13。
若式(2)的基团R7至R13中的彼此相邻的至少两个一起形成多环芳族基,则该多环芳族基中的稠合苯环数目可在例如2至8,优选2至6或2至4内变化。同样情况适用于式(3)中的基团R7至R13。
若多环芳族基由两个稠合苯环形成,则其衍生自萘,该萘可为经取代或未经取代的。
若多环芳族基由三个稠合苯环形成,则其衍生自菲或蒽,其中各自可为经取代或未经取代的。
若多环芳族基由四个稠合苯环形成,则其衍生自例如四苯(tetraphene,即苯并[a]蒽)、并四苯(tetracene)、三联苯(即9,10-苯并菲)、苯并[c]菲或芘,其中各自可为经取代或未经取代的。
若多环芳族基由五个稠合苯环形成,则其衍生自例如二苯并蒽如二苯并[a,h]蒽、二苯并[a,c]蒽、1,2:7,8-二苯并蒽(也称为二苯并[a,j]蒽)和1,2:5,6-二苯并蒽;苯并如苉(也称为苯并[a]和苯并[b]并五苯(pentacene);二苯并菲如1,2:3,4-二苯并菲和3,4,5,6-二苯并菲;苯并并四苯如苯并[a]并四苯;苯并芘如苯并[a]芘和苯并[e]芘;其中各自可为经取代或未经取代的。
若多环芳族基由六个稠合苯环形成,则其衍生自例如苯并苉,如苯并[s]苉;苯并并五苯;苯并苝;或二苯并芘;其中各自可为经取代或未经取代的。
或者,用于制备前体组件(其优选由片段SSWCNT和连接于片段SSWCNT的第二端E2的SWCNT帽构成)的多环芳族化合物可为例如式(4)化合物:
其中
基团R1至R4中的至少一个为经取代或未经取代的苯基或多环芳族基;或
基团R1与R2和/或基团R3与R4一起形成单环或多环芳族基。
关于例示性多环芳族基,可参考前面提供的说明。
若基团R1至R4中的一个或多个为经取代的苯基,则一或多个取代基可为例如苯基。
在式(4)中,基团R1和R2彼此独立地可为例如氢、经取代或未经取代的苯基或多环芳族基;且基团R3与R4可一起形成带有苯基或联苯取代基的苯环,或基团R3与R4可一起形成具有2至6个稠合苯环的多环芳族基,其可为经取代或未经取代的。
或者,用于制备前体组件(其优选由片段SSWCNT和连接于片段SSWCNT的第二端E2的SWCNT帽构成)的多环芳族化合物可为例如碗烯(corannulene)化合物。
可用于制备(6,6)扶手椅型SWCNT的一种例示性多环芳族化合物具有以下式(I)和式(II)之一:
可用于制备(9,9)扶手椅型SWCNT的一种例示性多环芳族化合物具有下式(III):
可用于制备(9,0)锯齿状SWCNT的一种例示性化合物具有下式(IV):
其中R为苯基(即,-C6H5)。
可用于制备(12,6)手性SWCNT的一种例示性化合物具有以下式(V)和式(VI)之一:
可用于制备(12,0)锯齿状SWCNT的一种例示性化合物具有以下式(VII)和式(VIII)之一:
可用于制备(12,3)手性SWCNT的一种例示性化合物具有下式(IX):
可用于制备(12,1)手性SWCNT的一种例示性化合物具有下式(X):
可用于制备(11,3)手性SWCNT的一种例示性化合物具有下式(XI):
可用于制备(12,2)手性SWCNT的一种例示性化合物具有下式(XII):
可用于制备(10,6)手性SWCNT的一种例示性化合物具有下式(XIII):
可用于制备(11,5)手性SWCNT的一种例示性化合物具有下式(XIV):
可用于制备(10,7)手性SWCNT的一种例示性化合物具有下式(XV):
可用于制备(11,4)手性SWCNT的一种例示性化合物具有下式(XVI):
可用于制备(8,2)手性SWCNT的一种例示性化合物具有下式(XVII):
可用于制备(7,4)手性SWCNT的一种例示性化合物具有下式(XVIII):
可用于制备(11,0)锯齿状SWCNT的一种例示性化合物具有下式(XIX):
可用于制备(10,2)手性SWCNT的一种例示性化合物具有下式(XX):
可用于制备(7,7)扶手椅型SWCNT的一种例示性化合物具有下式(XXI):
可用于制备(8,8)扶手椅型SWCNT的一种例示性化合物具有下式(XXII):
可用于制备(9,5)扶手椅型SWCNT的一种例示性化合物具有下式(XXIII):
在任意这些化合物(I)至(XXIII)中,至少一些氢原子可被卤素原子(如Cl、Br或I)替代。
若进行分子内环化,则多环芳族化合物(I)至(XXIII)中的每一个形成这样的前体组件,该前体组件由SWCNT片段SSWCNT和连接于该片段的一端的SWCNT帽组成。如上文所论述,各前体组件的片段SSWCNT由至少一个由邻位稠合苯环形成的环构成且具有开放端E1。
若多环芳族化合物用于提供前体组件,则所述多环芳族化合物可通过技术人员原则上已知的有机合成程序来制备。
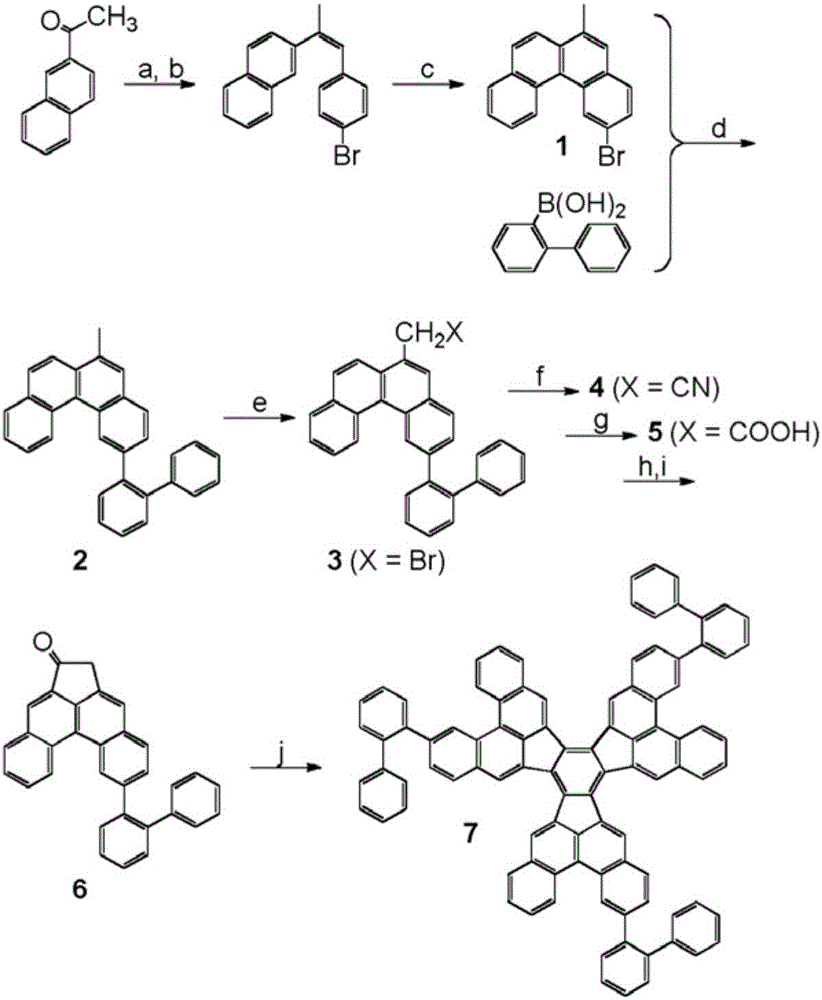
仅作为举例,多环芳族化合物(I)和(II)的合成途径示于图2和图3中。
图2中所示的制备式(I)的多环芳族化合物(在图2中:化合物7)的合成途径包括以下步骤:a)PPh3、甲苯,回流,95%;b)BrPh3PCH2PhBr、KOtBu、EtOH,回流,81%;c)I2、hv、环氧丙烷、环己烷,72%;d)Pd(PPh3)4、Cs2CO3、甲苯/MeOH,110℃,79%;e)NBS、DBPO、CCl4,回流,70%;f)NaCN、DMSO,RT,40%;g)H2SO4、H2O、HOAc,回流,98%;h)SOCl2,65℃;i)AlCl3、CH2Cl2,RT,57%;j)丙酸、TsOH、o-DCB,180℃,65%。
图3中所示的制备式(II)的多环芳族化合物(在图3中:化合物19)的合成途径包括以下步骤:a)Pd(PPh3)4、K2CO3、甲苯/MeOH,110℃,86%;b)I2、hv、环氧丙烷、环己烷,52%;c)NBS、DBPO、CCl4,回流,87%;d)PPh3、甲苯,回流,71%;e)P(OEt)3,160℃,60%;f)C10H7COCH3、KOtBu、THF,50℃,31%;g)I2、hv、环氧丙烷、环己烷,24%;h)NBS、DBPO、CCl4,回流,60%;i)NaCN、DMSO,RT,75%;j)H2SO4、H2O、HOAc,回流,73%;k)SOCl2,65℃;1)AlCl3、CH2Cl2,RT,36%;m)TiCl4、o-DCB,180℃,35%。
前体组件可通过已知方法如分子内环化由多环芳族化合物制备。优选的分子内环化反应为环化脱氢、环化脱卤和Bergman环化。
当多环芳族化合物进行转化步骤如环化脱氢时,多环芳族化合物的芳环经由分子内反应获得连接且芳族化合物(其可典型地视为或多或少平坦的分子)转化成由管片段SSWCNT和SWCNT帽构成的“碗状”SWCNT前体组件,其中SWCNT帽将管片段的一端闭合而管片段的另一端保持开放。
若分子内环化如环化脱氢用于将多环芳族化合物转化成前体组件,则其优选为表面辅助(也称为“表面催化的”)分子内环化。适当方法条件和其上可进行多环芳族化合物的分子内环化(如环化脱氢)的表面是技术人员已知的。在上下文中,可参考例如K.Amsharov等人,Angew.Chem.Int.Ed.2010,第9392-9396页,以及G.Otero等人,Nature,第454卷,2008,第865-868页。
在一个优选的实施方案中,表面辅助分子内环化在含金属的催化剂的表面上,更具体而言在含金属的催化剂的金属表面上进行。优选地,金属为Pd、Pt、Ru、Ir、Rh、Au、Ag、Fe、Co、Cu、Ni或其任意混合物或合金。
多环芳族化合物可通过适合于将有机化合物沉积于表面上的任何方法沉积于催化剂表面上。该方法可为例如真空沉积(升华)方法;基于溶液的方法,如旋涂、喷涂、浸涂、印刷、电喷雾沉积、脉冲喷射沉积;干接触转印或干压印;激光诱导的解吸附方法或制备型质谱法(尤其用于沉积仅以不纯混合物可得的化合物)。
若多环芳族化合物包含可围绕单键旋转的基团(例如在分子外周的苯基或联苯基团)并因此显示了构象异构,则沉积于催化剂表面上的分子可具有不同构象。然而,只有以适当构象吸附在催化剂表面上的那些分子将形成(例如通过环化脱氢)SWCNT前体组件,其继而将在步骤(ii)的气相反应中生长为最终SWCNT。任何以不适当构象吸附的分子将不会形成可随后在步骤(ii)中生长的SWCNT前体组件。因此,在步骤(ii)中仅真正形成具有预定手性的那些SWCNT。换言之,通过本发明的方法,可获得极高纯度(即纯度为约100%)的具有预定手性的SWCNT。
优选地,多环芳族化合物在条件下沉积以便实现对于使前体组件和因此SWCNT的产率优化而言足够高但对于尽可能地抑制相邻化合物之间的接触而言足够低的表面密度(即每单位面积催化剂表面沉积的化合物数目)。若二或更多相邻多环芳族化合物接触,则在转化步骤(i)之后获得的所限定的前体组件的产率可能降低。
为了使多环芳族化合物与催化剂表面之间的接触面积最大化,从而提高环化脱氢步骤的效率,含金属的催化剂粒子的平均直径应保持足够大。
优选地,步骤(i)的含金属的催化剂为具有满足以下关系的平均直径dcat的粒子形式:dcat>2×dSWCNT,更优选dcat>4×dSWCNT,甚至更优选dcat>10×dSWCNT或dcat>20×dSWCNT或dcat>50×dSWCNT,或为连续膜形式。
步骤(i)中的含金属的催化剂的粒子的平均粒度可为例如至少5nm,更优选至少10nm,甚至更优选至少20nm或至少50nm或至少100nm,条件为dcat>2×dSWCNT。
平均粒度可通过图像分析(例如由透射电子显微术(TEM)或扫描电子显微术(SEM)获取的图像)测定。在该基于图像的粒度分析中,测定TEM(或SEM)图像上所示的粒子的粒度分布。单独粒子的粒度为可由图像获得的其最大延伸。粒度分布曲线的峰值为平均粒度。
术语“连续膜形式的催化剂”意指由催化剂提供且多环芳族化合物沉积其上的表面的尺寸远超过芳族化合物的分子大小。优选地,此类膜形式的催化剂提供平坦表面,从而提高多环芳族化合物与催化剂表面之间的相互作用且提高分子内环化效率。
步骤(i)中所用的催化剂可部分或全部为结晶的。然而,也可能的是,步骤(i)中的催化剂为无定形的。
可在步骤(i)中用于分子内环化的催化剂是技术人员通常已知的且为市售的或可通过通常已知的标准方法制备。
如技术人员所知,SWCNT的直径可根据以下等式计算:
dSWCNT=(n2+m2+nm)1/2x 0.0783nm
其中n和m为定义手性向量且因此SWCNT结构的整数。此外,如上文已讨论,通过选择适当的多环芳族化合物,获得具有特征性n,m-整数的特定SWCNT。根据这些已知整数n和m,可计算SWCNT的直径。
在步骤(i)中将多环芳族化合物转化成SWCNT前体组件的适当方法条件原则上为技术人员所知。
进行分子内环化的温度可在宽范围内变化。优选地,步骤(i)在100℃至1000℃,更优选200℃至800℃或200℃至600℃的温度T1下进行。
或者,也可使用在两端均为开放的纳米管片段SSWCNT,而非具有开放端E1及闭合端E2的纳米管片段SSWCNT。
此类在两端均为开放的短纳米管片段可以化学方式,例如经由Pure Appl.Chem.,2012,第84卷,No.4,第907-916页中所述的化学反应来制备。或者,将SWCNT切成较小片段,例如电子束光刻和氧等离子体离子蚀刻(参见例如Nano Letters,2009,第9卷,No.4,第1673-1677页)。
如上文所指出,本发明的方法包括步骤(ii),该步骤(ii)包括通过与碳源化合物在含金属的催化剂的表面,优选金属表面上的气相反应使前体组件生长,其中前体组件与含金属的催化剂的表面经由片段SSWCNT的开放端E1接触,且含金属的催化剂为具有满足以下关系的平均直径dcat的粒子形式:dcat>2×dSWCNT,或为连续膜形式。
碳源化合物(即含有一或多个碳原子的化合物)可选自典型地用于通过化学气相沉积(CVD)制造碳纳米管的那些化合物。此类碳源化合物是技术人员通常已知的。
可用于本发明的步骤(ii)的碳源化合物为例如烷烃,如C1-4烷烃(优选甲烷或乙烷);烯烃,如乙烯;炔烃,如乙炔;醇,如C1-4醇(优选甲醇或乙醇);芳族化合物,如苯;一氧化碳;含氮有机化合物(例如胺);含硼有机化合物或其任意混合物。碳源化合物可与可使SWCNT掺杂有杂原子的含杂原子的非碳化合物(例如NH3等)混合。
除了碳源化合物之外,可使用一或多种载气,如H2或H2O。在经由气相反应的碳纳米管制造过程中使用此类载气是技术人员通常已知的。
作为含金属的催化剂的金属,可提及典型地用于通过化学气相沉积(CVD)制造碳纳米管的那些金属。优选地,金属为Pd、Pt、Ru、Ir、Rh、Au、Ag、Fe、Co、Cu、Ni或其任意混合物或合金。
可使步骤(i)中制备的SWCNT前体组件与含金属的催化剂的表面接触并通过技术人员通常已知的方法将其沉积于含金属的催化剂的表面上。若在步骤(i)中使用含金属的催化剂且其不同于步骤(ii)的含金属的催化剂,则可通过通常已知的方法将前体组件转移至步骤(ii)的含金属的催化剂的表面。然而,如下文将进一步详细讨论,优选在步骤(i)和步骤(ii)中使用相同的含金属的催化剂。在该优选实施方案中,不需要将SWCNT前体组件由第一催化剂转移至第二催化剂。换言之,在该优选实施方案中,步骤(ii)包括通过与碳源化合物在步骤(i)的含金属的催化剂的表面上的气相反应使前体组件生长。
优选地,步骤(ii)的含金属的催化剂为具有满足以下关系的平均直径dcat的粒子形式:dcat>4×dSWCNT,更优选dcat>10×dSWCNT或dcat>20×dSWCNT或dcat>50×dSWCNT。
如上文所提及,技术人员已知,SWCNT的直径可根据以下等式计算:
dSWCNT=(n2+m2+nm)1/2x 0.0783nm
其中n和m为定义手性向量且因此SWCNT结构的整数。此外,通过选择适当的多环芳族化合物,获得具有特征性n,m-整数的特定SWCNT。根据这些已知整数n和m,可计算SWCNT的直径。
步骤(ii)中的含金属的催化剂的粒子的平均粒度可为例如至少5nm,更优选至少10nm,甚至更优选至少20nm或至少50nm或至少100nm,条件为dcat>2×dSWCNT。
平均粒度可通过图像分析(例如由透射电子显微术(TEM)或扫描电子显微术(SEM)获取的图像)测定。在该基于图像的粒度分析中,测定TEM(或SEM)图像上所示的粒子的粒度分布。单独粒子的粒度为可由图像获得的其最大延伸。粒度分布曲线的峰值为平均粒度。
术语“膜形式的催化剂”意指由催化剂提供且多环芳族化合物沉积其上的表面的尺寸远超过芳族化合物的分子大小。优选地,此类膜形式的催化剂提供平坦表面,从而提高多环芳族化合物与催化剂表面之间的相互作用且提高分子内环化效率。
步骤(i)中所用的催化剂可部分或全部为结晶的。然而,也可能的是,步骤(ii)中的催化剂为无定形的。
在一个优选实施方案中,步骤(i)中也使用含金属的催化剂,且步骤(i)的含金属的催化剂与步骤(ii)的含金属的催化剂相同。
步骤(ii)的温度T2可在宽范围内变化,例如100℃至1000℃。
在本发明中出人意料地实现,当步骤(ii)在低温下进行时,SWCNT的产率可甚至得到进一步提高。因此,在一个优选实施方案中,步骤(ii)在700℃或更低,更优选600℃或更低、甚至更优选550℃或更低的温度下进行。适当温度范围为例如300℃至700℃,更优选350℃至600℃,甚至更优选350℃至550℃。
优选地,步骤(ii)的温度保持足够低以便在步骤(ii)中避免任何自发形成CNT。换言之,步骤(ii)的温度优选足够低以使得碳源化合物仅引发步骤(i)中已提供的SWCNT前体组件的生长但不在催化剂表面上产生另外的CNT,因为这些另外的CNT将不具有明确限定的均匀手性。若步骤(ii)的催化剂粒子相当大,则此可能已经足以用于抑制在步骤(ii)中形成另外的CNT。若步骤(ii)中使用较小催化剂粒子(但仍符合关系dcat>2×dSWCNT),则可能存在自发形成非均匀手性的CNT的剩余风险。然而,由于本发明中所用的SWCNT前体组件可与碳源化合物甚至在低温下反应,因此步骤(ii)可在对于抑制另外的CNT自发形成而言足够低但对于引发步骤(i)中所提供的SWCNT前体组件的生长而言足够高的温度下进行。
步骤(ii)中进行气相反应(以及因此SWCNT生长)的压力可在宽范围内变化。气相反应可在相当低的压力下进行,如小于10-4毫巴或小于10-5毫巴或甚至小于10-6毫巴,但仍提供足够的生长率。若在低压下工作,则可提高最终SWCNT的纯度。然而,也可在较高压力下进行气相反应同时仍获得具有高品质和极高异构纯度的SWCNT。
原则上,步骤(ii)可继续直至SWCNT已生长至所需长度。此外,若需要,SWCNT端帽可在后期移除以便获得开端型SWCNT。
若需要,本发明的SWCNT可通过技术人员通常已知的方法与含金属的催化剂的表面分离。
根据另一方面,本发明提供可通过上述方法获得的单壁碳纳米管。
如上文所提及,可通过本发明的方法以极高纯度获得具有均匀结构或手性(即扶手椅型或锯齿状或手性)的SWCNT。因此,在一个优选实施方案中,单壁碳纳米管的异构纯度为至少90%,更优选至少95%或甚至至少98%,最优选100%。异构纯度为例如95%意指,100个SWCNT中的95个SWCNT由相同整数n和m表征。
由于所述制备方法,本发明的单壁碳纳米管可提供于含金属的基底的表面上,含金属的基底对应于上述含金属的催化剂且为连续膜形式或为具有满足以下关系的平均直径d基底的粒子形式:d基底>2×dSWCNT。
可用于制备本发明的前体组件的一些多环芳族化合物尚未描述于先前技术中。因此,根据另一方面,本发明提供具有式(I)至式(XXIII)之一的多环芳族化合物:
其中R为苯基(即,-C6H5)
根据另一方面,本发明涉及式(I)至式(XXIII)的多环芳族化合物制备单壁碳纳米管的用途。
现将通过以下实施例进一步详细说明本发明。
实施例
表征方法
在恒定电流模式和77K样品温度下使用低温扫描隧道显微镜进行STM测量。将系统固持在基础压力为1×10-11毫巴的独立UHV腔室中。在Bruker Senterra仪器中记录拉曼光谱,光谱分辨率为4cm-1,使用532和782nm的激光,功率为20mW。在Carl Zeiss Orion Plus仪器中以30keV束能量和0.4pA的束电流进行He离子扫描显微术测量。
多环芳族化合物的制备
制备式(I)和式(II)的多环芳族化合物。
式(I)化合物通过根据图2中所示的合成途径的多步有机合成来获得。通过将2-乙酰萘进行标准Wittig烯烃化成相应的茋,随后进行利用Katz改进(Katz-improvement)的Mallory光环化反应,获得2-溴-6-甲基苯并菲(1)。1与2-二苯基硼酸的Suzuki偶联得到2。用N-溴琥珀酰亚胺进行苄型溴化,随后在DMSO中用氰化钠处理得到化合物4。接着将氰基化合物水解成相应乙酸并以两个步骤转化成6。首先与亚硫酰氯反应得到酰氯,接着用于Friedel-Crafts酰化以实现闭环。最终合成步骤,即转化成式(I)化合物(图2中称为7),使用TiCl4或布朗斯台德酸条件通过羟醛环化三聚进行。两种情况均以令人满意的产率获得所需化合物。
式(II)化合物通过根据图3中所示的合成途径的多步有机合成来获得。通过联苯-2-硼酸与4-溴甲苯的Suzuki偶联产生8,随后进行使用Katz改进的Mallory光环化反应,制得2-甲基三联苯。用N-溴琥珀酰亚胺对9进行苄型溴化得到2-溴甲基三联苯,随后将其转化成相应Wittig盐11或膦酸盐12。尽管不可通过11与乙酰萘的Wittig反应获得茋13,但使用12和13的Horner-Emmons-Wadsworth反应是成功的。接下来的Mallory光环化反应导致形成所需的化合物14。接着用N-溴琥珀酰亚胺对14进行苄型溴化,随后在CH2Cl2中,在四丁基溴化铵存在下用氰化钾处理并最终水解,获得相应乙酸17。接着通过用亚硫酰氯处理将17转化成酰氯,随后使用Friedel-Crafts酰化实现闭环,获得酮18。对于18的三聚合,在o-DCB中在180℃下进行与TiCl4的反应。获得式(II)化合物(图3中称为19)。
通过多环芳族化合物的分子内环化制备(6,6)单壁碳纳米管的前体组件
式(I)和式(II)的多环芳族化合物分别在来自Knudsen单元型蒸发器的UHV中以速率在预先清洁的Pt(111)表面上在RT下进行蒸发。
将由表面制备实验室(SPL)获得的Pt单晶用作环化脱氢催化剂。通过用能量为1KeV的Ar离子首先在室温下且接着在1100K下进行标准溅射,随后在1370K下进行无离子轰击的最终快速退火来清洁表面。
在约200℃下进行后退火。在约500℃下的进一步退火诱导分子的完全表面催化环化脱氢,从而形成要制备的(6,6)SWCNT的所需前体组件。
前体组件由SWCNT片段和连接于SWCNT片段的一端的SWCNT帽构成,而SWCNT片段的另一端保持开放。SWCNT片段由通过邻位稠合苯环形成的片段环(或者说两个或更多环)构成。前体组件的结构示于图1中。SWCNT片段由灰色阴影部分表示,而闭合SWCNT片段的一端的帽由无阴影部分表示。
图4示出了在RT下(图4(a))和在退火之后在500℃下(图4(b))沉积于Pt表面上的多环芳族分子的STM图像。由于Pt单晶和其表面远大于沉积分子的尺寸,Pt表面反映了或多或少连续和平坦膜的情况,其上施加了多环芳族分子。
图5示出了多环芳族分子在环化脱氢之前的线轮廓(实线)和在环化脱氢之后获得的前体组件的线轮廓(点线)。如由图5可以看出,高度由约0.2nm(或多或少平坦芳族化合物的高度)增加至约0.45nm的高度,后者为“碗状”前体组件的高度。
在图6中示出了,式(II)的多环芳族化合物存在不同构象异构体。然而,这些构象异构体中仅一种形成由SWCNT片段(在此特定情况下为(6,6)-SWCNT片段)和SWCNT帽构成的弯曲碗状元件。仅此具有“正确”构象的特定碗状元件将随后在CVD步骤中生长。在图6中,最左侧的构象异构体形成前体组件(由两个不同视角示出),其由(6,6)-SWCNT片段和连接于该管片段的一端的帽组成。
通过与碳源分子的气相反应使前体组件生长为异构纯的(6,6)SWCNT
用于使前体组件生长为所需(6,6)SWCNT的碳源化合物分别为乙烯(C2H4)和乙醇(C2H5OH)。在腔室中维持1×10-7毫巴的压力。将基底在1h期间在400℃或500℃下退火。为了在低剂量实验中获得控制,使用1×10-8毫巴的压力。
对于CVD生长步骤,将前体组件留在已在步骤(i)中使用的环化脱氢催化剂的表面上。因此,步骤(ii)的含金属的催化剂与步骤(i)中所用的相同。如上文已提及,Pt单晶催化剂和其表面远大于沉积分子和由其制备的前体组件的尺寸,因此Pt表面反映了或多或少连续和平坦催化剂膜的情况,在其上制造异构纯SWCNT。
图7示出了SWCNT前体组件在与碳源化合物反应之前的线轮廓(实线)和在已通过馈入碳源化合物引发生长期之后的线轮廓(虚线:1朗缪尔(Langmuir)剂量;虚线:5朗缪尔剂量)。如由图7所说明的,由(6,6)-SWCNT片段和连接于该管片段的一端的帽组成的前体组件将通过气相反应生长为所需SWCNT。
图8示出了长度为300nm的SWCNT的扫描He离子显微图像。
图9示出了SWCNT拉曼光谱,该图与单分散异构纯(6,6)-SWCNT相符。拉曼光谱呈现(6,6)SWCNT在预期位置处的定义非常明确的谱带。在295cm-1处的极窄的谱带与(6,6)管的径向呼吸模式(RBM)相关。RBM为正切平面外声学模式,其频率很大程度上取决于纳米管直径。极其重要的是要指出,光谱未显示RBM范围(200-400cm-1)内的任何另外的谱带,甚至在以λ=782nm红激光照射下也不显示,因此证实了该方法的极高选择性。另外,在通过使用较低放大物镜照射大得多的面积时,拉曼光谱呈现相同谱带。该图的插图示出了通过照射明80μm2面积所记录的RBM谱带,因此确保了大量SWCNT的测量且因此说明了该方法的高选择性。与石墨烯晶格的平面内光学振动相关的G谱带在1518cm-1和1591cm-1处呈现为双峰。小直径SWCNT的显著曲率使G峰迁移至较低频率,尤其是与横向(垂直于管轴)原子位移(G-)相关的振动,其呈现相对于sp2石墨烯晶格振动的向下位移。所观测到的分裂与具有相似直径和手性的SWCNT一致。先前已观测到400-1200cm-1范围内的额外峰且将其用作扶手椅型SWCNT存在的证据,因为在半导体管中没有该范围内的峰存在。
本文中报导的方法的极高清洁度产生预定手性和无缺陷的SWCNT。一个证据是在拉曼光谱中不存在任何D谱带。
SWCNT结构的高分辨率STM图片示于图10中,其进一步证明了在本发明中制备的SWCNT的真正单手性。在图10中,可观测到SWCNT呈现在管轴方向上的较高对比度线中的内部结构。将SWCNT的模型叠加在STM图像上以进一步证实那些较高对比度线实际上为石墨烯结构在(6,6)SWCNT中的碳位置。管直径和石墨烯晶格的周期性的突出一致性说明了观察到的1-D结构为预期的(6,6)SWCNT。
步骤(ii)中温度对产物品质和产率的影响
图11示出了步骤(ii)中在500℃(实线)和400℃(点线)的温度下制备的(6,6)-SWCNT的拉曼光谱。
两种光谱都与异构纯(6,6)-SWCNT一致。然而,通过比较D/G谱带与RBM/G之间的相对强度,可判断当在较低温度下操作时可以较高产率获得所需的预定SWCNT。
- 还没有人留言评论。精彩留言会获得点赞!