一种组成型表达碱性β‑甘露聚糖酶的重组菌的制作方法与工艺
一种组成型表达碱性β-甘露聚糖酶的重组菌技术领域本发明涉及生物技术领域,尤其涉及一种组成型表达碱性β-甘露聚糖酶的重组菌。
背景技术:
β-甘露聚糖酶,又称β-1,4-D-甘露聚糖酶(β-1,4-D-mannanmannohydrolase;EC.3.2.1.78)是一类能够水解β-1,4-D-甘露糖苷键的甘露寡糖、甘露多糖的内切水解酶,属半纤维素酶类。β-甘露聚糖酶广泛存在于动物、植物和微生物体内,来源丰富。β-甘露聚糖酶按其最适pH值的不同有酸性、中性、碱性之分,其中碱性β-甘露聚糖酶的研究相对较晚,发现主要由嗜碱芽孢杆菌产生,某些地衣芽孢杆菌也产生碱性β-甘露聚糖酶,如杨文博等报道了一株地衣芽孢杆菌NK-27,其最适pH值为9.0。一般情况下枯草芽孢杆菌,放线菌等产生中性β-甘露聚糖酶,而霉菌产生酸性β-甘露聚糖酶。第一个碱性甘露聚糖酶由Akino等发现。马延和等于199l年报道了嗜碱芽孢杆菌N16-5的三个胞外碱性甘露聚糖酶的纯化及酶学性质,并于2004年报道了其中一种嗜碱甘露聚糖酶的基因序列。Takeda等于2004年发现了芽孢杆菌属的一个菌株JAMB-602产碱性甘露聚糖酶,此酶属于GH家族5,最适pH为9。目前所发现的嗜碱甘露聚糖酶的最适pH值最高为10,由Takeda等于2004年报道。碱性β-甘露聚糖酶不仅可以生产用作双歧杆菌促生长因子的低聚糖,而且在需碱性环境的纸浆漂白等工业方面具有广泛的应用前景。但到目前为止,碱性β-甘露聚糖酶的制备仍旧是限制其工业应用的瓶颈,前人的报道中无论是使用碱性β-甘露聚糖酶的原产菌株还是使用重组菌株生产,其产量都比较低。如马延和报道了由AlkaliphilicBacillussp.N16-15菌株经培养优化后,产量达到500U/mL(专利公开号CN1266096)。来源于BacilluslicheniformisstrainDSM13的碱性β-甘露聚糖酶在大肠杆菌中重组表达,酶活力为50U/mL(参见SongsiriritthigulC:MicrobCellFact.,11:9-20,2010)。来源于AlkaliphilicBacillussp.N16-5的碱性β-甘露聚糖酶在PichiapastorisGS115中重组表达,酶活力为32.2IU/mL(参见HeXP:EnzyMicrobTechnol,43(1):13-18,2008)。来源于Aspergillussulphureus的碱性β-甘露聚糖酶在Pichiapastoris中重组表达,酶活力为96U/mL(参见XiaolingChen:JBiotechnol,12(8):452–461,2007)。吕红等用鹰嘴豆克鲁维酵母表达来自AlkaliphilicBacillussp.N16-5突变体基因,最终碱性甘露聚糖酶酶活达1000U/mL。目前已经用甲醇营养型酵母毕赤酵母诱导表达来源于Bacillussp.N16-15的碱性甘露聚糖酶,经发酵罐高密度发酵培养,酶产量水平可达到6000U/mL以上,满足工业化生产要求。但该技术存在的不足之处在于:1、蛋白表达需要添加甲醇诱导。在生产规模时,整个诱导发酵过程甲醇用量很大,甲醇的存放和运输存在安全隐患;2、发酵周期较长,整个发酵过程需要6天以上。
技术实现要素:
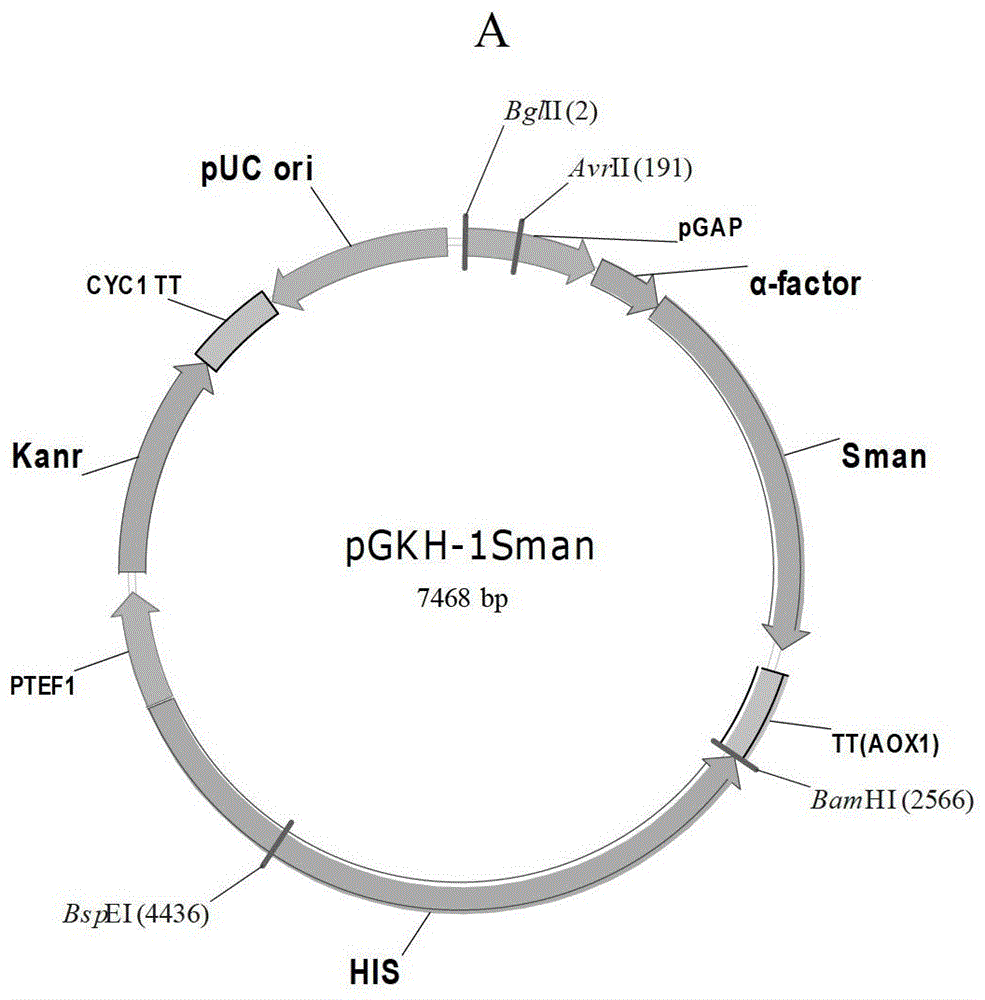
本发明的一个目的是提供一种制备重组菌B的方法。本发明提供的制备重组菌B的方法,包括如下步骤:为将调控因子基因和碱性β-甘露聚糖酶编码基因表达盒导入宿主菌中,得到重组菌B;所述调控因子为转录激活因子HAC1或蛋白质二硫键异构酶Pdi;所述碱性β-甘露聚糖酶编码基因表达盒中的启动子为组成型启动子。上述方法中,所述碱性β-甘露聚糖酶编码基因表达盒包括组成型启动子、信号肽、碱性β-甘露聚糖酶编码基因和终止子;所述组成型启动子具体为甘油醛3-磷酸脱氢酶启动子PGap;所述碱性β-甘露聚糖酶编码基因表达盒为1个或多个。上述方法中,所述转录激活因子HAC1(又称为Hac1p调控因子)来源于酵母或霉菌,也可以是来自于其他真核系统如酵母和霉菌中的同源蛋白,它们具有相同或相似的功能;所述蛋白质二硫键异构酶Pdi(又称为Pdi调控因子)来源于毕赤酵母,也可以是来自于其他真核系统的同源蛋白,它们具有相同或相似的功能;所述转录激活因子HAC1氨基酸序列为序列表中的序列2;所述转录激活因子HAC1基因的核苷酸序列具体为序列表中序列3;所述蛋白质二硫键异构酶Pdi氨基酸序列为序列表中的序列4;所述蛋白质二硫键异构酶Pdi基因的核苷酸序列具体为序列表中序列5;1个所述碱性β-甘露聚糖酶编码基因表达盒的核苷酸序列为序列表中的序列1。上述方法中,所述碱性β-甘露聚糖酶编码基因表达盒为3个;所述转录激活因子HAC1通过重组载体1导入所述宿主菌;所述蛋白质二硫键异构酶Pdi通过重组载体2导入所述宿主菌;3个所述碱性β-甘露聚糖酶编码基因表达盒通过重组载体B导入所述宿主菌;所述重组载体1为将所述转录激活因子HAC1基因插入表达载体,得到重组载体1;所述重组载体2为将所述蛋白质二硫键异构酶Pdi基因插入表达载体,得到重组载体2;所述重组载体B按照如下方法制备:1)将所述碱性β-甘露聚糖酶编码基因插入表达载体中,得到含有1个碱性β-甘露聚糖酶编码基因表达盒的中间载体1;2)将1个所述碱性β-甘露聚糖酶编码基因表达盒插入所述中间载体1中,得到含有2个碱性β-甘露聚糖酶编码基因表达盒的中间载体2;3)将1个所述碱性β-甘露聚糖酶编码基因表达盒插入所述中间载体2中,得到含有3个碱性β-甘露聚糖酶编码基因表达盒的重组载体B。上述方法包括如下步骤:1)将所述重组载体B导入宿主菌中,得到重组菌A;2)再将所述重组载体1或所述重组载体2导入重组菌A中,得到重组菌B;所述宿主菌具体为毕赤酵母菌。由上述方法制备的重组菌B也是本发明保护的范围。本发明的另一个目的是提供一种制备重组菌A的方法。本发明提供的方法,包括如下步骤:将上述制备重组菌B方法中的碱性β-甘露聚糖酶编码基因表达盒导入宿主菌中,得到重组菌A。上述方法中,所述碱性β-甘露聚糖酶编码基因表达盒为1个或多个;所述方法具体为如下1)-3)中任一种:1)所述方法为将3个碱性β-甘露聚糖酶编码基因表达盒通过所述含有3个碱性β-甘露聚糖酶编码基因表达盒的重组载体B导入宿主菌,得到重组菌A;2)所述方法为将1个碱性β-甘露聚糖酶编码基因表达盒通过所述含有1个碱性β-甘露聚糖酶编码基因表达盒的中间载体1导入宿主菌,得到重组菌A;3)所述方法为将2个碱性β-甘露聚糖酶编码基因表达盒通过所述含有2个碱性β-甘露聚糖酶编码基因表达盒的中间载体2导入宿主菌,得到重组菌A;所述宿主菌具体为毕赤酵母菌。由上述方法制备的重组菌A也是本发明保护的范围。上述的重组菌B或上述的重组菌A在制备碱性β-甘露聚糖酶中的应用也是本发明保护的范围;本发明还提供了一种制备碱性β-甘露聚糖酶的方法。本发明提供的方法,为发酵上述的重组菌B或上述的重组菌A,收集发酵产物,即得到碱性β-甘露聚糖酶。本发明的实验证明,本发明通过将碱性β-甘露聚糖酶编码基因表达盒或者该表达盒和调控因子共同导入宿主菌中,得到重组菌;且碱性β-甘露聚糖酶编码基因表达盒中的启动子为组成型启动子。本发明构建的这些重组菌,可以无需甲醇诱导,保证生产安全;且可大量组成型分泌表达碱性β-甘露聚糖酶,满足工业需要;另外发酵周期比现有技术水平显著缩短,可提高生产效率;能够实现更好的工业化制备碱性β-甘露聚糖酶。附图说明图1为分泌型表达载体pGAPKH的构建示意图图2为重组表达载体pGKH-1Sman、pGKH-2Sman、pGKH-3Sman的构建示意图图3为重组表达载体pGapZ-Hac1p的构建示意图图4为重组表达载体pGapZ-Pdi的构建示意图具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例1、重组菌的制备一、重组表达载体的制备1、单拷贝碱性β-甘露聚糖酶分泌表达载体的构建1)、碱性β-甘露聚糖酶基因的克隆根据GenBank中芽孢杆菌来源的碱性β-甘露聚糖酶基因,设计PCR引物如下:引物1:5’ATATCTCGAGAAAAGATCCCCATCAGAAGC3’引物2:5’ATTGGCGGCCGCTATTCTTAAAGTAACATTAT3’在α-mating因子信号肽编码序列下游有一个XhoI酶切位点(CTCGAG,对应氨基酸为Leu-Glu),其后紧跟一个Kex2蛋白酶识别位点(AAAAGA,对应氨基酸序列Lys-Arg),因此,引物1的设计是在甘露聚糖酶基因之前引入XhoI位点和Kex2蛋白酶识别位点序列。通过该设计,即可将甘露聚糖酶基因直接连接在信号肽序列之后,这样蛋白翻译后,在毕赤酵母自身Kex2蛋白酶的作用下,即可切表达出氨基酸不变的目的蛋白(而不会因酶切位点的原因引入额外的氨基酸残基)。引物2包含终止密码子TAG,并引入一个NotI酶切位点GCGGCCGC。以嗜碱芽孢杆菌N16-5(Ma,Y.andY.Xue,etal.(2004)."Characterizationandgenecloningofanovelβ-mannanasefromalkaliphilicBacillussp.N16-5."Extremophiles8(6):447-54.公众可从中国科学院微生物研究所获得。)的总DNA为模板,加入引物1、引物2、HiFiTaqDNA聚合酶及dNTP混合物,进行降落PCR(touchdownPCR),反应条件如下:94℃5min;94℃30s,60-50℃30s,72℃90s,每个循环降1℃,共10个循环;94℃30s,55℃30s,72℃90s,共25个循环。PCR扩增得到1400bp的片段,命名为Sman,用Omega纯化试剂盒纯化后,连接入pMD18-T载体(购自TaKaRa公司),得到重组载体pMD18-Sman,将pMD18-Sman送生工生物工程(上海)有限公司进行测序,测序结果表明PCR扩增得到的片段Sman即为碱性β-甘露聚糖酶的编码基因,该片段具有序列表中序列1自5’末端第748-2145位核苷酸,为GenBank号为AY534912的自5’端的第73-1479位所示的核苷酸序列。2)、单拷贝碱性β-甘露聚糖酶分泌表达载体的构建将上述1)得到的pMD18-Sman采用XhoI和NotI双酶切,得到约为1400bp的片段,与同样经过XhoI和NotI双酶切的6000bppGAPKH(结果示意图见图1,载体pGAPKH的核苷酸序列为序列表中的序列6)表达载体骨架连接,形成单拷贝碱性β-甘露聚糖酶分泌重组表达载体pGKH-1Sman(见图2A)。经过测序,pGKH-1Sman为将序列表中序列1自5’末端第748-2145位核苷酸插入pGAPKH载体的XhoI和NotI酶切位点间得到的载体。pGKH-1Sman中含有1个Sman表达盒,Sman表达盒包括组成型启动子甘油醛3-磷酸脱氢酶启动子(PGap)(序列表中序列1自5’末端第1-483位核苷酸)、alpha-factor(α-mating因子信号肽,序列表中序列1自5’末端第493-747位核苷酸)、碱性β-甘露聚糖酶基因(序列表中序列1自5’末端第748-2145位核苷酸)和终止子(序列表中序列1自5’末端第2230-2564位核苷酸)。2、两拷贝碱性β-甘露聚糖酶分泌表达载体的构建将步骤1得到的重组表达载体pGKH-1Sman用BamHI和BglII双酶切,回收2500bpBamHI-1Sman表达盒-BglII片段,与经过BamHI酶切的7500bp重组表达载体pGKH-1Sman连接,得到两拷贝Sman的表达载体pGKH-2Sman(见图2B)。经过测序,pGKH-2Sman为将含有1Sman表达盒的DNA分子(该DNA分子的核苷酸序列为序列表中序列1)插入pGKH-1Sman载体的BamHI酶切位点间得到的载体。pGKH-2Sman中含有2个Sman表达盒,每个Sman表达盒的启动子均为组成型启动子甘油醛3-磷酸脱氢酶启动子。3、三拷贝碱性β-甘露聚糖酶分泌表达载体的构建将步骤1得到的重组表达载体pGKH-1Sman用BamHI和BglII双酶切,回收2500bpBamHI-1Sman表达盒-BglII片段,与步骤2得到的经过BamHI酶切的10000bp的重组表达载体pGKH-2Sman连接,得到三拷贝Sman的表达载体pGKH-3Sman(见图2C)。经过测序,pGKH-3Sman为将含有1Sman表达盒的DNA分子(序列表中序列1)插入pGKH-2Sman载体的BamHI酶切位点间得到的载体。pGKH-3Sman中含有3个Sman表达盒,每个Sman表达盒的启动子均为组成型启动子甘油醛3-磷酸脱氢酶启动子。4、组成型表达载体pGapZ-Hac1p的构建调控因子Hac1p(也称为转录激活因子HAC1)的氨基酸序列为序列表中的序列2,调控因子Hac1p的基因Hac1p的核苷酸序列为序列表中序列3。人工合成Hac1p基因序列3,并在序列5’端和3’端各引入一个EcoRI和NotI位点,经EcoRI和NotI酶切处理后,将Hac1p基因连接到同样经EcoRI和NotI酶切的pGapZA载体(Invitrogen,目录编为V20020)上,即可得到组成型表达载体pGapZ-Hac1p(见图3)。将得到的重组表达载体pGapZ-Hac1p转化大肠杆菌DH5α感受态细胞,用含有25μg/mL博来霉素(Zeocin)的LB培养基(5g/L氯化钠、10g/L蛋白胨、5g/L酵母粉,pH7.4-7.6)筛选出所需重组子并进行测序分析,经过测序鉴定,pGapZ-Hac1p组成型表达载体为将序列表中序列3所示的核苷酸所示的Hac1p基因插入pGapZA载体(Invitrogen,目录编为V20020)的EcoRI和NotI酶切位点间得到的载体,证明重组表达载体pGapZ-Hac1p的序列和结构均正确。5、组成型表达载体pGpaZ-Pdi的构建调控因子Pdi(蛋白质二硫键异构酶)的氨基酸序列为序列表中的序列4,调控因子Pdi的基因Pdi的核苷酸序列为序列表中序列5。人工合成Pdi基因序列5,并在序列5’端和3’端各引入一个EcoRI和NotI位点,酶切处理后,将Pdi基因连接到同样经EcoRI和NotI酶切的pGapZA载体(Invitrogen,目录编为V20020)上,即可得到pGapZ-Pdi组成型表达载体(见图4)。将得到的重组表达载体pGapZ-Pdi转化大肠杆菌DH5α感受态细胞,用含有25μg/mL博来霉素(Zeocin)的LB培养基(5g/L氯化钠、10g/L蛋白胨、5g/L酵母粉,pH7.4-7.6)筛选出所需重组子并进行测序分析,经过测序鉴定,pGapZ-Pdi为将序列表中序列5所示的核苷酸插入pGapZA载体的EcoRI和NotI酶切位点间得到的重组表达载体,证明重组表达载体pGapZ-Pdi的序列和结构均正确。二、表达碱性β-甘露聚糖酶的重组酵母菌株的获得1、电转化巴斯德毕赤酵母细胞的制备接种毕赤酵母(Pichiapastoris)菌株GSl15(Invitrogen,Ca.18100)于500mLYPD培养基(10g/L酵母膏、20g/L蛋白胨、20g/L葡萄糖、15g/L琼脂糖),30℃,200r/min培养至OD600=1.3-1.5。4℃,1,500g离心5min收集菌体,分别用500mL、250mL预冷的灭菌水和20mL预冷的1mol/L山梨醇各洗一次。每次洗后均在4℃,1,500g下离心5min收集菌体,最后用1mL预冷的1mol/L山梨醇悬浮,即获得电击感受态细胞。2、重组酵母菌GS-1Sman、GS-2Sman、GS-3Sman的获得重组表达载体pGKH-1Sman、pGKH-2Sman、pGKH-3Sman电转化酵母细胞,具体如下:将一构建好的重组表达载体pGKH-1Sman、pGKH-2Sman和pGKH-3Sman各取约10μg用BspEI酶切线性化,乙醇沉淀回收线性DNA并溶解于10μL无菌水中。将上述线性化DNA分别与80μLGS115感受态细胞混合,转入预冷的0.2cm电转杯中。采用美国BIO-RAD公司电转化仪,根据所使用电转化仪预设的毕赤酵母参数进行电击。电击结束后立即加入1mL预冷的1mol/L山梨醇至电击杯中,然后将电击杯中的溶液全部转移至一无菌离心管中,37℃孵育2h,取500μL涂布MD(2g/L葡萄糖、13.4g/LYNB、4×10-4g/L生物素、1.5g/L琼脂)平板,于30℃倒置培养2-4天直至菌落出现。至此,得到不同拷贝Sman的重组毕赤酵母菌株,分别命名为GS-1Sman、GS-2Sman、GS-3Sman。将GS-1Sman、GS-2Sman、GS-3Sman分别提取质粒验证,分别得到质粒pGKH-1Sman、pGKH-2Sman和pGKH-3Sman,说明重组毕赤酵母菌株构建成功。3、重组酵母菌GS-3Sman-H和GS-3Sman-P的获得将一构建好的重组表达载体pGapZ-Hac1p和pGapZ-Pdi分别电转化三拷贝重组酵母细胞GS-3Sman,得到重组酵母菌GS-3Sman-H和GS-3Sman-P;具体如下:将一构建好的重组表达载体的pGapZ-Hac1p、pGapZ-Pdi各约10μg用AvrII酶切线性化,上述2的方法进行电转化到三拷贝重组酵母细胞GS-3Sman,电击结束后立即加入1mL预冷的1mol/L山梨醇至电击杯中,然后将电击杯中的溶液全部转移至一无菌离心管中,于30℃温浴2小时,然后取500μL菌液涂布在含有50μg/mL博来霉素(Zeocin)的YPD平板上。30℃倒置培养4天直至菌落出现,分别命名为GS-3Sman-H以及GS-3Sman-P。将GS-3Sman-H和GS-3Sman-P分别提取质粒验证,分别得到质粒pGapZ-Hac1p和pGapZ-Pdi,说明重组毕赤酵母菌株构建成功。实施例2、重组酵母菌株在制备碱性β-甘露聚糖酶中的应用一、摇瓶发酵1、发酵培养将实施例1获得的表达碱性β-甘露聚糖酶的单拷贝Sman的菌株GS-1Sman、两拷贝Sman的菌株GS-2Sman、三拷贝SmanGS-3Sman的菌株以及含有调控因子的GS-3Sman-H、GS-3Sman-P菌株分别接种于25mLYPD液体培养基(10g/L酵母膏、20g/L蛋白胨、20g/L葡萄糖和水组成),于30℃、200r/min摇床培养72h,得到发酵产物。同时以诱导型高产菌株GSG4SMAN(CGMCCNO.4095)作为对照。巴斯德毕赤酵母GSG4SMAN已于2010年8月18日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称CGMCC,地址为:北京市朝阳区北辰西路1号院3号),保藏号为CGMCCNo.4095,建议的分类命名为Pichiapastoris。2、胞外碱性β-甘露聚糖酶酶活的测定反应液为0.5%的槐豆胶:0.5g槐豆胶(购自Sigma、产品目录号为G0753,主要成分为甘露聚糖)放入250mL烧杯中,加入90mL在70℃预热的pH9.0,0.05mol/L甘氨酸-氢氧化钠缓冲液(50mL0.2mol/L甘氨酸+8.8mL0.2mol/L氢氧化钠,加水稀释至200mL),磁力搅拌混匀,然后定容到500mL,得到反应液。DNS:称取酒石酸钾钠182.0g,溶于500mL蒸馏水中,加热(不超过50℃),于热溶液中依次加入3,5-二硝基水杨酸6.3g,NaOH21.0g,苯酚5.0g,无水亚硫酸钠5.0g,搅拌至溶解完全,冷却后用蒸馏水定容至1000mL,贮存于棕色瓶中,室温保存吸取上述1得到的发酵产物1mL于离心管中,以9178g离心10min,吸取上清液于试管中,得到样品溶液。加入0.01mL适当稀释的上述上清液和0.19mL反应液,37℃保温10min;再加入0.2mLDNS,煮沸5min;离心取上清液在540nm处测定吸光值。以灭过活的酶液做空白对照,空白对照的处理方法与样品的处理一样。采用不同浓度的葡萄糖为标准品进行测定,以所得的OD540值与葡萄糖浓度做标准曲线,获得的标准曲线为y=0.015x-0.220(R2=0.998),线性关系较好,其中y为OD540值,x为葡萄糖含量。酶活定义:在37℃、pH值为9.0的条件下,每分钟从浓度为3mg/mL的甘露聚糖(SigmaG0753)溶液中降解释放1μmol还原糖所需要的酶量为一个酶活力单位U。发酵72h后,各菌株摇瓶发酵液酶活见表1。表1不同拷贝菌株的酶活可以看出,与诱导型高产菌株GSG4SMAN相比,组成型单拷贝Sman的菌株GS-1Sman、两拷贝Sman的菌株GS-2Sman、三拷贝SmanGS-3Sman的菌株以及含有调控因子的GS-3Sman-H、GS-3Sman-P菌株的酶活都有显著提高,且不需要甲醇诱导,其中最高的是GS-3Sman-H。二、发酵罐发酵种子液摇瓶培养:从甘油管中接200μL由实施例1得到的重组酵母菌GS-3Sman-H的培养液(在YPD液体培养基中培养10h得到的培养液)于10mLYPD中进行培养12h后按照1%的接种量接种到50mLYPD中,于30℃、220r/min继续培养24h得到种子液;发酵罐发酵培养:按5%(v/v)的接种量将种子液接入1L全自动发酵罐,发酵罐装发酵培养基的量为600mL,以50%氨水控制pH为6.0,温度设定为30℃,调节搅拌转速和通气量维持溶氧30%以上,发酵72h。采用上述的方法检测酶活,结果为重组酵母菌GS-3Sman-H发酵后的胞外碱性β-甘露聚糖酶酶活可达到2200U/mL。同时以诱导型高产菌株GSG4SMAN作为对照,72h后,测得发酵后的胞外碱性β-甘露聚糖酶酶活为16.3U/mL。上述发酵培养基按照如下方法制备:将如下组分按照如下浓度与水混合得到:硫酸铵5g/L,硫酸钙0.46g/L,硫酸钾9.1g/L,七水硫酸镁14.9g/L,磷酸二氢钾6.8g/L,甘油40g/L,蛋白胨20g/L,酵母粉10g/L,PTM(五水硫酸铜6g/L,碘化钾0.08g/L,硫酸锰2.68g/L,硼酸0.02g/L,二水钼酸钠0.2g/L,七水硫酸锌20g/L,七水硫酸亚铁65g/L,六水氯化钴0.916g/L,硫酸5mL/L,生物素0.2g/L)14mL/L。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1