一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法与流程
本发明属于陶瓷制备技术领域,尤其涉及一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法。
背景技术:
陶瓷纳米粉体的定义是直径小于100nm。一方面由于其具有高比表面积和高活性,可以明显降低烧结温度,陶瓷材料的烧结温度一般比较高,如果能有效降低烧结温度是非常有实际价值的;另一方面由于其粉体粒径非常小(小于100nm),使得其烧结后陶瓷晶粒的尺寸也很小,因为陶瓷晶粒的尺寸很大程度上取决于原料陶瓷粉体的尺寸。更小的(亚微米级)的陶瓷晶粒可以明显减小微观缺陷(如气孔)的尺度,也可以明显提高陶瓷材料的机械强度和韧性以及其他的特性(如电学、磁学、光学等),如果可以达到纳米尺度的晶粒获得具有纳米晶粒的烧结体材料,还可以产生纳米效应。随着材料科学技术的发展,人们越来越意识到纳米材料在陶瓷领域应用的重要性,合成纳米材料的化学和物理法非常多,也得到很大的发展,但是如何制备易烧结的陶瓷纳米粉体的一直是国内制粉产业的瓶颈问题。
很多商业化的纳米粉体其烧结性能并不好,一方面主要是因为纳米粉体中存在团聚体;另一方面纳米粉体的形貌不是近球形,比表面积极大(适合于做催化剂,比表面积达到几十到一百多m2/g),表面缺陷非常多。这种粉体作为陶瓷材料十分难成型,或成型的密度很低,这样严重影响试样烧结的致密度或提高烧结温度。如何解决比表面积巨大(大于30m2/g)、颗粒形貌异形、表面缺陷非常多的陶瓷纳米粉体的致密低温烧结。通常情况下比表面积在小于20m2/g陶瓷粉体才具有比较好的成型性和烧结性,对于流延成型其比表面积要求更小,最好要小于10m2/g,如果高了,会明显增大流延浆料的粘度,降低固含量,使得流延坯片的密度降低或坯片开裂(无法成型),进而影响烧结温度和烧结密度等。比表面积巨大的商业纳米粉体,其表面缺陷多,微晶的尺寸都非常小,粉体的结晶程度较低。粉体微晶尺寸如果小于100nm时,粉体的xrd(x光衍射谱)的峰就会展宽,通过scherrer公式可以计算出粉体的微晶尺寸。因此粉体除了粒径外,其比表面积和微晶尺寸也是非常重要的指标。但是由于制备工艺原因,很多的陶瓷纳米粉体都具有较高的比表面积,粉体的微晶尺寸很小(小于20nm),所以普遍存在难以实现低温烧结,往往是烧结不致密,重要原因是成型的坯体密度无法达到基本的致密度。
一般的纳米粉体可以通过造粒的方法改善其成型性能。造粒法是将纳米粉体的一次粒子分散后通过一定量的粘结剂粘结粉体的一次颗粒获得球形的尺寸较大的(亚微米级)二次颗粒。这种造粒的方法处理的粉体仅仅适用于干法成型,对于湿法成型(如流延法、注浆法等)是不适合的,因为造粒的粉体会在溶剂中化开仍然以一次颗粒的形式存在于溶剂中。因此特别对于采用湿法成型的纳米粉体,通过造粒方法处理粉体是不合适的。但是如果是简单的煅烧来提高纳米粉体的颗粒的微晶尺寸,其结果是大大降低粉体的烧结活性,明显提高粉体的烧结温度。
技术实现要素:
有鉴于此,本发明的目的在于提供一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法。本发明提供的方法既能保持陶瓷纳米粉体的高活性,同时提高陶瓷纳米粉体的成型性及烧结性。
为了实现上述发明目的,本发明提供以下技术方案:
一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法,包括以下步骤:
将陶瓷纳米粉体与小分子有机分散剂进行第一湿法球磨,得到湿法球磨物料,所述陶瓷纳米粉体的比表面积大于20m2/g,所述陶瓷纳米粉体的微晶尺寸小于20nm;
将所述湿法球磨物料与粘结剂混合进行第二湿法球磨,得到分散球磨物料;
将所述粉体进行煅烧。
优选地,所述陶瓷纳米粉体的微晶尺寸由xrd测得并由scherrer公式计算获得。
优选地,所述小分子有机分散剂为聚丙烯酸铵、柠檬酸、乙酰丙酮或三乙醇胺。
优选地,所述小分子有机分散剂的用量为陶瓷纳米粉体质量的1~6%。
优选地,所述陶瓷纳米粉体、球磨介质和球磨溶剂的质量比为1:2:0.5~1:3:2,所述球磨介质为氧化锆磨球,所述氧化锆磨球的直径为0.3~0.8mm。
优选地,所述第一湿法球磨包括依次进行的快速球磨和慢速球磨,所述快速球磨的时间为10~60min,速度为300~400转/分,所述慢速球磨的时间为2~16h,速度为100~250转/分;所述第二湿法球磨的速度为100~250转/分,时间为2~24h。
优选地,所述粘结剂为聚乙烯醇、聚乙烯吡咯烷酮或聚乙烯醇缩丁醛。
优选地,所述造粒时的压强为10~60mpa。
优选地,所述造粒后粉体的粒径为41~250μm。
优选地,所述煅烧的温度为600~1000℃,在最高煅烧温度保温的时间为1~10h。
本发明提供了一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法,包括以下步骤:将陶瓷纳米粉体与小分子有机分散剂进行第一湿法球磨,得到湿法球磨物料,所述陶瓷纳米粉体的比表面积大于20m2/g,所述陶瓷纳米粉体的微晶尺寸小于20nm;将所述湿法球磨物料与粘结剂混合进行第二湿法球磨,得到分散球磨物料;将所述分散球磨物料进行造粒,得到粉体;将所述粉体进行煅烧。本发明中,由于陶瓷纳米粉体自聚能力较大,存在很多的软(硬)团聚体,采取球磨分散的方法进行第一湿法球磨,使粉体一次粒子表面吸附小分子有机分散剂后,陶瓷纳米粉体的一次粒子在溶剂中能够分散稳定存在,接着加入粘结剂,进行第二湿法球磨,粘结剂分子将几个一次粒子缠绕在一起,造粒进一步将一次粒子紧密地粘结在一起,煅烧的目的是将几个紧密粘结的纳米晶粒通过高温形成晶界得到多晶粉体颗粒(尺寸为亚微米级)或者长大的单个晶粒,使多表面缺陷(表面凹凸不平)的微晶变成表面较光滑的粉体颗粒,降低了陶瓷纳米粉体的比表面积。本发明将造粒与煅烧相结合来处理陶瓷纳米粉体,一方面既保持陶瓷纳米粉体的纳米尺寸和高活性(高的晶界面积具有高的烧结催动力),另一方面可控制地将多个一次粒子煅烧在一起组成二次颗粒(多晶或单个晶粒),这样陶瓷纳米粉体的颗粒变大了,易于成型。同时煅烧使得陶瓷纳米粉体颗粒的结晶程度提高,颗粒表面的缺陷减少,原来多表面缺陷(表面凹凸不平)的微晶转变成光滑的表面微晶,比表面积降低,达到比较高的固含量,大大提高坯体的密度,进而可以在较低温度下获得致密烧结体,可有效解决陶瓷纳米粉体很难烧结的瓶颈问题。且本发明方法简单、低成本,不仅在实验室可以实施,而且在工业上也可以实施。实施例的数据表明,本发明提供的提高陶瓷纳米粉体的湿法成型性及烧结性的方法可以在1300℃烧成,保温3h后的体积密度为7.02g/cm3,相对密度达到97%。
附图说明
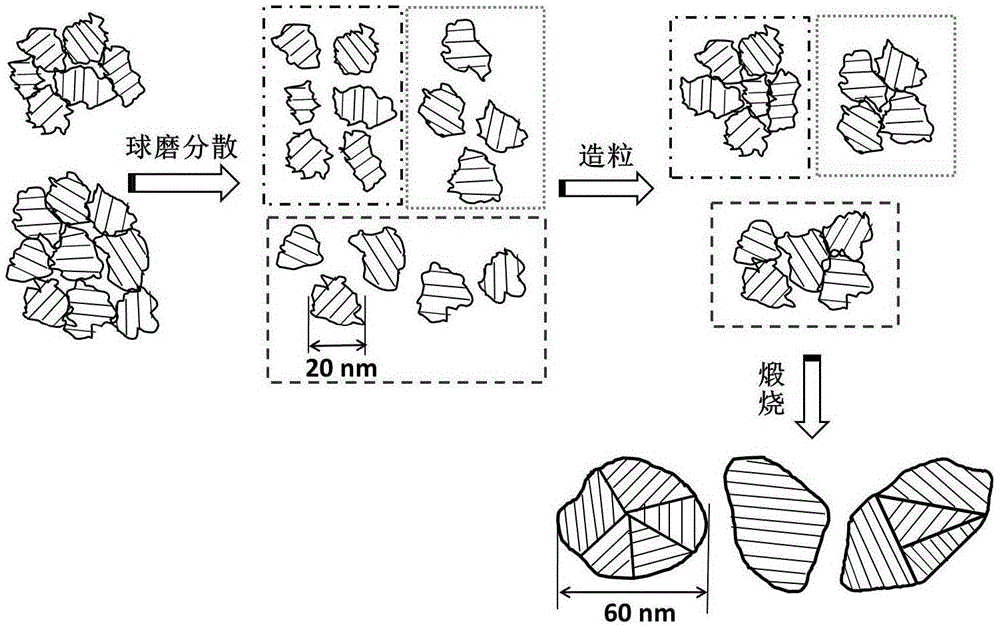
图1为本发明提高陶瓷纳米粉体的湿法成型性及烧结性的方法的流程图;
图2为实施例1中900℃煅烧前后的gdc粉体xrd谱图;
图3为实施例1中900℃煅烧后的gdc粉体在不同放大倍数下的tem图;
图4为实施例1中1300℃烧成后gdc断面sem图;
图5为实施例1中1300℃烧成后的gdc基片实物图;
图6为实施例2中800℃煅烧前后的nio粉体xrd图谱;
图7为实施例2煅烧前的nio粉体在不同放大倍率下的tem图;
图8为实施例2中1400℃烧成后的sofc阳极实物图;
图9为实施例2中1400℃烧成后的sofc阳极在不同放大倍率下的sem图;
图10为实施例2未处理的nio粉体为原料流延的阳极坯片实物照片。
具体实施方式
本发明提供了一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法,包括以下步骤:
将陶瓷纳米粉体与小分子有机分散剂进行第一湿法球磨,得到湿法球磨物料,所述陶瓷纳米粉体的比表面积大于20m2/g,所述陶瓷纳米粉体的微晶尺寸小于20nm;
将所述湿法球磨物料与粘结剂混合进行第二湿法球磨,得到分散球磨物料;
将所述分散球磨物料进行造粒,得到粉体;
将所述粉体进行煅烧。
本发明将陶瓷纳米粉体与小分子有机分散剂进行第一湿法球磨,得到湿法球磨物料,所述陶瓷纳米粉体的比表面积大于20m2/g,所述陶瓷纳米粉体的微晶尺寸小于20nm。
在本发明中,所述陶瓷纳米粉体的微晶尺寸优选由xrd测得并由scherrer公式计算获得。
在本发明中,所述陶瓷纳米粉体的比表面积优选为25~75m2/g。
在本发明中,所述陶瓷纳米粉体优选为纳米gd0.1ce0.9o1.95粉体或纳米nio粉体。本发明对所述陶瓷纳米粉体的来源没有特殊的限定,采用本领域技术人员熟知的市售商品即可。
在本发明中,所述小分子有机分散剂优选为聚丙烯酸铵、柠檬酸、乙酰丙酮或三乙醇胺。
在本发明中,所述小分子有机分散剂的用量优选为陶瓷纳米粉体质量的1~6%。
在本发明中,所述陶瓷纳米粉体、球磨介质和球磨溶剂的质量比优选为1:2:0.5~1:3:2,所述球磨介质优选为氧化锆磨球,所述氧化锆磨球的直径优选为0.3~0.8mm。在本发明中,所述球磨溶剂优选为去离子水、乙醇、异丙醇或丙酮。
在本发明中,所述第一湿法球磨优选包括依次进行的快速球磨和慢速球磨,所述快速球磨的时间优选为10~60min,速度优选为300~400转/分;所述慢速球磨的时间优选为2~16h,更优选为5h,速度优选为100~250转/分,更优选为200转/分。在本发明中,所述第一湿法球磨的作用是打开陶瓷纳米粉体的软团聚体,使粉体一次粒子表面吸附小分子有机分散剂后,粉体的一次粒子在溶剂中能够分散稳定存在。本发明中先快速球磨和慢速球磨,使得分散剂能够很好地在一次粒子的表面吸附。
得到湿法球磨物料后,本发明将所述湿法球磨物料与粘结剂混合进行第二湿法球磨,得到分散球磨物料。
在本发明中,所述粘结剂优选为聚乙烯醇(pva)、聚乙烯吡咯烷酮(pvp)或聚乙烯醇缩丁醛(pvb)。在本发明中,所述粘结剂的用量优选为陶瓷纳米粉体质量的1~7%。
在本发明中,所述第二湿法球磨的速度优选为100~250转/分,更优选为200转/分,时间优选为2~24h,更优选为5h。在本发明中,所述第二湿法球磨的过程中,粘结剂分子将几个一次粒子缠绕在一起,造粒过程中一次粒子紧密地粘结在一起。
得到分散球磨物料后,本发明将所述分散球磨物料进行造粒,得到粉体。本发明对所述造粒的具体方式没有特殊的限定,具体的可采用喷雾干燥或玛瑙研钵的方法。
在本发明中,所述造粒时的压强优选为10~60mpa。
在本发明中,所述造粒前优选进行烘干处理,所述烘干处理在工业上优选采用喷雾干燥的方法,实验室优选采用玛瑙研钵-干压的方法。在本发明中,所述喷雾干燥优选将分散球磨物料喷雾到干燥塔内,喷雾液滴所含的水分遇热瞬间被蒸发后形成干的粉体颗粒的一种干燥方法;所述玛瑙研钵-干压的方法具体操作优选如下:先将上述干燥的粉体压饼,压强的范围为10~60mpa,然后用玛瑙研钵将压饼碾碎成粉体。将造粒后的粉体采用筛上和筛下来控制粒径范围,控制在60目筛之下,325目筛之上(60目的孔径为250μm;325目的孔径为41μm),得到粉体。
在本发明中,所述造粒后粉体的粒径优选为41~250μm,更优选为41~94μm或优选为75~250μm。
在本发明中,优选采用筛上和筛下来控制粉体的粒径范围,控制在60目筛(60目的孔径为250μm)之下,325目筛(325目的孔径为41μm)之上。
得到粉体后,本发明将所述粉体进行煅烧。在本发明中,所述煅烧的温度优选为600~1000℃,更优选为800~900℃,煅烧的时间优选为1~10h,更优选为4h。在本发明中,所述煅烧的目的是将几个紧密粘结的纳米晶粒通过高温形成晶界得到多晶粉体颗粒(尺寸为亚微米级)或者长大的单个晶粒,使多表面缺陷(表面凹凸不平)的微晶变成表面较光滑的粉体颗粒,降低了陶瓷纳米粉体的比表面积。
煅烧完成后,本发明优选将煅烧产物进行球磨。在本发明中,所述球磨的目的是将煅烧时可能产生的团聚体打碎,获得分布较均匀的粉体,所述球磨的具体工艺优选为一般球磨工艺。
下面结合实施例对本发明提供的一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
图1为本发明提供的一种提高陶瓷纳米粉体的湿法成型性及烧结性的方法的流程图,将陶瓷纳米粉体与小分子有机分散剂进行第一湿法球磨,得到湿法球磨物料,所述陶瓷纳米粉体的比表面积大于20m2/g;将所述湿法球磨物料与粘结剂混合进行第二湿法球磨,得到分散球磨物料,所述分散球磨物料中有转变成多晶颗粒,也有转变成较大尺寸的一个晶体颗粒;将所述分散球磨物料进行造粒,得到粉体;将所述粉体进行煅烧。
实施例1
将比表面积为75m2/g的纳米gdc(gd0.1ce0.9o1.95)粉体100g加入200g无水乙醇中,加入1g乙酰丙酮,400转/分球磨60min后200转/分球磨5h,加入3gpvb后继续在200转/分球磨12h(氧化锆磨球,直径为0.3mm)。将球磨后的浆料在80℃下干燥后采用研钵造粒(造粒时采用的压强为30mpa),粒径为75~250μm(60目~200目筛网)。造粒后的粉体在900℃煅烧4h。然后采用一般球磨工艺球磨煅烧的粉体消除煅烧时可能产生的团聚体,获得改性后的纳米gdc粉体。通过xrd的数据和scherrer公式计算出900℃煅烧前后的gdc晶粒分别为11nm和30nm。通过tem可知,gdc粉体由多个纳米gdc晶粒组成的多晶颗粒团聚组成。煅烧后的gdc粉体通过流延成型,在1300℃烧成(比较低的烧结温度,属于纳米粉体的烧结温度),保温3h后的基片的体积密度为7.02g/cm3,相对密度达到97%。
图2为900℃煅烧前后的gdc粉体xrd图谱,通过该图谱可以计算出gdc粉体在900℃煅烧前后的晶粒尺寸分别为11nm和30nm,gdc各晶面的衍射峰强度增大,粉体结晶程度提高。
图3为900℃煅烧后的gdc粉体在不同放大倍数下的tem谱图,采用本发明提出的方法可以将gdc粉体的晶粒尺寸从11nm长大到30nm,如图3所示gdc粉体的晶粒尺寸约为30nm(比较均匀)。煅烧后gdc粉体的比表面积为15m2/g(由氮吸附法测定)。可知,本方法使得陶瓷纳米粉体的晶粒尺寸增大,比表面积减小,提高了该陶瓷纳米粉体的湿法成型性和烧结性。该粉体通过流延成型,在较低的温度下1300℃烧结致密,体积密度为7.02g/cm3,相对密度达到97%。
图4为1300℃烧成后gdc断面sem谱图,由图4可知致密度较高,晶粒尺寸较小且均匀。图5为1300℃烧成后的gdc基片实物图(直径约为2cm,厚度为1mm)。在未处理前该gdc粉体无法进行湿法成型,浆料的固含量很小,浆料粘度较大,流延后,坯片的生坯密度极低,即使提高烧成温度也无法烧结致密。
实施例2
将比表面积为25m2/g的纳米nio粉体100g加入120g无水乙醇中,加入1g乙酰丙酮,400转/分球磨20min后200转/分球磨5h,加入2gpvb后继续在200转/分球磨5h(氧化锆磨球,直径为0.8mm)。将球磨后的浆料在80℃下干燥后采用研钵造粒,粒径为41~94μm(160目~325目筛网)。造粒后的粉体在800℃煅烧4h。然后采用一般球磨工艺球磨煅烧的粉体消除煅烧时可能产生的团聚体,获得改性后的nio粉体。通过xrd的数据和scherrer公式计算煅烧前后的nio晶粒分别为18nm和530nm。煅烧后的nio和5ysz粉体及造孔剂(pmma,聚甲基丙烯酸甲酯)按一定比例混合通过流延成型,nio、5ysz和造孔剂的百分含量分别是50wt%、40wt%和10wt%。在1400℃烧成,保温3h,得到多孔sofc(固体氧化物燃料电池)阳极。
图6为800℃煅烧前后的nio粉体xrd图谱,通过该图谱可以计算出nio粉体在800℃煅烧前后的晶粒尺寸分别为18nm和530nm,nio各晶面的衍射峰强度明显增大,粉体结晶程度提高。采用本发明提出的方法可以将nio粉体的晶粒尺寸从18nm长大到530nm,nio粉体的晶粒尺寸约为18nm(团聚)。煅烧后nio粉体的比表面积为9m2/g(由氮吸附法测定)。图7为煅烧前的nio粉体在不同放大倍率下的tem谱图,可知本方法使得粉体的晶粒尺寸增大,比表面积减小,分散性提高,明显提高了该纳米粉体的湿法成型性和烧结性。
图8为1400℃烧成后的sofc阳极实物图,尺寸为10cm×10cm×0.4mm。
图9为1400℃烧成后的sofc阳极在不同放大倍率下的sem谱图,从图9中可见,nio和5ysz晶粒紧密烧结在一起,提高了阳极机械强度。在未处理前该nio粉体无法进行湿法成型,浆料的固含量很小,浆料粘度较大,流延后,坯片的开裂,如图10所示,以未处理的nio粉体为原料流延的阳极坯片干燥后严重开裂。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!