聚恶唑啉抗体药物结合物的制作方法
小分子药物长期以来是治疗多种疾病(包括但不限于癌症)的标准。此类小分子药物通常以液体、药丸、胶囊等形式口服施用或者以可注射制剂或静脉内制剂的形式肠胃外施用。虽然在治疗许多病症方面有效,但是仍然存在许多挑战。这些挑战包括但不限于控制药物递送的速率、将化合物靶向递送至所需作用部位以及使化合物在循环中的半衰期最大化。例如,许多化合物在到达作用部位之前由于代谢而表现出功效和治疗益处降低。另外,许多化合物相对不溶于水溶剂中,需要使用复杂的制剂以便施用。在一些情况下,这可能限制其在临床上的有用性,即使它们在临床前研究中有效。
药物递送领域中的一个目的是优先将化合物递送至所需作用部位。此外,另外的一个目的是一旦化合物到达作用部位至少部分地控制化合物的递送。更进一步地,另外的一个目的是利用递送系统实现前述目的中的一个或多个,所述递送系统在化合物在体内递送时使其稳定、延长化合物在体内的半衰期并且帮助溶解化合物,特别是那些不溶于水溶剂的化合物。
为了解决本领域的缺点,已经探索出了抗体-药物结合物(ADC)。大量的研究和开发已经表明ADC在提供动物模型以及人类中的癌细胞的靶向破坏方面是有效的(P.D.Senter和E.L.Sievers,《自然生物技术(Nature Biotechnology)》,30,631-637(2012);C&EN,《抗体药物结合物(Antibody Drug Conjugates)》,2012年6月18日,第12-18页;J.D.Thomas等人,《生物结合物化学(Bioconjugate Chem.)》,23,2007-2013(2012);以及其中的参考文献)。虽然这个概念已经讨论了许多年,但是这个概念的实现花了几十年才实现。第一个“武装抗体”直到2011年才被批准(P.D.Senter和E.L.Sievers,《自然生物技术(Nature Biotechnology)》,30,631-637(2012))。在其最常见的形式中,使用被设计成将药剂释放到肿瘤块或肿瘤细胞中的连接化学物质将药物或其它化合物(在本文中称为“药剂”)的一至四个分子与抗体偶联。这种方法的一个限制是不充分的毒素可以附着到抗体上以杀死某些肿瘤,特别是当使用威力较小的毒素时或者当肿瘤细胞表面上存在减少的抗原密度时。而且,增加抗体上毒素附着点的数量可能损害抗体结合能力并且降低其杀死癌细胞的能力。另外,这种方法对于难以溶解的化合物可能是有问题的。最后,一些毒素当附着到抗体上时是很疏水的,使得它们被独立于抗体的结合区的靶外组织摄取,从而限制它们的有效治疗潜力。
另一种方法是使用可生物降解的聚合物,例如衍生自氧化葡聚糖的聚缩醛(Yurokovetskiy等人,美国专利8,685,383)。在这种方法中,聚合物-ADC可以含有与聚合物主链连接的药剂的多个拷贝,其中所述聚合物本身与抗体连接。当单独与非靶向聚合物药物结合物相比时,大于1的药物-抗体比例(DAR)使得细胞毒性结果得以改善。可生物降解的聚合物,例如上述聚缩醛,具有降解并因此避免体内积累的优点。然而,它们还具有在制备期间或在注射后在体内比预期降解更快的缺点。该方法可能导致细胞毒素剂在循环过程中过早释放,从而在其到达靶细胞上的抗原之前限制了其有效性。第二,许多此类聚合物,包括上述聚缩醛,具有生物来源,并且这可能导致与去除危险杂质有关的制造挑战,并且当施用于受试者时可增加不良反应的风险。
另外的一种方法是将抗体附着到生物相容性聚合物(称为聚合物-ADC)上。一种简单的方法是将线性聚合物(例如聚乙二醇(PEG))附着在抗体与药物之间。这由Riggs-Sauthier等人(US 2014/0088021)证明,其中首先通过马来酰亚胺化学将2kDa或20kDa的一个线性异双功能的PEG附着到HER2抗体上的一个巯基上。然后通过酯连接将PEG抗体复合物与一种细胞毒性小分子偶联。该方法可能具有明显的缺点,因为酯连接在体内不稳定,并且将在其到达靶细胞上的抗原之前水解将细胞毒素剂释放在血液中。当使用2kDa或20kDa聚合物时,该项工作中的体内功效研究不能证明聚合物-ADC的主动靶向。所证明的功效可能是由于PEG部分引起的细胞毒素剂的药代动力学半衰期延长引起的。另外,当单独与药物相比时,PEG聚合物-ADC在细胞毒性测定方面丧失生物活性。在这种情况下,药物与抗体比例(DAR)仅为一,与非聚合物-ADC方法相比没有优势。利用该方法不能实现在线性PEG聚合物上配备药物或抗体的多个拷贝。
考虑到前述情况,该领域需要一种解决上述限制的ADC,特别是聚合物-ADC。本申请通过提供聚合物-ADC提供了对这些问题的解决方案,所述聚合物-ADC是合成制造的、使药剂更多地加载到靶向抗体上(更高的DAR值)、使结合物在体内的半衰期增加,并且基本上不干扰靶向剂(即,抗体)的结合活性。此外,本发明的聚合物-ADC结合物容易构建,并且它们可以增强待递送的化合物的溶解度。最后,本发明的聚合物-ADC结合物直到它们到达靶细胞上的抗原并且被内在化才释放药物,其中所述药剂在从聚合物裂解后释放。在本申请中,使用HBL-2、MEC-1和拉莫斯细胞系(Ramos cell line)(人B细胞淋巴瘤衍生的细胞系)作为实例。
附图说明
图1显示了R11POZ生物素与HBL-2和MEC-1细胞的流式细胞术曲线图。
图2显示了在提纯之前、期间和之后CD79b的POZ MMAE结合物的SDS-PAGE。
图3显示了通过ELISA测定天然的且聚恶唑啉结合的R11抗体与ROR1抗原的结合。
图4显示了MMAE和脱乙酰基秋水仙碱(DC)的CD79b POZ药物结合物的浓度对拉莫斯细胞的生存力的影响。
图5显示了MMAE和脱乙酰基秋水仙碱(DC)的CD79b POZ药物结合物的浓度对MEC-1细胞的生存力的影响。
图6显示了MMAE的R11POZ药物结合物的浓度对HBL-2和MEC-1细胞的生存力的影响。
技术实现要素:
在本发明中,描述了聚合物结合物,以及此类聚合物用于治疗人类疾病的用途。在一个实施例中,聚合物结合物是聚合物-ADC。在一个实施例中,聚合物结合物是生物稳定的。在另一个实施例中,所有或某些连接基团是生物稳定的。聚合物-ADC被设计成含有大量的聚合物结合剂(高DAR)。这些聚合物-ADC还可以在识别部分(例如但不限于抗体)与聚合物之间具有单个附着点或多个附着点,其中附着点可以远离识别部分的结合位点。在某些实施例中,单个识别部分可以结合本文所描述的不止一种聚合物-ADC。所描述的聚合物-ADC相对于传统ADC具有改善的药代动力学和溶解度。另外,这些聚合物-ADC具有在制备期间或在递送期间不降解的优点。此外,聚合物ADC的聚合物部分是合成的,并且不含生物杂质。可以定制药剂与聚合物之间的连接基团以在作用部位(例如肿瘤细胞)在所需位点且在所需条件下提供毒素的释放。本发明的聚合物-ADC的另一个特征是,提纯部分(例如但不限于生物素)可以附接到聚合物主链上,以使复合物结合物容易提纯。
定义
如本文所使用的,术语“药剂”是指具有治疗或诊断应用的任何分子,其中所述药剂能够与聚合物上的官能团或附着到聚合物上的连接基团形成连接,所述药剂包括但不限于治疗剂(例如但不限于药物)、诊断剂或有机小分子。在一个具体的实施例中,药剂可用于治疗癌症。
如本文所使用的,术语“连接”或“连接基团”,当针对本文所描述的聚合物或药剂或其组分使用时,是指通常由于化学反应而形成的基团或键。在某些实施例中,具体的连接是共价连接。在某些实施例中,具体的连接是可水解的。
如本文所使用的,术语“可水解连接基团”或“可释放的可水解连接基团”是指在含有可水解连接基团的本发明的结合物已经施用于受试者之后在受试者的特定生理条件下在受试者体内可裂解的化学连接。在进一步的实施例中,可水解连接基团可以是生物稳定的。可水解连接基团可以含有如本文所描述的可水解部分;在某些情况下,可水解连接基团可以含有不止一个可水解部分。在一个实施例中,可水解连接基团可在递送至作用部位(例如但不限于肿瘤细胞)时裂解。在一个实施例中,可水解连接基团仅可在被细胞(例如但不限于肿瘤细胞)摄取(例如通过内吞作用)时裂解。在一个实施例中,可水解连接基团可在溶酶体或核内体中裂解。在一个实施例中,可水解连接基团通过化学反应裂解。在该实施例的一个方面,裂解通过还原易于还原的基团,例如但不限于二硫化物或自我扩散反应。在一个实施例中,可水解连接基团被天然存在或被诱导存在于受试者中的物质裂解。在该实施例的一个方面,这样的物质是酶或多肽。因此,在一个实施例中,可水解连接基团通过酶促反应裂解。在一个实施例中,可水解连接基团通过前述的组合裂解。在一个实施例中,包含在可水解连接基团内的可水解部分在相同条件下并且通过相同的机制裂解。
如本文所使用的,术语“生物稳定的”是指当施用于受试者(包括人受试者)时抗裂解的聚合物或聚合物结合物(包括其中含有的任何连接基团或连接,例如但不限于可水解连接基团),当施用于受试者(包括人类受试者)时抗裂解,直到聚合物或聚合物结合物到达作用部位(例如但不限于肿瘤细胞)。“抗裂解”是指在聚合物到达作用部位之前至少80%、90%、95%、98%或99%或更多的聚合物或聚合物结合物没有被裂解。在一个实施例中,生物稳定的聚合物或聚合物结合物在受试者的血流中抗裂解,使得超过80%、90%、95%、98%、99%或更多的聚合物或聚合物结合物在血流中没有被裂解。术语“生物稳定的”还可以指连接基团或连接(例如但不限于可水解连接基团),使得连接基团或连接在施用于受试者(包括人受试者)时抗裂解,直到连接的连接基团被置于作用部位(例如但不限于肿瘤细胞)。因此,在一个实施例中,生物稳定的连接基团或连接在受试者的血流中抗裂解,使得超过80%、90%、95%、98%、99%或更多的连接基团或连接在血流中没有被裂解。
如本文所使用的,术语“烷基”,无论单独使用或作为取代基的一部分使用,包括包含1至20个碳原子的直链烃基。因此,所述短语包括直链烷基,例如甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基等。所述短语还包括直链烷基的支链异构体,包括但不限于通过实例提供的以下各项:-CH(CH3)2、-CH(CH3)(CH2CH3)、-CH(CH2CH3)2、-C(CH3)3、-C(CH2CH3)3、-CH2CH(CH3)2、-CH2CH(CH3)(CH2CH3)、-CH2CH(CH2CH3)2、-CH2C(CH3)3、-CH2C(CH2CH3)3、-CH(CH3)CH(CH3)(CH2CH3)、-CH2CH2CH(CH3)2、-CH2CH2CH(CH3)(CH2CH3)、-CH2CH2CH(CH2CH3)2、-CH2CH2C(CH3)3、-CH2CH2C(CH2CH3)3、-CH(CH3)CH2CH(CH3)2、-CH(CH3)CH(CH3)CH(CH3)CH(CH3)2、-CH(CH2CH3)CH(CH3)CH(CH3)(CH2CH3),以及其它。所述短语还包括环烷基,例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基,以及被如上定义的直链和支链烷基取代的这些的环。所述短语还包括多环烷基,例如但不限于金刚烷基、降冰片基和双环[2.2.2]辛基,以及被如上定义的直链和支链烷基取代的这些的环。
如本文所使用的,术语“烯基”,无论单独使用或作为取代基的一部分使用,包括在任何两个相邻碳原子之间具有至少一个双键的烷基。
如本文所使用的,术语“炔基”,无论单独使用或作为取代基的一部分使用,包括在任何两个相邻碳原子之间具有至少一个三键的烷基。
如本文所使用的,术语“未取代的烷基”、“未取代的烯基”和“未取代的炔基”是指不含杂原子的烷基、烯基和炔基。
如本文所使用的,术语“取代的烷基”、“取代的烯基”和“未取代的炔基”是指如上定义的烷基、烯基和炔基,其中与碳或氢键合的一个或多个键被与非氢或非碳原子键合的键置换,所述非氢或非碳原子例如但不限于:基团(例如烷氧基和芳氧基)中的氧原子;基团(例如烷基和芳基硫基、砜基、磺酰基和亚砜基等)中的硫原子;基团(例如三烷基甲硅烷基、二烷基芳基甲硅烷基、烷基二芳基甲硅烷基和三芳基甲硅烷基)中的硅原子;以及各种其它基团中的其它杂原子。
如本文所使用的,术语“未取代的芳烷基”是指如上定义的未取代的烷基或烯基,其中未取代或取代的烷基或烯基的氢或碳键被与如上定义的取代或未取代的芳基键合的键置换。例如,甲基(CH3)是未取代的烷基。如果甲基的氢原子被与苯基键合的键置换,例如如果甲基的碳与苯的碳键合,则该化合物是未取代的芳烷基(即苄基)。
如本文所使用的,术语“取代的芳烷基”就未取代的芳烷基而言,具有和取代的芳基就未取代的芳基而言相同的含义。然而,取代的芳烷基还包括其中基团的烷基部分的碳或氢键被与非碳或非氢原子键合的键置换的基团。
如本文所使用的,术语“未取代的芳基”是指在环部分具有6至12个碳原子的单环或双环芳香族烃基,例如但不限于苯基、萘基、蒽基、联苯基和二苯基,其不含杂原子。尽管短语“未取代的芳基”包括含有稠环(例如萘)的基团,但其不包括具有与一个环成员键合的其它基团(例如烷基或卤素基团)的芳基,因为芳基(例如甲苯基)在本文中被认为是如下所述的取代的芳基。然而,未取代的芳基可以与母体化合物中的一个或多个碳原子、氧原子、氮原子和/或硫原子键合。
如本文所使用的,术语“取代的芳基”就未取代的芳基而言,具有和取代的烷基就未取代的烷基而言相同的含义。然而,取代的芳基还包括其中一个芳香族碳与非碳或非氢原子之一键合的芳基,所述非碳或非氢原子例如但不限于上述关于取代的烷基描述的那些原子;并且还包括其中芳基的一个或多个芳香族碳与如本文定义的取代和/或未取代的烷基、烯基或炔基键合的芳基。这包括其中芳基的两个碳原子与烷基或烯基的两个原子键合以限定稠环体系(例如二氢萘基或四氢萘基)的键合排列。因此,短语“取代的芳基”包括但不限于甲苯基和羟苯基等。
如本文所使用的,术语“未取代的杂环基”是指芳香族和非芳香族环化合物,其包括含有3个或更多个环成员的单环、双环和多环化合物,其中所述环成员中的一个或多个是杂原子,例如但不限于N、O和S。尽管短语“未取代的杂环基”包括稠合杂环(例如苯并咪唑基),但其不包括具有与一个环成员键合的其它基团(例如烷基或卤素基团)的杂环基,因为化合物(例如2-甲基苯并咪唑基)是如下定义的“取代的杂环基”。杂环基的实例包括但不限于:含有1至4个氮原子的不饱和3至8元环、含有1至4个氮原子的稠合不饱和杂环基、含有1至2个氧原子和1至3个氮原子的不饱和3至8元环、含有1至2个氧原子和1至3个氮原子的饱和3至8元环、含有1至2个氧原子和1至3个氮原子的不饱和稠合杂环基、含有1个至3个硫原子和1至3个氮原子的不饱和3至8元环、含有1至2个硫原子和1至3个氮原子的饱和3至8元环、含有1至2个硫原子的饱和和不饱和3至8元环、含有1至2个硫原子和1至3个氮原子的不饱和稠合杂环、含有氧原子的不饱和3至8元环、含有1至2个氧原子的不饱和稠合杂环、含有氧原子和1至2个硫原子的不饱和3至8元环、含有1至2个氧原子和1至2个硫原子的饱和3至8元环、含有1至2个硫原子的不饱和稠合环,以及含有氧原子和1至2个氧原子的不饱和稠合杂环。杂环基还包括以上描述的其中环中的一个或多个S原子与一个或两个氧原子双键键合的那些(亚砜和砜)。
如本文所使用的,术语“取代的杂环基”就未取代的杂环基而言,具有和取代的烷基就未取代的烷基而言相同的含义。然而,取代的杂环基还包括其中一个碳与非碳或非氢原子之一键合的杂环基,所述非碳或非氢原子例如但不限于上面关于取代的烷基和取代的芳基描述的那些原子;并且还包括其中杂环基的一个或多个碳与如本文定义的取代和/或未取代的烷基、烯基或芳基键合的杂环基。这包括其中杂环基的两个碳原子与烷基、烯基或炔基的两个原子键合以限定稠环体系的键合排列。实例包括但不限于2-甲基苯并咪唑基、5-甲基苯并咪唑基、5-氯苯并噻唑基、1-甲基哌嗪基和2-氯吡啶基等。
如本文所使用的,术语“未取代的杂环基烷基”是指如上定义的未取代的烷基或烯基,其中未取代的烷基或烯基的氢或碳键被与如上定义的取代或未取代的杂环基键合的键置换。例如,甲基(CH3)是未取代的烷基。如果甲基的氢原子被与杂环基键合的键置换,例如如果甲基的碳与吡啶的碳2(与吡啶的N键合的碳之一)或吡啶的碳3或4键合,则所述化合物是未取代的杂环基烷基。
如本文所使用的,术语“取代的杂环基烷基”就未取代的杂环基烷基而言,具有和取代的芳基就未取代的芳基而言相同的含义。然而,取代的杂环基烷基还包括其中非氢原子与杂环基烷基的杂环基中的杂原子(例如但不限于哌啶基烷基的哌啶环中的氮原子)键合的基团。
为简单起见,在本领域技术人员清楚的适当结构情况下,化学部分始终限定和指代的可以是单价化学部分(例如烷基、芳基等)或多价部分。例如,“烷基”部分可以指一价基团(例如,CH3-CH2-),或在其它情况下,二价连接部分可以是“烷基”,在这种情况下,本领域技术人员将烷基理解为相当于术语“亚烷基”的二价基(例如-CH2-CH2-)。类似地,在其中需要二价部分并且被称为“烷氧基”、“烷基氨基”、“芳氧基”、“烷硫基”、“芳基”、“杂芳基”、“杂环基”、“烷基”、“烯基”、“炔基”、“脂肪族基团”或“环烷基”的情况下,本领域技术人员将理解,所述术语指相应的二价部分。
如本文所使用的,术语“治疗”是指在疾病或病症的症状、方面或特征发作后开始的作用过程(例如施用结合物或药物组合物),以便消除或减少这种症状、方面或特征。这种治疗不一定是绝对有用的。
如本文所使用的,术语“需要治疗”是指由护理者做出的患者需要治疗或将从治疗受益的判断。该判断是基于护理者专业领域中的各种因素而做出的,但是包括患者由于疾病或病症而患病或将患病的知识,所述疾病或病症可通过本发明的方法或化合物治疗。
如本文所使用的,术语“需要预防”是指由护理者做出的患者需要预防或将从预防受益的判断。该判断是基于护理者专业领域的各种因素而做出的,但是包括患者由于疾病或病症将患病或可能会患病的知识,所述疾病或病症可通过本发明的方法或化合物预防。
如本文所使用的,术语“个体”、“受试者”或“患者”是指任何动物,包括哺乳动物,例如小鼠、大鼠、其它啮齿类动物、兔、狗、猫、猪、牛、羊、马或灵长类动物,以及人类。所述术语可以指定男性或女性或两者,或者排除男性或女性。
如本文所使用的,术语“治疗有效量”是指单独或作为药物组合物的一部分的结合物的量,所述结合物能够对疾病或病症的任何症状、方面或特征具有任何可检测的积极作用。这种作用不一定是绝对有益的。
聚恶唑啉聚合物
聚恶唑啉(POZ)是由2-取代-2-恶唑啉单体制备的聚合物。这些聚合物是水溶性的,并且已经报道在哺乳动物模型体系中是无毒的。POZ通常通过适当化学计算量的2-烷基-2-恶唑啉与亲电子引发剂(例如三氟甲磺酸甲酯(CH3-OSO2-CF3)或强酸(例如三氟甲磺酸或对甲苯磺酸))反应、随后通过用亲核药剂(例如但不限于氢氧化物、硫醇或胺)终止而制备。所产生的聚合物用由最左侧基团表示的引发基团和由最右侧基团表示的终止基团简化方便地描述,其中2-烷基-2-恶唑啉组分在中间。因此,当在本说明书中使用该简化描述时,除非另有指定,否则意在名称的左侧给出“引发剂端”,以及名称的右侧给出“终止端”。
例如,当2-烷基-2-恶唑啉是2-甲基-2-恶唑啉时,使用三氟甲磺酸甲酯作为引发剂,并且使用氢氧化物作为终止剂,产生以下POZ:
CH3-[N(COCH3)CH2CH2]n-OH。
以上聚合物用简化符号方便地描述为M-PMOZ-OH,其中甲基引发剂由最左侧的M表示,PMOZ表示聚甲基恶唑啉,其中重复单元的甲基由PMOZ的M表示,并且终止羟基由-OH表示。
另一种常用的单体是2-乙基-2-恶唑啉,其利用三氟甲磺酸甲酯引发和氢氧化物终止将提供以下POZ聚合物:
CH3-[N(COCH2CH3)CH2CH2]n-OH。
以上聚合物用简化符号方便地描述为M-PEOZ-OH,其中甲基引发剂由最左侧的M表示,PEOZ表示聚甲基恶唑啉,其中重复单元的乙基由PEOZ的E表示,并且终止羟基由-OH表示。
针对良好表征的聚合物,聚合度n的范围可以为大约3至超过1000。
聚合过程被称为活性阳离子聚合,因为利用亲电子药剂引发产生恶唑啉鎓阳离子,然后在链式反应中所述恶唑啉鎓阳离子与另外的单体单元反应产生生长的“活性”阳离子。
可以通过假设活性阳离子可以以下面的非环状形式(用于由三氟甲磺酸甲酯引发的2-甲基-2-恶唑啉的聚合)表示来预测终止的产物,尽管事实上环状形式当然是最重要的:
CH3-[N(COCH3)CH2CH2]n-N(COCH3)CH2CH2+。
在本讨论中,我们将这个阳离子表示为M-PMOZ+。如上所述,该POZ阳离子可以通过与亲核药剂(例如氢氧化物、硫醇或胺)反应而“终止”。
恶唑啉聚合也可以用官能性亲电子药剂引发。例如,亲电子引发剂3-溴丙酸乙酯已经用于引发2-乙基-2-恶唑啉聚合。用氢氧化物终止得到以下聚合物:
HO2C-CH2CH2-[N(COCH2CH3)CH2CH2]n-OH。
制备具有官能团的聚恶唑啉的又一种途径是使单体(例如2-乙基-2-恶唑啉)与在2-位具有官能团的恶唑啉单体共聚(F.C.Gaertner,R.Luxenhofer,B.Blechert,R.Jordan和M.Essler,《控制释放杂志(J.Controlled Release)》,2007,119,291-300)。例如,Jordan和同事已经利用乙炔和在2-位上保护的醛、羧酸和胺制备了恶唑啉。这些官能单体与2-乙基-2-恶唑啉的共聚产生具有多个侧基或侧链官能团的无规共聚物。例如,利用三氟甲磺酸甲酯引发2-乙基-2-恶唑啉和2-戊炔-2-恶唑啉的聚合,随后用哌嗪(NHC4H8NH)终止,得到以下无规共聚物:
CH3-{[N(COCH2CH3)CH2CH2]n-[N(COCH2CH2CH2-CCH)CH2CH2]m}ran-NC4H8NH。
下标“ran”表示聚合物是无规共聚物。n的值通常在20-30左右,而m在2-5左右。
具有侧基官能团和末端官能团的这些共聚物是有用的,因为侧基官能团和末端官能团可以是“化学正交的”官能团。化学正交的官能团是不会彼此反应但将与其它官能团选择性反应的那些官能团。例如,以上分子具有两个官能团,末端仲胺和侧基乙炔。乙炔将不与胺反应,但将例如与叠氮基(-N3)反应。类似地,胺将不与乙炔或叠氮化物反应,但将与例如异硫氰酸盐基(-NCS)反应。Jordan已经使用该共聚物将叠氮化物官能化的RGD肽与乙炔基耦联,并且将异硫氰酸盐官能化的金属螯合剂与胺耦联。已知RGD肽靶向肿瘤,并且诊断或治疗性放射性核素可以与螯合基团结合。最终的结合物可以用于成像或治疗肿瘤(R.Luxenhofer、M.Lopez-Garcia、A.Frannk、H.Kessler和R.Jordan,《美国化学学会会议记录(Proceedings of the American Chemical Society)》,PMSE Prepr.2006,95,283-284)。
阻碍现有技术的聚恶唑啉聚合物(包括上面提到的哌嗪-或哌啶-封端的聚恶唑啉)的使用的一个问题是它们难以提纯。出现这种困难是因为在终止期间存在的污染水导致水的亲核攻击并且随后形成仲胺杂质(O.Nyken、G.Maier、A.Gross、Macromol.Chem.Phys.197,83-95,1996)。由于通过哌嗪和哌啶终止的产物总是含有叔胺,所以不能使用离子交换色谱法来去除污染的仲胺。
上面说明的侧基聚恶唑啉的另一个限制是这些化合物具有单个末端官能团。因此,这种结构构型限制了可以附着到末端的药物或靶向部分的数量,而此类化合物有效用于治疗性诊断和靶向应用可能需要多次加载这些部分。本发明的聚合物通过提供两个化学反应性和正交官能团中的每一个的多个拷贝来避免这种限制。
聚恶唑啉聚合物-抗体结合物
许多聚恶唑啉聚合物可用于本发明中。在美国专利号8,110,651、8101,706和8,383,093和PCT申请号PCT/US2012/063088(所述文献中的每一篇通过引用并入本文用于此类教导)中描述了此类聚合物。在一个实施例中,用于所描述的ADC中的聚恶唑啉聚合物携带2种或更多种药剂,例如但不限于细胞毒素剂。在一个实施例中,用于所描述的ADC中的聚恶唑啉聚合物携带5种或更多种药剂,例如但不限于细胞毒素剂。在另一个实施例中,用于所描述的ADC中的聚恶唑啉聚合物携带8种或更多种药剂,例如但不限于细胞毒素剂。在一个实施例中,用于所描述的ADC中的聚恶唑啉聚合物携带10个或更多种药剂,例如但不限于细胞毒素剂。在前述实施例的每一个中,所述药剂可以相同或所述药剂可以不同(例如,在抑制癌症生长的途径中依次作用的两种细胞毒性化合物)。在一个实施例中,所述药剂是相同的。本文描述了代表性的药剂。在一个实施例中,聚恶唑啉聚合物可以是携带3种或更多种、5种或更多种、8种或更多种、20种或更多种或者40种或更多种药剂的杂官能聚合物,例如但不限于如所描述的细胞毒素剂。
在一个实施例中,聚恶唑啉-抗体结合物包含与识别部分连接的聚恶唑啉,其中识别部分包含结合残基,聚恶唑啉包含多种药剂,并且聚恶唑啉在结合残基处与识别剂连接。还提供了包含聚恶唑啉-抗体结合物和药学上可接受的载体的组合物。
在一个实施例中,识别部分是抗体或抗体片段,例如但不限于单链抗体。在进一步的实施例中,识别部分是抗体或抗体片段,例如但不限于单链抗体,并且结合残基是化学计量结合残基,其中聚恶唑啉聚合物和抗体以1:1的比例在化学计量结合残基处结合。在进一步的实施例中,识别部分是抗体或抗体片段,例如但不限于单链抗体,并且结合残基是硒代半胱氨酸残基,其中聚恶唑啉聚合物和抗体以1:1的比例在硒代半胱氨酸残基处结合。在进一步的实施例中,识别部分是单链抗体,并且结合残基是硒代半胱氨酸残基,其中聚恶唑啉聚合物和单链抗体以1:1的比例在硒代半胱氨酸残基上结合。在某些实施例中,多个聚恶唑啉聚合物可以与如所描述的单个识别部分结合。在某些实施例中,2、4、6、8或10个聚恶唑啉聚合物在不同位置与单个识别部分结合。在某些实施例中,2、3、4或5个聚恶唑啉聚合物在不同位置与单个识别部分结合。
在一个实施例中,聚恶唑啉结合物由以下通用结构表示:
R1-{[N(COX)CH2CH2]o-[N(COR2)CH2-CH2)]n-[N(COY)CH2-CH2)]m}a-R20
其中:
R1是引发基团;
R2是非反应性侧基部分;
针对每个重复单元,X是第一侧基部分,其中至少一个第一侧基部分含有药剂或提纯部分并且将所述药剂或所述提纯部分与聚合物主链连接;
针对每个重复单元,Y是第二侧基部分,其中每个第二侧基部分任选地含有药剂或提纯部分并且将所述药剂或所述提纯部分与聚合物主链连接;
R20是含有识别部分并且将所述识别部分与聚合物主链连接的识别剂连接部分;
a是表示无规共聚物的ran或表示嵌段共聚物的block;
n是0至1000的整数;以及
o和m各自是独立地选自0-50的整数,
条件是o和m不都是0,并且n、o和m的总和至少为30并且存在至少一种药剂。
在所述的聚合物结合物中,可以针对每个重复单元独立地选择药剂,使得在聚合物结合物中存在不止1种类型的药剂。同样,可以针对每个重复单元独立地选择提纯部分,使得在聚合物结合物中存在不止1种类型的提纯部分。在某些实施例中,仅存在1种类型的药剂和1种类型的提纯部分。在某些实施例中,存在2种类型的药剂和1种类型的提纯部分。如本文所讨论的,在某些实施例中,药剂和/或提纯部分(其可以包括第一和第二侧基部分的一部分)可以独立地合成并且与聚合物结合物连接,其中所连接的这些侧基部分的数量可以由侧基部分的摩尔比控制,从而允许在单个反应中添加多种类型的药剂和/或提纯部分。其它组合也在本发明的范围内。
在一个实施例中,o和m中的一个为0,并且o或m中的另一个为3、5、8、10、15、20、30、40或更多至50,并且o、m和n的总和为20至1000、30至500、30至250或30至100。在一个实施例中,o和m中的一个为0,并且o或m中的另一个大于或等于5且小于或等于20,并且o、m和n的总和为20至1000、30至500、30至250或30至100。在一个实施例中,o和m中的一个为0,并且o或m中的另一个大于或等于5且小于或等于15,并且o、m和n的总和为20至1000、30至500、30至250或30至100。在一个实施例中,o和m中的一个为0,并且o或m中的另一个大于或等于5且小于或等于10,并且o、m和n的总和为20至1000、30至500、30至250或30至100。在一个实施例中,前述任何一个中,m并非为0,而可以为1至50、1至30、1至20、1至15、1至10或1至5。在一个实施例中,前述任何一个中,m并非为0,而可以为1至50、1至30、1至20、1至15、1至10或1至5,并且第二侧基部分的全部或一部分没有药剂或提纯部分。
在一个实施例中,存在1至10、1至4或1至2个提纯部分。如本文所讨论的,此类提纯部分可以包含在X和/或Y中。提纯部分可以相同或者它们可以不同。在一个实施例中,存在2至20、2至15、2至10或2至7种药剂。在一个实施例中,存在2、3、4、5、6、7、8、9或10种药剂。在一个实施例中,存在多达40种药剂。如本文所讨论的,此类药剂可以包含在X和/或Y中。药剂可以相同或者它们可以不同。在本文所公开的某些结构中,结合物中存在的提纯部分的数量由o1标识表示,以及结合物中存在的药剂的数量由o2标识指示。
与结合物连接的药剂和/或提纯部分的数量小于或等于o和m的和。例如,如果o和m的和为15,则与聚合物连接的药剂和/或提纯部分的最大数量为15。在某些实施例中,与结合物连接的药剂和/提纯部分的数量小于o和m的和,意味着在某些实施例中存在游离的第一和/或第二侧基部分。因此,不是每个侧基部分都需要含有药剂和/或提纯部分。在某些实施例中,结合物具有2至40、2至20、5至20、10至20或20至40的DAR。在某些实施例中,结合物具有2、3、4、5、6、7、8、9或10的DAR。
第一和第二侧基部分的性质可以变化,只要基团具有用于将药剂和/或提纯部分与聚合物连接的结构。本文描述了第一和第二侧基部分的代表性结构。在一个实施例中,第一和第二侧基部分各自含有官能团(即,第一和第二官能团)。官能团参与到如本文所描述的药剂和/或提纯部分与聚合物的连接中。在一个实施例中,第一和第二官能团彼此化学正交,使得能够使用不同的反应化学将药剂和/或提纯部分与聚合物连接。第一和第二官能团包括但不限于炔、胺、羟基胺、醛、酮、缩醛、缩酮、马来酰亚胺、酯、羧酸、活化的羧酸(例如但不限于N-羟基琥珀酰亚胺基(NHS)和1-苯并三嗪活性酯)、活性碳酸酯、氯甲酸酯、醇、叠氮化物、乙烯基砜或邻吡啶基二硫化物(OPSS)。另外,第一和第二侧基部分可以含有将官能团与聚合物主链连接的其它基团,例如但不限于未取代或取代的烷基、未取代或取代的烯基、未取代或取代的芳烷基或者未取代或取代的杂环基烷基。在某些实施例中,第一和/或第二官能团是炔。在某些实施例中,第一和/或第二侧基部分中的至少一个不与药剂或提纯部分连接。
在某些实施例中,聚合物结合物是生物稳定的聚合物结合物。在某些实施例中,第一和/或第二侧基部分是生物稳定的。在某些实施例中,第一和/或第二侧基部分包含含有可水解部分的可水解连接基团,并且可水解连接基团和可水解部分是生物稳定的。
R2是非反应性侧基部分。因此,R2没有官能团;在一个实施例中,对于每个重复单元,R2独立地选自未取代或取代的烷基、未取代或取代的烯基、未取代或取代的芳烷基或者未取代或取代的杂环基烷基。在另一个实施例中,R2是长度为1至10或1至5个碳的未取代或取代的烷基。
示范性的引发基团包括但不限于氢、烷基、取代的烷基、芳烷基或取代的芳烷基。在一个特定的实施例中,引发基团是甲基。在一个实施例中,R1基团被选择为没有官能团。在PCT申请号PCT/US2008/078159中公开了另外的示范性引发基团,所述文献通过引用并入本文用于此种教导。
在一个实施例中,聚合物-结合物可以由以下通用结构表示:
其中所述药剂是本文所描述的任何药剂,X表示第一侧基部分,以及Y表示第二侧基部分。在前述的一个实施例中,o为3-15,m为1-45,并且n、m和o的总和为30-500(或者其中或本文所描述的任何子范围)。在该实施例中,第二侧基部分Y显示为没有与药剂或提纯部分附着。此外,为了简单起见,该实施例中的非反应性侧基显示为乙基,应当理解,针对R2,可以存在所描述的任何基团。当侧基部分没有与药剂或提纯部分(例如上述Y)连接时,侧基部分可以是炔(以及本文所描述的其它基团)。例如,当Y是炔时,聚合物结合物可以具有以下结构:
在一个实施例中,聚合物结合物可以由以下通用结构表示:
其中所述药剂是本文所描述的任何药剂,PM表示提纯部分,X表示第一侧基部分,以及Y表示第二侧基部分。在前述的一个实施例中,o1为1至4、为3至15,m为1至45,并且n、m和o的总和为30至500(或者其中或本文所描述的任何子范围)。在该实施例中,第二侧基部分Y显示为没有与药剂或提纯部分的连接。此外,为了简单起见,该实施例中的非反应性侧基显示为乙基,应当理解,针对R2,可以存在所描述的任何基团。当侧基部分没有与药剂或提纯部分(例如上述Y)连接时,侧基部分可以是炔(以及本文所描述的其它基团)。例如,当Y是炔时,聚合物结合物可以具有以下结构:
可通过调节如实例中所描述的中间体来控制结合物的合成期间药剂和提纯部分的数量。在一个实施例中,用于加入药剂和提纯部分(如果存在)的反应化学是相同的。在另一个实施例中,反应化学是不同的。例如,如果反应化学是相同的并且在最终结合物中需要2个提纯部分和6种药剂,则在结合反应期间,将2摩尔当量的含有提纯部分的侧基部分和6摩尔当量的含有药剂的侧基部分加入到反应混合物中,得到所需的结构。当药剂和提纯部分存在时,侧基连接部分的性质可以相同或者可以不同。换句话说,第一侧基连接基团的性质可以与如本文所描述的相同或不同。此外,如本文所描述的,X和Y侧基连接部分的性质可以使得所存在的反应性基团提供正交官能团,使得药剂和提纯部分通过不同的反应化学附着。
本领域已知的任何提纯部分可以用于本文所描述的聚合物结合物中。在一个实施例中,提纯部分是生物素。在一个实施例中,结合物中存在1至3个生物素分子。在一个实施例中,存在2个生物素分子。
在一个实施例中,所述药剂通过可释放的可水解连接(即,第一和/或第二侧基部分是可水解连接基团)与聚(恶唑啉)聚合物连接。在这种情况下,第一和/或第二侧基部分含有允许在将聚(恶唑啉)聚合物结合物施用于受试者后使所述连接裂解的至少一个可水解部分。第一和/或第二侧基部分可以含有单个可水解部分或不止一个可水解部分。可水解部分可以通过酶或通过化学反应裂解。在实例中,在存在不止一个可水解部分的情况下,一个可水解部分可以被酶裂解,并且一个可水解部分可以通过化学反应裂解,或者全部可以通过相同的机制裂解。在一个实施例中,第一和/或第二侧基部分含有1个可水解部分。在另一个实施例中,第一和/或第二侧基部分含有2个可水解部分。在另一个实施例中,第一和/或第二侧基部分含有2个可水解部分,其中1个部分可被酶裂解。
在一个实施例中,提纯部分通过稳定的(即使在作用部位,所述连接不裂解)或可水解连接基团与聚(恶唑啉)聚合物连接。因此,当药剂和提纯部分各自通过相同的反应化学与聚合物连接时(即,使用相同的反应化学,使用第一侧基部分将药剂和提纯部分连接),第一侧基部分的一部分可以含有可水解部分,并且第一侧基部分的一部分可以含有稳定的连接。在另一个实施例中,提纯部分通过如本文所描述的可释放的可水解连接与聚恶唑啉聚合物连接。在某些实施例中,可水解连接基团可以是生物稳定的。因此,当药剂和提纯部分各自通过相同的反应化学与聚合物连接时(即,使用相同的反应化学,使用第一侧基部分将药剂和提纯部分连接),药剂和提纯部分的所有连接含有可水解部分。然而,如果需要,可水解部分的性质可以在药剂与提纯部分之间变化。
许多酶可用于裂解可水解部分。在一个实施例中,在血液或血浆中不存在负责裂解的酶。在一个实施例中,在细胞区室中存在负责裂解的酶,例如但不限于溶酶体或核内体。在一个特定的实施例中,酶是肽酶,例如但不限于组织蛋白酶。代表性的组织蛋白酶是组织蛋白酶B。在一个特定的实施例中,可水解连接和可水解部分在血液或血浆中是生物稳定的,使得药剂和/或提纯部分在血流中不被释放到显著量。在一个实施例中,被酶裂解的可水解部分是酰胺键,并且这种酰胺键可以包含在氨基酸序列中。
示范性的可水解部分包括但不限于羧酸酯(-C(O)-O-)、碳酸酯(-O-C(O)-O-)、氨基甲酸酯(-O-C(O)-NH-)、二硫化物(-S-S-)、硫化物(-S-)、缩醛(-CH(OR')(OR”))、半缩醛(-CH(OR')(OH))、磷酸酯(-O-P(O)(OH)-(O)-)、膦酸酯(-O-P(O)(OR')-(O)-)和酰胺(-C(O)-NH-);在本文中讨论了其它可水解部分,其中R'和R”是H或者取代或未取代的烷基。在一个特定的实施例中,可水解部分是酯。在另一个特定的实施例中,可水解部分是酰胺。另外,可释放的可水解连接可以包含在天然存在的氨基酸、非天然存在的氨基酸或与一个或多个天然存在和/或非天然存在的氨基酸连接的聚合物中。
在一个实施例中,可释放的可水解连接是在R3或R4基团之一中含有可水解部分的二取代三唑。在一个实施例中,可水解部分在R4基团中。在一个具体的实施例中,二取代三唑具有以下结构:
在另一个实施例中,二取代三唑具有以下结构:
在前述结构的每一个中:
R3是将三唑部分与聚合物链连接的连接基团。在一个实施例中,R3(至少部分地)由聚合物链上的官能团限定。在一个实施例中,R3是-R5-或-C(O)-R5-,其中R5不存在或是长度为1至10个碳的取代或未取代的烷基。在一个实施例中,R5是长度为1至10个碳的未取代的烷基。在一个实施例中,R5是长度为1至5个碳的未取代的烷基。
R4形成三唑部分与药剂之间的连接。R基团的性质可以变化,只要连接功能以可释放的方式完成即可(即,所述连接含有可水解部分)。在一个实施例中,R4含有烷基部分。在另一个实施例中,R4含有聚合物部分。在另一个实施例中,R4含有一个或多个氨基酸。在另一个实施例中,R4含有聚合物部分和一个或多个氨基酸,其中可水解部分是氨基酸部分中的酰胺键。合适的聚合物包括本领域已知或本文所描述的任何水溶性聚合物。在一个实施例中,水溶性聚合物是由-(O-CH2-CH2)S-表示的聚乙二醇(PEG)聚合物,其中s是1至15、1至10或1至5的整数。s的具体值包括但不限于2、3、4和5。
在一个实施例中,R4是-R6-R7-R8-,其中
R6是取代或未取代的烷基、取代或未取代的芳烷基、聚合物或Ul-(pol)b-U2-([NR16-C(R13)(R14)-C(O)]c)-NR17-Ar-(CH2)S(如下所定义)并且任选地含有可水解部分;
R7是连接基团,任选地含有可水解部分或可水解部分的一部分;以及
R8不存在或者是O、S、CRc或NRc,其中Rc是H或者取代或未取代的烷基,条件是R6和R7中的至少一个含有可水解部分。
在一个实施例中,在R4中存在不止一个可水解部分。在一个实施例中,在R4中存在单个可水解部分。在一个实施例中,R7和R8组合形成可水解部分。在一个实施例中,R7含有可水解部分。在一个实施例中,R6含有可水解部分。在一个实施例中,在R6和R7中都存在可水解部分。此类可水解部分可以通过相同的机制或通过不同的机制裂解。
在前述的一个实施例中,R6是直链取代或未取代的C1-C16烷基或支链取代或未取代的C1-C16烷基。在前述的一个实施例中,R6是直链取代或未取代的C1-C5烷基或支链取代或未取代的C1-C5烷基。在一个实施例中,R6是-(CH2)p-,其中p为1至16、1至10或1至5。
在另一个实施例中,R6是聚合物。合适的聚合物包括本领域已知或本文所描述的任何水溶性聚合物。在一个实施例中,水溶性聚合物是由-(O-CH2-CH2)b-表示的聚乙二醇(PEG)聚合物,其中b是1至30、1至15、1至10或1至5的整数。b的具体值包括但不限于2、3、4和5。在另一个实施例中,聚合物是氨基酸聚合物。合适的氨基酸聚合物包括但不限于长度为1至10个氨基酸的聚甘氨酸聚合物和聚丙氨酸聚合物。这种聚合物,包括PEG或氨基酸聚合物,可以含有由连接产生的加成末端基团。
在另一个实施例中,R6含有与氨基酸部分连接的直链取代或未取代的、支链取代或未取代的烷基或聚合物(各自如上所述)(在聚合物是氨基酸聚合物的情况下,两个氨基酸部分将被连接)。氨基酸部分可以含有1至6个氨基酸、1至4个氨基酸或1至2个氨基酸。合适的氨基酸包括天然存在的氨基酸、非天然存在的氨基酸或前述的组合。在某些具体的实施例中,氨基酸可以是缬氨酸、瓜氨酸、赖氨酸和苯丙氨酸。在某些实施例中,氨基酸部分是缬氨酸和瓜氨酸或赖氨酸和苯丙氨酸。
在一个实施例中,R7是-Ra-O-C(O)-Rb-、-Ra-O-C(O)-O-Rb-、-Ra-O-C(O)-NH-Rb-、-Ra-CH(OH)-Rb-、-Ra-S-S-Rb-、-Ra-S-Rb-、-Ra-O-P(O)(OR11)-Rb-(其中R11是H或者取代或未取代的C1-C5烷基),或-Ra-C(O)-NH-Rb-,其中Ra和Rb各自独立地不存在或者是取代或未取代的烷基。在另一个实施例中,Ra和Rb各自独立地不存在或者是C1-C16取代或未取代的烷基。在另一个实施例中,Ra和Rb各自独立地不存在或者是C1-C5取代或未取代的烷基。在另一个实施例中,Ra和Rb各自不存在。
在某些实施例中,R7是-Ra-C(O)-Rb-、-Ra-O-C(O)-Rb-,Ra和Rb各自不存在并且R8是O,R7是-Ra-O-C(O)-Rb-或者-Ra-C(O)-Rb,Ra和Rb各自不存在并且R8是NH,以及R7是-Ra-S-S-Rb-、-Ra-S-Rb-,Ra和Rb各自不存在并且R8不存在。
在某些具体的实施例中,R7是-Ra-O-C(O)-Rb-、-Ra-O-C(O)-O-Rb-,Ra和Rb各自不存在并且R8不存在。
在一个实施例中,R6由U1-(pol)b-U2-([NR16-C(R13)(R14)-C(O)]c)-NR17-Ar-(CH2)S表示,其中
U1表示任选的连接基团;
pol表示聚合物部分;
U2表示任选的连接基团;
R17、R16和R13各自独立地是H或者取代或未取代的C1-C5烷基;
R14是天然存在或非天然存在的氨基酸上的侧链基团;
Ar表示芳基;
b是1至15的整数;
c是1至10的整数;以及
s是0至4的整数;
合适的聚合物包括本领域已知或本文所描述的任何水溶性聚合物。在一个实施例中,水溶性聚合物是由-(O-CH2-CH2)b-表示的聚乙二醇(PEG)聚合物,其中s是1至15、1至10或1至5的整数。s的具体值包括但不限于2、3、4和5。在另一个实施例中,聚合物是氨基酸聚合物。合适的氨基酸聚合物包括但不限于长度为1至10个氨基酸的聚甘氨酸聚合物和聚丙氨酸聚合物。
在某些实施例中,U1是长度为1-10、1-8、1-5或1-3个碳的取代或未取代的烷基。在一个实施例中,U1是长度为1-5个碳的未取代的烷基。在某些实施例中,U1不存在。
在某些实施例中,U2由-(CH2)t-C(O)-、-C(O)-或-NH-表示。在某些实施例中,U2不存在。在前述内容中,t是1-10、1-8、1-5或1-3。
在某些实施例中,R16和R13各自是H。
在某些实施例中,R14表示来自缬氨酸、瓜氨酸、赖氨酸或苯丙氨酸的侧链。在某些实施例中,c等于2,并且R14是来自缬氨酸和瓜氨酸或赖氨酸和苯丙氨酸的侧链。在某些实施例中,可水解部分是在基团-([NR16-C(R13)(R14)-C(O)]c)-内的酰胺键。
Ar基团可以含有单环或可以是多环的。在某些实施例中,Ar基团选自由以下组成的群组:苯、吡啶、吡嗪、咪唑、吡唑、恶唑、噻吩、萘、蒽和菲。在某些实施例中,Ar基团是苯。
在某些实施例中,s是1。
在一个实施例中,R6是Ul-(pol)b-U2-([NR16-C(R13)(R14)-C(O)]c)-NR17-Ar-(CH2)s,R7是-O-C(O)-,R8不存在。在一个实施例中,R6是U1-(pol)b-U2-([NR16-C(R13)(R14)-C(O)]c)-NR|7-Ar-(CH2)s,R7是-O-C(O)-,R8不存在,U1是取代或未取代的C1-C10烷基或不存在,U2是-(CH2)t-C(O)-、-C(O)-或-NH-或不存在,pol是聚乙二醇聚合物,以及Ar是苯。在一个实施例中,R6是Ul-(pol)b-U2-([NR16-C(R13)(R14)-C(O)]c)-NR17-Ar-(CH2)s,R7是-O-C(O)-,R8不存在,U1是取代或未取代的C1-C10烷基或不存在,U2是-(CH2)t-C(O)-、-C(O)-或-NH-或不存在,pol是聚乙二醇聚合物,Ar是苯,b是4,c是2以及s是1。
另外,当在结合物上存在提纯部分时,R3和R4部分没有可水解部分。
在一个实施例中,R3和R4部分没有可水解部分,R4由U3-(pol)b-U4表示,其中
U3表示任选的连接基团;
pol表示聚合物部分;以及
U4表示任选的连接基团;以及
b是1至15的整数。
合适的聚合物包括本领域已知或本文所描述的任何水溶性聚合物。在一个实施例中,水溶性聚合物是由-(O-CH2-CH2)b-表示的聚乙二醇(PEG)聚合物,其中s是1至15、1至10或1至5的整数。s的具体值包括但不限于2、3、4和5。在另一个实施例中,聚合物是氨基酸聚合物。合适的氨基酸聚合物包括但不限于长度为1至10个氨基酸的聚甘氨酸聚合物和聚丙氨酸聚合物。
在某些实施例中,U3是长度为1-10、1-8、1-5或1-3个碳的取代或未取代的烷基。在一个实施例中,U3是长度为1-5个碳的未取代的烷基。在某些实施例中,U3不存在。
在某些实施例中,U4由-C(O)-或-NH-表示。在某些实施例中,U4不存在。
在某些实施例中,R8不存在。
在某些实施例中,R3是-(CH2)3-或-C(O)-(CH2)3-,并且R4是-CH2-C(O)-O-、-CH2-CH2-C(O)-O-或-CH2(CH2)-C(O)-O-。
在一个特定的实施例中,R3是-(CH2)3-或-C(O)-(CH2)3,并且R4是
在一个特定的实施例中,R3是-(CH2)3-或-C(O)-(CH2)3,并且R4是
在一个特定的实施例中,R3是-(CH2)3-或-C(O)-(CH2)3,并且R4是
在另一个实施例中,可释放的可水解连接具有结构R9-Y-R10,其中Y是可水解部分,并且R9和R10各自是将Y与聚合物结合物和药剂分别连接的基团。R9和R10可以相同或不同。在一个实施例中,R9和R10各自独立地不存在或者是取代或未取代的烷基。在另一个实施例中,R9和R10各自独立地不存在或者是C1-C16取代或未取代的烷基。在一个实施例中,R9和R10各自不存在。
在前述的一个实施例中,连接基团是-R9-C(O)-O-R10-、-R9-O-C(O)-O-R10-、-R9-O-C(O)-NR18-R10-(其中R18是H或者取代或未取代的C1-C5烷基)、-R9-CH(OH)-O-R10-、R9-CH(OR12)-O-R10-(其中R12是H或者取代或未取代的C1-C5烷基)、-R9-S-S-R10-、-R9-S-R10-、-R9-O-P(O)(OR12)-O-R10-(其中R12是H或者取代或未取代的C1-C5烷基)、-R9-C(O)-NR18-R10-(其中R18是H或者取代或未取代的C1-C5烷基)或-R9-[NR18-C(R13)(R14)-C(O)]c-R10-(其中R18是H或者取代或未取代的C1-C5烷基,R13是H或C1-C5烷基,R14是天然存在或非天然存在的氨基酸上的侧链基团,并且c是1-10)。
在一个实施例中,可水解部分的裂解速率由可释放的可水解连接的性质控制。在前述的每一个中,可释放的可水解连接的可水解部分可以被裂解以释放药剂。在一个实施例中,可释放的可水解连接的可水解部分在施用于受试者后在受试者的生理条件下被化学裂解。在一个实施例中,可释放的可水解连接的可水解部分被在施用于受试者后在受试者的生理条件下天然存在或诱导存在于受试者中的物质裂解。在一个实施例中,这种物质是酶或多肽。在一个实施例中,可释放的可水解连接的可水解部分被前述的组合裂解。
在一个实施例中,聚合物-ADC可以由以下通用结构表示:
其中所述药剂是本文所描述或本领域已知的任何药剂,PM是本文所描述或本领域已知的任何提纯部分,q是1至10的整数,b和d独立地是1至30的整数,并且其余的变量如在当前说明书中所定义的。
在前述的一个实施例中,o是3-15,并且m和o的总和是1至50,并且n是1-1000或1-500。在某些实施例中,m是0。在另一个实施例中,o1、o2和m的和小于或等于50,o1是1至5的整数,o2是3至45的整数,m是1至50的整数,以及n是1至1000或1至500。
在一个实施例中,R20是用于将识别部分与聚合物连接的连接基团。在一个实施例中,R20含有与识别部分上的结合残基反应的反应性基团。在一个实施例中,反应性基团具体地与识别部分上的化学计量结合残基以1:1的比例反应。在一个特定的方面,识别部分是抗体,例如但不限于单链抗体,化学计量结合残基是如本文所描述的硒代半胱氨酸部分,并且反应性基团是马来酰亚胺基团。在一个特定的方面,识别部分是抗体,例如但不限于单链抗体,化学计量结合残基是如本文所描述的硒代半胱氨酸部分,并且反应性基团是碘代乙酰亚胺基团。在一个方面,R20包含间隔物以提供识别部分和聚恶唑啉聚合物的分离。在一个方面,R20可以通过使本发明的聚恶唑啉聚合物与终止亲核药剂反应并且任选地使所述结构进一步进行化学反应而形成。
在一个特定的实施例中,R20可以由以下结构表示:
R21-Z-R22,其中
R21选自由以下组成的群组:S、O或N;
Z是连接基团;以及
R22是含有识别部分的部分。
在一个实施例中,Z由-(CH2)r-表示,其中r是1至10。
在另一个实施例中,Z由-(CH2)r1-R23-R24-(CH2)r2-表示,其中R23不存在、是-C(O)-或-N-R25-,其中R25是H或者取代或未取代的烷基,R24不存在、是-O-或-N-R26-,其中R26是H或者取代或未取代的烷基,r1和r2各自独立地是0至10的整数。
在前述的一个方面,R23是-C(O)-。在一个方面,R24是-O-或-NH-。在一个方面,r1是1至4的整数并且r2是0至4的整数。在一个方面,R23是-C(O)-,R24是-O-,r1是2并且r2是0。在一个方面,R23是-C(O)-,R24是-NH-,r1是2并且r2是2。
在前述的一个方面,Z由-(CH2)r1-R23-R24-(CH2)r2-表示,R22是-NH-C(O)-CH2-S-Ab、-NH-C(O)-CH2-Se-Ab、其中Ab表示抗体。
在另一个实施例中,Z由-(CH2)r1-R23-R24-(CH2)r2-R27-R28-(CH2)r3-表示,其中R23不存在、是C=O或N-R25,其中R25是H或者取代或未取代的烷基,R24不存在、是O或N-R26,其中R26是H或者取代或未取代的烷基,R27不存在、是N-R29,其中R29是H或者取代或未取代的烷基或-C(O)-,R28不存在、是-C(O)-或N-R30,其中R30是H或者取代或未取代的烷基,并且r1、r2和r3各自独立地是0至10的整数。
在前述的一个方面,R23是-C(O)-。在一个方面,R24是-O-或-NH-。在一个方面,r1是1至4的整数,r2是0至4的整数,并且r3是0至4的整数。
在一个方面,R23是-C(O)-,R24是-NH-,R27是-NH-,以及R28是-C(O)-,r1是2,r2是2,并且r3是1。
在前述的一个方面,其中Z由-(CH2)r1R23-R24-(CH2)r2-R27-R28-(CH2)r3-表示,R22是S-Ab或Se-Ab,其中Ab表示抗体。
在一个方面,R22含有用于将识别部分与聚合物连接的反应性基团中的所有或一部分。代表性的连接基团包括但不限于与识别部分连接的碘乙酰胺、溴乙酰胺、氯乙酰胺、马来酰亚胺或丙烯酰胺基团。在一个方面,R22是与识别部分连接的马来酰亚胺基团或碘乙酰胺基团。在另一个实施例中,R22没有这样的部分。
在进一步的方面,识别部分是抗体,并且结合残基是半胱氨酸残基。在进一步的方面,识别部分是sc-抗体,并且结合残基是半胱氨酸残基。
在进一步的方面,识别部分是抗体,并且结合残基是硒代半胱氨酸残基,其中聚恶唑啉聚合物和抗体以1:1的比例结合在硒代半胱氨酸残基处。
在进一步的方面,识别部分是sc-抗体,并且结合残基是硒代半胱氨酸残基,其中聚恶唑啉聚合物和sc-抗体以1:1的比例结合在硒代半胱氨酸残基处。
在这种方面,R22基团可以由以下结构表示。尽管显示了代表性的反应性基团和结合残基的结构,但是本申请的公开内容不应限于所公开的实例。当马来酰亚胺是用于将抗体与聚合物连接的反应性基团时,R22可以具有以下结构:
当碘乙酰胺是用于将抗体与聚合物连接的反应性基团时,R22可以具有以下结构:
因此,代表性的R20基团包括但不限于(应当理解,以下结构中的硒(Se)可以被硫(S)或其它结合残基置换):
以及
在一个实施例中,本发明提供了以下结合物,其中o1、o2的和小于或等于50,o1是1至5的整数,o2是1至45的整数,m是1至50的整数,a表示无规或嵌段共聚物,药剂表示所述药剂,BR表示抗体上的结合残基,Ab表示抗体,以及PM表示提纯部分:
以及
在一个实施例中,Ab表示sc-抗体。在一个实施例中,Ab表示IgG、IgM、IgA、IgE或IgD抗体。Ab项可以表示本文所描述或本领域已知的抗体中的任何一种。在一个实施例中,BR表示硒代半胱氨酸。在一个实施例中,BR表示硫。BR项可以表示可以存在于所描述的抗体上的任何结合残基。在一个实施例中,药剂是秋水仙碱(例如但不限于脱乙酰基秋水仙碱)或奥瑞斯他汀(auristatin)(例如但不限于单甲基奥瑞斯他汀E、单甲基奥瑞斯他汀F或去甲基奥瑞斯他汀F)。所述药剂可以是本文所描述或本领域已知的任何药剂。
聚恶唑啉聚合物的合成方法
在一个实施例中,本发明的聚合物通过含有官能团的适当的2-取代-2-恶唑啉的共聚而制备。例如,除了简单的2-烷基-2-恶唑啉外,可以使用以下恶唑啉单体合成含有炔基和缩醛基的水溶性共聚物:
所述聚合由亲电子药剂(例如但不限于三氟甲磺酸甲酯、甲苯磺酸甲酯、对甲苯磺酸或三氟甲磺酸)引发。所述共聚可以通过如本文所讨论的亲核药剂终止。如果需要末端官能度,可以使用官能化的终止剂,例如但不限于甲基硫代乙酸酯。对于非反应性终止末端,可以使用亲核药剂,例如烷基硫醇。类似地,终止羟基对许多药剂也不具有反应性,并且因此可用于本申请中。用于聚合的优选溶剂是氯苯或乙腈。优选的温度范围为约40℃至约120℃。聚合所需的时间取决于温度、所需的分子量和溶剂,并且范围可以为约1小时至约100小时。在某些实施例中,期望将聚合反应限制在基本上完成聚合反应所需的时间。在一个实施例中,使用MALDI和/或GPC监测聚合反应的进展。
聚合可以以几种方式进行。在一个实施例中,可以将适当的恶唑啉组分的混合物与引发剂在优选的溶剂中在搅拌下反应。当所述恶唑啉组分中的一种或多种对彼此较不具有反应性时,当恶唑啉组分与彼此或与嵌段共聚物具有相同的反应性时,该反应产生无规共聚物。在一个替代的实施例中,聚合物也可以通过使用仅一种恶唑啉组分在优选的溶剂中用适当的引发剂引发聚合而以嵌段形式合成。可以利用MALDI或GPC监测反应以确定反应何时基本上完成。当第一嵌段的聚合完成时,加入第二恶唑啉组分以在聚合物链的末端用初始活性阳离子重新引发聚合;聚合反应的监测可以如所描述的那样进行。所述溶剂可以与第一聚合反应中相同或不同;反应温度和其它变量也可以根据需要进行调节。在第二嵌段的聚合完成时,加入第三恶唑啉组分以在聚合物链的末端用活性阳离子重新引发聚合;聚合反应的监测可以如所描述的那样进行。所述溶剂可以与第一或第二聚合反应中相同或不同;反应温度和其它变量也可以根据需要进行调节。当第三聚合步骤完成时,可以通过加入终止剂来终止聚合。恶唑啉组分可以含有官能团,或者可以没有官能团。连续聚合可以以任何顺序进行。在另一个实施例中,也可以进行两种恶唑啉单体的无规共聚,随后继续第三嵌段的聚合,以制备具有无规和嵌段构型的聚合物。在一个实施例中,所述恶唑啉单体中的一种没有官能团。
一旦所需的聚合过程完成,聚合物例如在乙醚中沉淀数次并且在真空下干燥。聚合物可以通过标准技术(例如但不限于MALDI、NMR和GPC)进一步表征。
对聚乙二醇有效已经表明,在靶分子的修饰中经常需要利用分子量(MW)为20,000Da或更高且分子量分布或多分散性(PD)小于1.1的聚合物。已经有大量的工作表明,针对POZ链,利用常规技术不能实现在上述范围内的MW和PD。如本领域已知的,PD值将随MW变化;通常,随着分子量的增加,PD值也增加。通常看到,随着生长POZ链的分子量达到大约5,000Da,多分散性明显增加。副反应(包括但不限于链转移)开始在重要性方面增长。当用于产生高MW的POZ链时,上述现有技术产生具有不可接受的PD值的POZ衍生物。本发明的聚恶唑啉衍生物可以使用聚恶唑啉链产生,所述聚恶唑啉链使用新的方法制造,所述新的方法导致具有低PD值和由不想要的副反应(例如但不限于链转移)产生的减少量的杂质的聚恶唑啉衍生物。在一个实施例中,制造聚恶唑啉链以使不想要的副反应(例如但不限于链转移)最小化,使得产生具有低PD值、提高纯度的POZ衍生物。因此,本发明的POZ衍生物可以被产生具有适合用于药物应用的提高纯度和低PD值。在PCT申请号PCT/US2008/078159中描述了此类方法,所述文献通过引入并入本文用于此种教导。
在一个实施例中,使用点击化学将药剂与聚恶唑啉聚合物连接。点击化学方法涉及炔基与叠氮基之间的反应。在一个方面,点击化学反应涉及药剂上的叠氮基酯和聚恶唑啉聚合物上的炔的反应。在该方面的一个特定的实施例中,叠氮基酯基团通过与药剂上的化学基团(例如但不限于羟基)进行合适的化学反应而形成。示范性的反应将是通过用叠氮化钠取代来自卤酸的卤化物以形成叠氮酸,随后用所述药剂(在此示例为罗替戈汀)上的羟基酯化叠氮基酸来制备叠氮基酯。
然后将叠氮基酯与存在于聚恶唑啉聚合物上的炔官能团连接。在一个特定的实施例中,炔官能团是存在于聚恶唑啉聚合物的侧位的乙炔官能团。
虽然可以使用上述方法,但是可以使用其它方法形成可水解部分。例如,还可以通过在聚恶唑啉聚合物上产生叠氮官能团(例如在聚恶唑啉聚合物上的侧基)、在药剂上产生炔基(例如乙炔酯),以及使叠氮基和炔基反应以形成包含可水解部分的可释放的可水解连接(在这种情况下为酯键)来形成含有酯可水解部分的连接。
在另一种方法中,可以在聚恶唑啉聚合物上产生羧酸基团,例如在聚恶唑啉聚合物上产生侧基,并且通过直接酯化该药剂上的醇或酚基使羧酸基团反应来形成含有可水解部分的可释放的可水解连接(在这种情况下为酯键)。在一个实施例中,在聚恶唑啉聚合物上的羧酸基团通过在聚合反应中包括羧化单体而产生在聚恶唑啉聚合物的侧位处。
在本发明的聚恶唑啉聚合物结合物的制备中,聚恶唑啉聚合物上的药剂数量由存在于聚恶唑啉聚合物上的反应性基团的数量控制;在一个实施例中,反应性基团存在于聚恶唑啉聚合物上的侧位中。对于在侧位的反应性基团,存在于聚合物上的反应性基团的数量由具有能够与药剂或连接基团形成连接的官能化侧链(例如乙炔)的单体单元(例如单体恶唑啉)相对于在聚合中使用的具有非活性侧链(例如烷基)的单体单元的比例控制。另外,对于具有官能化侧链的单体单元的给定比例,可以控制聚合物长度,提供对加载到给定聚合物结合物上的药剂的数量的进一步控制。因此,可以控制附着于特定聚合物结合物的药剂的数量。如上所述,连接基团的性质、聚合物的尺寸和施用途径允许控制药剂从聚合物的释放动力学。
识别部分
本发明的ADC包含识别部分。如本发明中所使用的,术语“识别部分”是指识别并结合特异性表位/抗原/细胞表面标志物的分子,包括但不限于经历内化进入细胞质的蛋白质,在细胞质中所述蛋白质可以与催化附着的药剂的释放的核内体融合。示范性的识别部分包括但不限于抗体、抗体的片段、单链抗体、多肽或肽模拟物等。除了将ADC靶向特定细胞、组织或位置之外,识别部分还可以具有某些治疗效果(例如针对靶细胞或途径的抗增殖(即,细胞抑制和/或细胞毒性)活性)。识别部分包含或可以被工程化以包含至少一个用于与ADC的聚合物部分连接的化学反应性基团。在一个方面,反应性基团是硒代半胱氨酸部分。
在一个特定的实施例中,识别部分是抗体。
如本说明书中所使用的,术语抗体是指任何含多肽链的分子结构,该分子结构具有结合并识别靶细胞上的抗原/表位的特定形状,其中一个或多个非共价结合相互作用稳定含多肽链的分子结构与抗原/表位之间的复合。在一个实施例中,抗体分子是免疫球蛋白。因此,术语抗体包括来自所有来源(例如,人、啮齿类动物、兔、牛、羊、猪、狗、其它哺乳动物等)的所有类型的免疫球蛋白(IgG、IgM、IgA、IgE、IgD等)。抗体可以是单克隆抗体或多克隆抗体;在一个方面,抗体是单克隆抗体。产生抗体和产生单克隆抗体的方法是本领域技术人员已知的。抗体或抗原结合片段也可以通过遗传工程产生。
在一个方面,本发明的抗体被修饰以减少诱导受试者(例如人受试者)中免疫应答的可能性。因此,术语抗体包括人源化、嵌合或异种人抗体,当施用于人时它们产生较少的免疫应答。可选地,可以产生含有人可变区的单链抗体(sc-Fv,如下所述)。因此,术语抗体可以包括单链抗体(本文中也称为sc-抗体或sc-Fv片段)。
除了整个免疫球蛋白结构之外,可以使用包含抗原/表位结合位点的免疫球蛋白片段。因此,术语抗体包括免疫球蛋白片段。此类片段包括但不限于Fab'、F(ab')2或其它片段。此类抗体片段可以通过胃蛋白酶、木瓜蛋白酶或其它蛋白酶裂解从完整免疫球蛋白产生。也可以利用重组免疫球蛋白技术设计此类片段。例如,用于本发明中的sc-抗体“Fv”免疫球蛋白可以通过经由肽连接基团(例如,不形成α螺旋或β折叠基序的聚甘氨酸或另一种序列)将可变轻链区与可变重链区连接而制备。
术语抗体还包括功能等同物。术语“功能等同物”包括具有同源序列的抗体、嵌合抗体、人工抗体和修饰抗体。本领域普通技术人员将理解,在称为“抗体片段”的分子组和称为“功能等同物”的组中存在重叠。产生功能等同物的方法是本领域技术人员已知的。人工抗体包括scFv片段、双抗体、三抗体、四抗体和分子识别单位(mru),它们中的每一个具有抗原结合能力。在单链抗体中,抗体的VH和VL结构域通过柔性肽连接。通常,该连接基团肽约为15个氨基酸残基长。如果连接基团较小,例如5个氨基酸,则形成为二价scFv二聚体的双抗体。如果连接基团减少到少于三个氨基酸残基,则形成被称为三抗体和四抗体的三聚体和四聚体结构。抗体的最小结合单位是CDR,通常是重链的CDR2,其具有足够的特异性识别和结合以使其可以单独使用。这样的片段称为mru。几个这样的mru可以利用短连接基团肽连接在一起,因此形成具有比单个mru更高亲合力的人工结合蛋白。
功能等同物还包括修饰抗体,包括通过将任何类型的分子与抗体共价连接而修饰的抗体。例如,修饰抗体包括通过由已知的保护/阻断基团、蛋白水解裂解、与细胞配体或其它蛋白质的连接糖基化,乙酰化、磷酸化、酰胺化、衍生化而修饰的抗体。这些修饰可以通过已知技术(包括但不限于特异性化学裂解、乙酰化和甲酰化)进行。
功能等同物可以通过使在不同框架内不同链上的不同CDR互换来产生。因此,例如,针对给定的一组CDR,通过取代不同的重链,不同类别的抗体是可能的,由此可以产生例如1gG1-4、IgM、IgA1-2、IgD、IgE抗体类型和同种型。类似地,在本发明范围内的人工抗体可以通过在完全合成的框架内嵌入给定的一组CDR来产生。可以使用本领域已知的多种方法,通过在特定一组CDR侧翼的可变和/或恒定区序列内的突变、删除和/或插入容易地产生功能等同物。对于表位识别和抗体结合,CDR是最重要的。然而,可以对包含CDR的残基进行改变,而不干扰抗体识别和结合其同源表位的能力。例如,可以进行不影响表位识别但增加抗体对表位的结合亲和力的改变。因此,同样包括在本发明的范围内的是前述的改进形式,优选具有增加的亲和力。基于一级抗体序列的知识,基于其性质(例如结合和表达水平),几个研究已经调查了在抗体序列中的多个位置处引入一个或多个氨基酸改变的效果(Yang,W.P.等人,1995,J.Mol.Biol.,254:392-403;Rader,C.等人,1998,Proc.Natl.Acad.Sci.USA,95:8910-8915;Vaughan,T.J.等人,1998,《自然生物技术(Nature Biotechnology)》,16:535-539)。在这些研究中,通过使用例如寡核苷酸介导的定点诱变、盒式诱变、易错PCR、DNA改组或者大肠杆菌的增变株等方法改变CDR1、CDR2、CDR3或框架区中重链和轻链基因的序列,产生了一级抗体的等同物(Vaughan,T.J.等人,1998,《自然生物技术(Nature Biotechnology)》,16:535-539;Adey,N.B.等人,1996,第16章,第277-291页,在“肽和蛋白质的噬菌体展示(Phage Display of Peptides and Proteins)”中,Eds.Kay,B.K.等人,学术出版社(Academic Press))。这些改变一级抗体序列的方法导致抗体亲和力改善(Gram,H.等人,1992,Proc.Natl.Acad.Sci.USA,89:3576-3580;Boder,E.T.等人,2000,Proc.Natl.Acad.Sci.USA,97:10701-10705;Davies,J.和Riechmann,L,1996,《免疫技术(Immunotechnolgy)》,2:169-179;Thompson,J.等人,1996,J.Mol.Biol,256:77-88;Short,M.K.等人,2002,《生物化学杂志(J.Biol.Chem)》,277:16365-16370;Furukawa,K.等人,2001,《生物化学杂志》,276:27622-27628)。
在一个实施例中,此类氨基酸取代是这样的氨基酸取代:(1)降低对蛋白水解的易感性、(2)降低对氧化的易感性、(3)改变形成蛋白质复合物的结合亲和力,以及(4)赋予或修饰此类抗体的其它物理化学或功能性质。此类功能等同物可以包括来自天然存在的肽序列的序列的各种突变。例如,可以在天然存在的序列中(优选地在形成分子间接触的结构域外的多肽部分中)进行单个或多个氨基酸取代(优选地保守性氨基酸取代)。保守性氨基酸取代实质上不应改变亲本序列的结构特征(例如,替代氨基酸不应倾向于破坏亲本序列中出现的螺旋,或破坏表征亲本序列的其它类型的二级结构)。在《蛋白质、结构和分子原理(Proteins,Structures and Molecular Principles)》(Creighton,Ed.,W.H.Freeman and Company,New York(1984))中、《蛋白结构简介(Introduction to Protein Structure)》(C.Branden和J.Tooze,eds.,Garland Publishing,New York,N.Y.(1991)),以及Thornton等人,1991,《自然(Nature)》,354:105中描述了本领域公认的多肽二级和三级结构的实例,所述文献各自通过引用并入本文。
此外,前述抗体中的任何一种可以被修饰以含有一个或多个非经典氨基酸。在一个实施例中,非经典氨基酸是硒代半胱氨酸残基(例如,如在《生物化学(Biochemistry)》.2009年12月22日;48(50):12047-57中所描述的,所述文献通过引用并入本文用于此种教导)。在一个实施例中,将非经典氨基酸置于抗体的C-末端处,以使与抗原/表位结合的干扰最小化。
在一个特定的实施例中,识别部分是抗体。在进一步特定的实施例中,识别部分是单链抗体。抗体,特别是单克隆抗体和sc-抗体在预防、诊断和治疗人类疾病中的使用正在增加。超过20种单克隆抗体由食品和药物管理局批准,并且更多的单克隆抗体处于临床发展的各个阶段。另外,还正在探索ADC以开发抗体组分的特异性和化学组分/药剂(例如小分子或含药剂的聚合物)的作用。例如,尽管抗体分子提供特异性和亲和力,但是小分子提供成像能力或细胞毒性。
在一个实施例中,无论抗体是单链抗体还是其它形式,识别部分与受体酪氨酸激酶样孤儿受体(ROR)结合(综述于Front.Oncol.,2012年4月18日|doi:10.3389/fonc.2012.00034)。已知受体酪氨酸激酶(RTK)是正常细胞过程(例如分化、迁移、增殖和存活)的关键调节剂,但是它们在许多类型的人类癌症的发展和进展中也具有关键作用。因此,RTK及其配体已成为用于癌症的治疗性干预的有吸引力的分子靶。这些受体通常在发育期间以高水平表达,在骨骼和神经器官发生中起关键作用,但随后在成体组织中被抑制。RTK的ROR族由两个进化上保守的蛋白质(ROR1和ROR2)和将ROR1和ROR2鉴定为在各种癌症中过表达的签名基因之一的基因表达谱组成。例如,已经发现ROR1在多种造血癌症(包括B细胞慢性淋巴细胞性白血病,B细胞急性淋巴细胞性白血病,以及套细胞白血病)中过表达,同时已经发现ROR2在多种肉瘤和癌中过表达。
ROR2是在正常骨和软骨发育过程中通过其与Wnt5A糖蛋白的缔合而被非规范Wnt信号传导激活的膜键合的RTK。需要ROR2表达以在哺乳动物的腭发育期间介导细胞的迁移,并且已经显示ROR2基因中的突变引起例如短指症B型和常染色体隐性Robinow综合征的疾病。已经报道ROR2在某些细胞系中具有促肿瘤发生作用。已经发现ROR2在多种肉瘤和癌中过表达。
癌是最常见类型的人类癌症,其由已经形成上皮细胞的细胞学外观、组织结构或分子特征的细胞引起。癌是相当异质性的实体,反映了各种致癌启动子的广泛的种类、强度和效力。亚型包括腺癌、鳞状细胞癌、间变性癌、大细胞和小细胞癌及其混合。癌也根据其发生的部位分类,例如肺癌、乳腺导管癌、前列腺腺癌、结肠和直肠的腺癌或鳞状细胞癌等。肉瘤是超过60种肿瘤类型的异质组,其源自间充质细胞,并且占所有人类恶性肿瘤的大约1%。大多数肉瘤显示出局部侵袭性生长和转移的倾向。
硒代半胱氨酸结合
将抗体与化合物结合的许多常规方法是已知的。例如,在与药剂上的适当官能团的结合反应中可以使用赖氨酸(Lys)残基的ε-氨基或半胱氨酸(Cys)残基的硫醇基。然而,使用这些方法,所产生的ADC不是单一种类的分子,而是具有一系列抗体/药剂化学计量和附着点的分子的混合物。此外,所产生的ADC的特征在于基本的批次间可变性。这种结合的随机模型在许多情况下可能损害抗原/抗体结合反应。化学组分/药剂与抗体的位点特异性结合提供了限定的化学计量、降低的批次可变性和保持抗原/抗体结合反应的可能性。位点特异性结合的目的是向抗体中引入独特的化学反应性,而不显著影响抗体分子的结构和/或功能。
硒代半胱氨酸形式近来已经用于通过在抗体与化学组分之间提供工程界面来提供化学组分与抗体的位点特异性结合。硒代半胱氨酸提供化学组分在所需位置的靶向放置,并且提供1:1的化学计量。该过程已由Hofer及其同事描述(PNAS,105(34),12451-12456,2008;《生物化学(Biochemistry)》,48(50),12047-12057,2009;前述文献中的每一篇通过引用并入本文用于此类教导)。硒代半胱氨酸界面技术已经与完整抗体分子和抗体片段一起使用,而不影响抗体或抗体片段的生物学性质。与其它位点特异性抗体结合方法相比,硒代半胱氨酸界面方法:(a)涉及抗体结构的微小修饰,例如在C-末端,其不干扰结构或功能;(b)利用简单和有效的偶联化学;以及(C)导致抗体和化学组分的1:1化学计量。
药剂
药剂可以是用于治疗疾病或病症或诊断疾病或病症的任何药剂。在某些实施例中,药剂是诊断剂或治疗剂。在某些实施例中,治疗剂是有机小分子。此外,药剂可以是具有治疗或诊断应用的任何分子,其中药剂能够与本发明的聚合物(例如但不限于POZ聚合物)上的官能团或者与本发明的聚合物连接的连接基团形成连接。
用于本发明的药剂可以包括任何已知的药剂。在一个实施例中,药剂可用于治疗癌症。在一个实施例中,药剂是细胞毒素剂。在一个实施例中,药剂是微管抑制剂、DNA-损伤剂或聚合酶II抑制剂。微管抑制剂结合微管蛋白、使微管不稳定,并且引起G2/M阶段细胞周期停滞。奥瑞斯他汀和美登木素生物碱类美登素(maytansinoid)是两类微管抑制剂。DNA-损伤剂包括蒽环霉素、卡奇霉素(calicheamicin)、多卡霉素(duocarmycin)和吡咯并苯二氮呯类(PBD)。所有这些药物通过结合DNA的小沟和引起DNA断裂、烷基化或交联而起作用。
可用于本发明的结合物中的药剂类别包括但不限于奥瑞斯他汀(例如但不限于单甲基奥瑞斯他汀E、单甲基奥瑞斯他汀F、去甲基奥瑞斯他汀F)、蒽环霉素(例如但不限于柔红霉素、多柔比星、表柔比星、伊达比星、戊柔比星和米托蒽醌)、加利车卡奇霉素(例如但不限于加利车卡奇霉素和N-乙酰基-γ加利车卡奇霉素1,2-二甲基肼二氯化物)、美登木素生物碱类美登素(例如但不限于DM1、DM4和安丝菌素)、吡咯并苯并二氮杂苯并二氮呯(例如但不限于赤霉素、芝加霉素、DC-81、甲基氨茴霉素、新斯拉霉素A和B、波罗斯拉霉素、prothracarcin、西班米星(DC-102)、西伯利亚霉素和托马霉素)、吡咯并苯并二氮杂苯并二氮呯类的二聚体(例如但不限于SJG-136、SG 2285、DRG-16和ELB-21)、秋水仙碱类(例如但不限于秋水仙碱、脱乙酰基秋水仙碱)、多拉司他汀海兔毒素类(例如但不限于多拉司他汀海兔毒素10、15和16)、微管蛋白类(例如但不限于微管蛋白、A、B、D和M)、美登醇、多卡霉素类(例如但不限于多卡霉素A、B1、B2、C1、C2、D、SA和CC-1065和阿多来新、比折来新和卡折来新),奈莫柔比星和其它多柔比星类似物,以及隐藻素类(例如但不限于隐藻素8、24、52、55、296和309)、埃坡霉素类(例如但不限于埃坡霉素A、B、C、D、E、F、BMS 247550和BMS 310705)和番红菌素(例如但不限于番红菌素A和B)。前述的衍生物和类似物也包括在本发明的范围内。
药用组合物
本发明的聚合物结合物(包括聚合物-ADC)可以被配制用于人和兽医用途,并且可以包含聚合物-ADC和药学上可接受的载体。通常,除了一种或多种无活性剂,例如无菌的生物相容性载体(包括但不限于无菌水、盐水、缓冲盐水或葡萄糖溶液),药物组合物将包括本发明的聚合物-ADC。药物组合物可以单独施用或与其它治疗方案(包括其它化学治疗化合物、激素、疫苗和/或放射治疗)组合施用。“与……组合”并不意味着额外的方案必须同时施用或被配制用于一起递送,尽管这些递送方法在本发明的范围内。通常,每一种将以一定剂量和根据为该方案确定的时间表施用。另外,本发明包括与可以改善生物利用度、减少或改变代谢、抑制排泄或改变体内分布的药剂组合递送本发明的聚合物-ADC。可选地或另外,本发明的聚合物-ADC可以与一种或多种解决所治疗的疾病或病症的症状或病因或患者所患的任何其它疾病的其它化合物一起施用。尽管本发明的药物组合物可以用于治疗有需要的任何受试者(例如,任何动物),但是它们最优选用于人的治疗。
本发明的药物组合物可以通过多种途径(包括口服、静脉内、肌内、动脉内、皮下、心室内、经皮、直肠、阴道内、腹膜内、局部(如通过粉末、软膏或滴剂)、口腔,或作为口或鼻喷雾剂或气雾剂)施用于人和其它动物。通常,最适当的施用途径将取决于多种因素,包括化合物的性质(例如,其在胃肠道环境中的稳定性)、患者的状况(例如,患者是否能够忍受口服施用)。目前,静脉内途径最常用于递送本发明的聚合物-ADC。然而,本发明涵盖通过考虑药物递送科学中的可能进展的任何适当途径递送本发明的聚合物-ADC。
治疗方法
在某些实施例中,本发明的聚合物-结合物可以靶向特定的细胞或组织,例如癌细胞或肿瘤块。在该实施例的一个方面,聚合物-ADC可以用于靶向表达抗原(例如但不限于细胞表面受体)的特定细胞或组织,所述抗原能够被附着于聚合物的抗体结合。
在一个实施例中,公开了一种治疗疾病状态或病症的方法。此种方法包含向受试者施用一定量的本发明的聚合物结合物(包括聚合物-ADC结合物)的步骤,所述聚合物结合物结合靶细胞上的选定抗原并将药剂递送至细胞。在一个实施例中,所述药剂是细胞毒素剂。在一个实施例中,所述药剂是微管抑制剂、DNA-损伤剂或聚合酶II抑制剂。
提供了用于抑制表达由本发明的聚合物结合物上的抗体识别的抗原的细胞的生长的方法。此种方法包含向受试者施用一定量的本发明的聚合物结合物(包括聚合物-ADC结合物)的步骤,所述聚合物结合物结合靶细胞上的选定抗原并将药剂递送至细胞。在一个实施例中,所述药剂是细胞毒素剂。在一个实施例中,所述药剂是微管抑制剂、DNA-损伤剂或聚合酶II抑制剂。抑制所选细胞群体生长的方法可以在体外、体内或离体进行。如本文所使用的,“抑制生长”是指在短时间或长时间内减缓细胞的生长、降低细胞生存力、引起细胞死亡、裂解细胞和诱导细胞死亡。
因此,本发明包括被描述为药物的抗聚合物-ADC结合物的使用。
在所描述的方法中,聚合物结合物可以单独施用或作为如本文所描述的药物组合物的一部分施用。在一个实施例中,确定受试者需要这种治疗。在进一步的实施例中,聚合物结合物以治疗有效量施用。在本文所公开的方法中,受试者可以是哺乳动物。在某些实施例中,受试者是人。
在一个实施例中,疾病状态是癌症。本发明的聚合物-ADC可用于治疗或预防多种癌症,包括(但不限于)以下各项:癌,包括膀胱癌、乳腺癌、结肠癌、肾癌、肝癌、肺癌、卵巢癌、胰腺癌、胃癌、宫颈癌、甲状腺癌和皮肤癌;包括鳞状细胞癌;造血淋巴系肿瘤,包括白血病、急性淋巴细菌性白血病、急性淋巴细胞性白血病、B细胞淋巴瘤、T细胞淋巴瘤、伯基特氏淋巴瘤;髓系的造血肿瘤,包括急性和慢性髓细胞性白血病和早幼粒细胞性白血病;间质源性肿瘤,包括纤维肉瘤和横纹肌肉瘤;其它肿瘤,包括黑色素瘤、精原细胞瘤、畸形瘤、神经母细胞瘤和神经胶质瘤;中枢和周围神经系统的肿瘤,包括星形细胞瘤、神经母细胞瘤、神经胶质瘤和神经鞘瘤;间质源性肿瘤,包括纤维肉瘤、横纹肌肉瘤和骨肉瘤;以及其它肿瘤,包括黑色素瘤、着色性干皮病、角化棘皮瘤、精原细胞瘤、甲状腺滤泡状癌和畸胎癌。待治疗的癌症的性质可以通过与聚合物结合物结合的抗体的性质来确定。
提供了通过向有需要的患者施用本发明的聚合物-ADC来抑制ROR1和/或ROR2阳性细胞生长的方法,所述本发明的聚合物-ADC与ROR1和/或ROR2结合并且将药剂递送至ROR1和/或ROR2阳性细胞。在一个实施例中,所述药剂是细胞毒素剂。抑制所选细胞群体生长的方法可以在体外、体内或离体进行。如本文所使用的,“抑制生长”是指在短时间或长时间内减缓细胞生长、降低细胞生存力、引起细胞死亡、裂解细胞和诱导细胞死亡。因此,本发明包括被描述为药物的抗聚合物-ADC结合物的使用。
对于临床体内使用,聚合物-结合物将作为测试无菌性和内毒素水平的溶液提供。用于施用本发明的聚合物结合物的合适方案的实例如下:通过任何合适的途径每日、每半周、每周施用至少一周、至少2周、至少3周、至少4周或更久。关于施用途径的具体临床方案、赋形剂、稀释剂、剂量、时间等可以由本领域普通技术人员根据临床情况确定。
本发明提供了试剂盒,所述试剂盒包含本发明的聚合物结合物、包装材料,以及用于将前述施用于受试者以治疗疾病或病症(例如但不限于癌症)的说明书,基本上由其组成或由其组成。
具体实施方式
实例
材料的制备
为了说明本发明的教导,提供了聚恶唑啉-ADC结合物的制备,并且描述了这些结合物的生物活性。所使用的缩写如下:DC,脱乙酰基秋水仙碱;MMAE,奥瑞斯他汀;POZ,用丙酸封端的聚恶唑啉;VC,缬氨酸-黄晶;Phe-Lys,苯丙氨酸和赖氨酸;PAB,对氨基苄醇;DCM,二氯甲烷;TEA,三甲胺;DMAP,二甲基氨基吡啶;DIPEA,二异丙基乙胺;ACN,乙腈;NPC,硝基苯基碳酸酯,PEG,聚乙二醇;N3,叠氮化物或叠氮基。
材料:
秋水仙碱购自西格玛(Sigma)。二碳酸二叔丁酯、4-(二甲基氨基)吡啶DMAP、三乙胺(TEA)、0.5N甲醇钠甲醇溶液、三氟乙酸(TFA)、N,N’-二环己基碳二亚胺(DCC)、N-羟基琥珀酰亚胺(NHS)、N-Boc-乙二胺(Boc-EDA)、N,N-二异丙基乙胺(DIPEA)、Fmoc氯化物(Fmoc-Cl)、N-(2-氨基乙基)马来酰亚胺TFA盐、1-羟基苯并三唑水合物(HOBT)、碘化铜(I)和哌啶购自西格玛奥德里奇(Sigma-Aldrich)。N3-PEG4-VC-PAB-PNP、N3-PEG4-VC-PAB-MMAE和N3-PEG4-Phe-Lys(Trt)-PAB-NPC购自Concortis。二乙醚、二氯甲烷(DCM)和乙腈(ACN)购自EMD。生物素-PEG3-叠氮化物购自Chempep。L(+)-抗坏血酸钠盐和五水硫酸铜(CUSO4·5H2O)购自Fluka。M4195购自Supelco。二甲基甲酰胺(DMF)、四氢呋喃(THF)、二氯甲烷(DCM)、乙腈(ACN)、无水硫酸钠、无水硫酸镁和硅胶60(70-230目)购自EMD。琥珀酰亚胺基碘乙酸酯购自CovaChem。生物素-PEG3-叠氮化物购自Chempep。
实例1.
POZ 10p马来酰亚胺5K的制备
具有10个侧基炔基并且用羧酸封端的MW 5,000的POZ(POZ 10p酸5K)根据在美国专利号8,110,651和8,101,706(所述文献中的每一篇通过引用并入本文用于此类教导)中描述的程序合成。POZ 10p酸5K具有通过基质辅助激光解吸/电离(MALDI)质谱法(MS)测定的为5,500g/mol的Mn和为1.01的PDI。通过AKTA色谱法(DEAE柱)的聚合物的端基分析显示聚合物具有99.8%的酸端基。1H NMR谱与结构一致,并且表明聚合物中的2-戊炔基-2-恶唑啉和2-乙基-2-恶唑啉单元的比例为10:40。
作为琥珀酰亚胺基衍生物(POZ 10p SPA 5K)活化的POZ酸根据在美国专利号7,943,141中描述的程序合成。1H NMR谱与结构一致并且表明酸基团定量转化为琥珀酰亚胺基酯。
在25mL圆底烧瓶中将N-(2-氨基乙基)马来酰亚胺三氟乙酸盐(0.0794g,3.125×10-4mol,1.25当量)溶解于15mL的二氯甲烷中。将0.0871mL的三乙胺(0.0632g,6.25×10-4mol,2.5当量)加入到烧瓶中。然后将1.375g的POZ 10p SPA 5K(2.5×10-4mol,1当量)加入烧瓶中,并且将溶液在室温下在氩气下搅拌20小时。在该时间结束时,将溶液旋转蒸发至干。将残余物溶解在100mL的去离子水,10wt.%氯化钠中,并且萃取到二氯甲烷(3×25mL)中。分离二氯甲烷相并用25mL的10wt.%氯化钠溶液洗涤。再次分离二氯甲烷相,用硫酸钠干燥,过滤,浓缩至15mL,并且沉淀到225mL的二乙醚中。将聚合物通过玻璃烧结的玻璃料过滤并且在高真空下干燥过夜,得到1.00g的产物(产率:72%)。1H NMR(Varian,10mg/mL CDCl3,δ)在1.12ppm(s,3H,CH3CH2CO-)、2.31ppm(小s)和2.41ppm(大s)(总面积2H,CH3CH2CO-)和3.47ppm(s,4H,-NHCH2CH2NH-)处显示常见的主链峰。侧基峰出现在1.84ppm(m,2H,-CH2CH2CH2CC≡H)和2.00ppm(m,1H,CH2CH2CH2CC≡H)处。端基-CH2SCH2CH2CONH-峰出现在2.70-2.82ppm处。连接到马来酰亚胺的氮和马来酰亚胺的-CH基团的-CH2基团分别出现在3.68ppm和6.72ppm处。聚合物中2-戊炔基-2-恶唑啉和2-乙基-2-恶唑啉单元的比例为10.2:40。POZ 10p马来酰亚胺5K具有通过MALDI测定的为6400g/mol的Mn和为1.02的PDI。
POZ 2p马来酰亚胺5K的制备
POZ 2p酸5K是根据在美国专利号8,110,651和8,101,706中描述的程序合成的。POZ2p酸5K具有通过MALDI测定的为5,200g/mol的Mn和为1.02的PDI。通过AKTA(DEAE柱)的聚合物的端基分析显示聚合物具有98.6%的酸端基。1H NMR表明聚合物中2-戊炔基-2-恶唑啉和2-乙基-2-恶唑啉单元的比例为1.9:48。
POZ 2p SPA 5K是根据在美国专利号7,943,141中描述的方法合成的。1H NMR光谱与结构一致并且表明酸基团定量转化为NHS酯。
在100mL圆底烧瓶中将0.31g的N-(2-氨基乙基)马来酰亚胺三氟乙酸盐(1.2×10-3mol,1.25当量)溶解于50mL的二氯甲烷中。将三乙胺(0.24g,2.4×10-3mol,2.5当量)加入到烧瓶中。然后将POZ 2p SPA 5K(5.0g,9.6×10-4mol,1当量)加入烧瓶中。将溶液在室温下在氩气下搅拌24小时。用10wt%NaCl(0.1N HCl)溶液(3x 50mL)洗涤所述溶液。分离二氯甲烷相,用硫酸钠干燥,过滤,浓缩至50mL,并且沉淀到750mL的二乙醚中。将聚合物通过玻璃烧结的玻璃料过滤并且在高真空下干燥过夜,得到4.53g的产物(产率:90%)。1H NMR(Varian,10mg/mL CDCl3,δ)在1.13ppm(s,3H,CH3CH2CO-)、2.29ppm(小s)和2.41ppm(大s)(总面积2H,CH3CH2CO-)和3.47ppm(s,4H,-NHCH2CH2NH-)处显示常见的主链峰。侧基峰出现在1.84ppm(m,2H,-CH2CH2CH2CC≡H)和2.00ppm(m,1H,CH2CH2CH2CC≡H)处。端基-CH2SCH2CH2CONH-峰出现在2.28-2.82ppm处。连接到马来酰亚胺的氮和马来酰亚胺的-CH基团的-CH2基团分别出现在3.67ppm和6.72ppm处。聚合物中2-戊炔基-2-恶唑啉和2-乙基-2-恶唑啉单元的比例为1.9:48。POZ 2p马来酰亚胺5K具有通过MALDI测定的为4,850g/mol的Mn和为1.02的PDI。
实例2.叠氮基毒素与具有侧基戊炔基的POZ的偶联
该反应如在美国专利8,101,706中所描述的那样进行,所述文献通过引用并入本文用于此种教导。
实例3.脱乙酰基秋水仙碱·TFA(3)的合成
步骤1.N-Boc-秋水仙碱(1)的合成
根据L.Lebeaii等人,《合成化学(Syn.Comm.)》,27(2),293-296(1997)来合成脱乙酰基秋水仙碱(DC)。将秋水仙碱(2.00gm,4.76mmol)在乙腈(ACN)(20mL)中的溶液蒸发至干。然后将ACN(20mL)加入到固体中。向溶液中加入三乙胺(TEA)(0.74mL,5.34mmol)和二甲基氨基吡啶(DMAP)(0.65gm,5.33mmol),随后加入二碳酸二叔丁酯(6.23mL,27.1mmol)。将溶液在氩气气氛下在80℃下搅拌过夜。在冷却至室温后,将溶液在旋转蒸发器上蒸发至干。将粗残余物溶解于乙酸乙酯中,并使用乙酸乙酯作为流动相在Biotage Isolera系统上通过硅胶色谱法进行提纯。将产物馏分在旋转蒸发器上蒸发至干,得到1.47gm的固体(1.产率:59%)。通过HPLC测定纯度>99%。化合物1在CDCl3中的1H-NMR:7.573ppm,s,1H;7.198ppm,d,1H;6.758ppm,d,1H;6.524ppm,s,1H;5.139ppm,m,1H;3.966ppm,s,3H;3.930ppm,s,3H;3.894ppm,m,3H;3.657ppm,s,3H;2.497-2.672ppm,m,3H;2.277ppm,s,3H;1.961ppm,m,1H;1.558ppm,s,9H。
步骤2.N-Boc-脱乙酰基秋水仙碱(2)和脱乙酰基秋水仙碱·TFA(3)的合成
将N-Boc-秋水仙碱(1,1.47gm,2.94mmol)溶解于甲醇(35mL)中,在氩气气氛下将其冷却至0℃。向搅拌的溶液中逐滴加入0.5N甲醇钠的甲醇溶液(23.6mL,11.78mmol)。将溶液在0℃下搅拌。反应65分钟后,加入NH4Cl(0.63gm,11.78mmol)以在0℃下中和反应混合物。搅拌5分钟后,将澄清溶液在旋转蒸发器上蒸发至干。将残余物与DCM(30mL)一起搅拌,并且然后过滤以去除固体盐。将盐用更多的DCM(10mL)冲洗,并过滤到相同的滤液中。向粗N-Boc-脱乙酰基秋水仙碱(2)的DCM的滤液中加入TFA(4mL)。将溶液在室温下在氩气下搅拌。反应2.5小时后,将甲苯(35mL)加入到反应混合物中,随后蒸发至干(斜升至0托)。将粗残余物溶解于DCM(10mL)中,使用DCM-甲醇(9:1v/v)作为流动相在Biotage上经硅胶柱进行提纯。在247nm处监测洗脱。通过旋转蒸发将含产物的馏分(通过TLC测定Rf=0.34)蒸发至接近干燥,并进一步在真空中干燥过夜,得到1.2gm的黄色固体(3,产率:87%)。通过HPLC测定纯度>99%。3在DMSO-d6中的1H-NMR:8.447ppm,s,3H;7.105-7.189ppm,m,3H;6.823ppm,s,1H;3.910ppm,s,3H;3.844ppm,s,3H;3.813ppm,m,1H;3.780ppm,s,3H;2.655ppm,m,1H;2.267ppm,m,2H;1.929ppm,m,1H。
实例4.N-(N3-PEG4-VC-PABC)-脱乙酰基秋水仙碱(4)的合成
将脱乙酰基秋水仙碱·TFA(3,0.30gm,0.64mmol)溶解于无水DCM(20mL)中,并且在氩气气氛下搅拌。向黄色溶液中加入N3-PEG4-VC-PAB-PNP(0.66gm,0.76mmol),随后加入TEA(0.27mL,1.91mmol)、ACN(5mL)和DMF(5mL)。然后将DIPEA(0.44mL,2.55mmol)加入到反应混合物中。反应2天后,反应混合物的反相HPLC分析显示97%的转化率。将反应混合物在37℃下在真空下蒸发至干。将粗残余物溶解于乙酸乙酯-甲醇(10mL,10:1v/v)中,通过使用乙酸乙酯作为流动相A和甲醇作为流动相B在Biotage上用硅胶柱进行提纯。在247nm处监测洗脱。通过旋转蒸发将含产物的馏分蒸发至接近干燥,并且进一步在真空中干燥过夜,得到0.56gm的黄色固体(4,产率:84%)。化合物4在DMSO-d6中的1H-NMR:9.985ppm,s,1H;8.112ppm,m,2H;7.868ppm,d,1H;7.576ppm,d,2H;7.234ppm,d,3H;7.115ppm,d,1H;7.032ppm,d,1H;6.768ppm,s,1H;5.977ppm,t,未溶解,1H;5.411ppm,s,NH2-;4.903ppm,q,2H;4.379ppm,m,1H;4.230ppm,m,1H;4.147ppm,m,1H;3.879ppm,s,3H;3.833ppm,s,3H;3.789ppm,s,3H;3.592ppm,s,3H;3.531ppm,m,-CH2CH2O-;3.316ppm,t,2H;3.020ppm,m,1H;2.949ppm,m,1H;2.568ppm,m,1H;2.375ppm,m,1H;2.184ppm,m,1H;1.829ppm,m,1H;1.686ppm,m,1H;1.592ppm,m,1H;1.433ppm,m,1H;1.369ppm,m,1H;0.860ppm,d,3H;0.827ppm,d,3H。
实例5.N-(N3-PEG4-Phe-Lys(Trt)-PABC)-脱乙酰基秋水仙碱5
将脱乙酰基秋水仙碱·TFA(3,0.18gm,0.39mmol,1.0当量)和N3-PEG4-Phe-Lys(Trt)-PAB-NPC(0.50gm,0.46mmol,1.2当量)溶解于无水DMF(5mL)中。向反应混合物中加入TEA(162μL,1.16mmol,3.0当量)和DIPEA(202μL,1.16mmol,3.0当量)。将黄色溶液在室温下在氩气气氛下搅拌过夜。将反应混合物在37℃下在真空下蒸发至干。将粗产物溶解于乙酸乙酯中,使用乙酸乙酯作为流动相A和甲醇作为流动相B在Biotage上用硅胶柱进行提纯。在317nm处监测洗脱。通过旋转蒸发将含产物的馏分蒸发至接近干燥,并且进一步在真空中干燥过夜,得到0.47gm的黄色固体(5,产率:79%)。纯度98%(HPLC)。化合物5的1H NMR(Varian,10mg/mL DMSO-d6,δ):9.992ppm,s,1H;8.112ppm,m,2H;8.020ppm,d,1H;7.566ppm,d,2H;7.381ppm,d,6H;7.226-7.271ppm,m,14H;7.105-7.209ppm,m,4H;7.039ppm,d,1H;6.766ppm,s,1H;4.891ppm,dd,2H;4.557ppm,t,1H,未溶解;4.080ppm,m,1H;3.870ppm,s,3H;3.828ppm,s,3H;3.784ppm,s,3H;3.527ppm,m,-CH2CH2O-;3.519ppm,s,3H;2.996ppm,dd,1H;2.742ppm,7,1H;2.575ppm,m,1H;1.2-2.3ppm,多重峰。
实例6.POZ 10p-EDA-Boc 20K(7)的合成
将侧基POZ 10p酸20K(10gm,0.52mmol,1.0当量)和NHS(0.078gm,0.67mmol,1.3当量)溶解于无水ACN(200mL)中。通过旋转蒸发将溶液蒸发至干。将残余物溶解于无水DCM(100mL)中,随后加入DCC(0.138gm,0.67mmol,1.3当量)。将溶液在氩气气氛下搅拌过夜。通过缓慢加入到乙醚(1600mL)中来沉淀反应混合物。将沉淀物过滤,并且在真空下干燥过夜,得到8.8gm的白色粉末(6)。如通过离子交换色谱法所测定的,6上的琥珀酰亚胺基团的取代度为98%。化合物6的1H NMR(Varian,10mg/mL CDCl3,δ)在1.12ppm(s,3H,CH3CH2CO-)、2.31ppm(小s)和2.41ppm(大s)(总面积2H,CH3CH2CO-),以及3.47ppm(s,4H,-NHCH2CH2NH-)处显示常见的主链峰。侧基峰出现在1.84ppm(m,2H,-CH2CH2CH2CC≡H),以及2.00ppm(m,1H,CH2CH2CH2CC≡H)处。末端琥珀酰亚胺基团峰出现在2.86ppm(s,4H)处。
向BOC-EDA(0.10gm,0.62mmol,2.1当量)和TEA(0.17mL,1.2mmol,4.1当量)的无水DCM(100mL)溶液中加入化合物6(5.6gm,0.29mmol,1当量)。将溶液在室温下在氩气气氛下搅拌过夜。然后通过缓慢加入到二乙醚(800mL)中使反应混合物沉淀。将沉淀物过滤,并且在真空中干燥过夜,得到5.0gm的白色粉末(7)。如通过离子交换色谱法所测定的,在7上的Boc-EDA的取代度为91%。化合物7的1H NMR(Varian,10mg/mL CDCl3,8)在1.12ppm(s,3H,CH3CH2CO-)、2.31ppm(小s)和2.41ppm(大s)(总面积2H,CH3CH2CO-),以及3.45ppm(s,4H,-NHCH2CH2NH-)处显示常见的主链峰。侧基峰出现在1.84ppm(m,2H,-CH2CH2CH2CC≡H),以及1.94ppm(m,1H,CH2CH2CH2CC≡H)处。末端-O-C(CH3)3基团峰出现在1.43ppm(s,9H)处。
实例7.POZ 20K侧基[(生物素)2(PEG 4-VC-PABC-脱乙酰基秋水仙碱)6]a-碘乙酰胺(11)的合成
POZ 20k侧基生物素2-EDA-Boc(8)的结构
POZ 20K侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]a-EDA-Boc(9)的结构
POZ 20K侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]a-EDA(10)的结构
POZ 20K侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]a-碘乙酰胺(11)的结构
将叠氮化物-PEG3-生物素(98mg,0.22mmol,2.1当量)和POZ 10p-EDA-BOC 20K(7,2.000gm,0.10mmol,1.0当量)溶解于去离子水(25mL)中。将溶液用缓慢的氩气流鼓鼓泡15分钟。然后向烧瓶中加入L(+)-抗坏血酸钠盐(44mg,0.22mmol,2.1当量),随后立即加入CuSO4·5H2O(55mg,0.22mol,2.1当量)。将溶液在室温下在氩气气氛下搅拌过夜。反应混合物的反相HPLC分析表明反应完成。使溶液通过填充有M4195介质(20gm)的柱以去除铜。用另外的去离子水(175mL)洗脱所述柱。将NaCl(30gm)溶解在洗脱液(200mL)中。将溶液用DCM(3×100mL)萃取。将DCM溶液用无水硫酸镁(1.5gm)和硫酸钠(100gm)干燥。将混合物通过玻璃料过滤。通过旋转蒸发将滤液浓缩至接近干燥,并且在真空中进一步干燥过夜,得到1.7gm的白色固体(8)。化合物8的1H NMR(Varian,10mg/mL DMSO-d6,8)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.28ppm(大s)和2.33ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。剩余的侧基峰出现在1.65ppm(m,2H/-CH2CH2CH2CC≡H),以及2.75ppm(m,1H/CH2CH2CH2CC≡H)处。POZ末端基团峰出现在1.37ppm(s,9H,-O-C(CH3)3)、6.77ppm(t,未溶解,1H,-NH-Boc)、7.92ppm(t,未溶解,1H,-CONH-)处。侧基生物素相关峰包括3.79ppm(t,未溶解,2H/三唑-CH2CH2O-)、4.12ppm(m,在生物素上1H/-CONHCH(CH2S-)-)和4.30ppm(m,在生物素上1H/-CONHCH)、4.45ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)和6.40ppm(s)(2H/-NH-CO-NH-)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。
向50mL圆底烧瓶中的N-(N3-PEG4-YC-PABC)-脱乙酰基秋水仙碱(4,0.15gm,0.15mmol,7.5当量)中加入无水THF(10mL)。然后将POZ 20K侧基生物素2-EDA-Boc(8,0.40gm,0.020mmol,1当量)溶解在溶液中。将溶液在缓慢的氩气流下搅拌10分钟。然后加入CuI(23mg,0.12mmol,6.0当量),随后立即加入TEA(17μL,0.12mmol,6.0当量)。将DMF(1mL)加入到反应混合物中。将绿色溶液在氩气气氛下在40℃下搅拌过夜。反应混合物的反相HPLC分析表明,反应在搅拌过夜后完成。过滤反应混合物,并且通过旋转蒸发将滤液浓缩至接近干燥。在真空下进一步蒸发残余的DMF。向残余物中加入20mL的甲醇。通过使澄清的绿色溶液通过填充有M4195介质(20mL)的柱来对其进行提纯以去除铜。用另外的甲醇(80mL)洗脱所述柱。通过旋转蒸发将洗脱液(100mL)浓缩至干。将残余物溶解于DCM(10mL)中,通过缓慢加入二乙醚(150mL)使其沉淀。过滤沉淀物,并且然后在真空中干燥过夜,得到0.49gm的琥珀色粉末(9)。化合物9的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CTI2CO-)、2.28ppm(大s)和2.33ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。POZ末端基团峰出现在1.37ppm(s,9H,-O-C(CH3)3)、6.77ppm(t,未溶解,1H,-NH-Boc)、7.82ppm(t,未溶解,1H,-CONH-)处。侧基生物素相关峰包括3.79ppm(t,未溶解,2H/三唑-CH2CH2O-)、4.12ppm(m,在生物素上1H/-CONHCH(CH2S-)-)和4.30ppm(m,在生物素上1H/-CONHCH)、4.46ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)和6.40ppm(s)(2H/-NH-CO-NH-)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-VC-PABC-脱乙酰基秋水仙碱峰中的一些(下面H的数量是指每个侧基PEG4-VC-PABC-脱乙酰基秋水仙碱)包括:0.827ppm,d,3H;0.860ppm,d,3H;1.37ppm,m,1H;1.43ppm,m,1H;1.59ppm,m,1H;1.69ppm,m,1H;1.83ppm,m,1H;2.58ppm,m,1H;2.95ppm,m,1H;3.02ppm,m,1H;3.53ppm,m,-CH2CH2O-;3.59ppm,s,3H;3.79ppm,s,3H;3.83ppm,s,3H;3.88ppm,s,3H;4.15ppm,m,1H;4.23ppm,m,1H;4.38ppm,m,1H;4.45ppm,t,2H;4.90ppm,q,2H;5.41ppm,s,NH2-;5.98ppm,t,未溶解,1H;6.77ppm,s,1H;7.03ppm,d,1H;7.12ppm,d,1H;7.23ppm,d,3H;7.58ppm,d,2H;7.82ppm,t,1H;7.87ppm,d,1H;8.11ppm,m,2H;9.98ppm,s,1H。
将POZ 20K侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]a-EDA-BOC(9,0.480gm)溶解于DCM(3.2mL)中,随后加入TFA(3.2mL)。将溶液在室温下在氩气下搅拌3小时。然后通过在28℃下旋转蒸发将混合物蒸发至干。将残余物溶解于DCM(30mL)中。将溶液用15%盐水(2×20mL)洗涤。相分离后,用15%盐水(40mL,用1N NaOH将pH调节至12)洗涤DCM溶液。相分离后,收集DCM溶液,用无水硫酸钠(20gm)干燥。将混合物通过玻璃料过滤。将滤液蒸发至干。将残余物溶解于DCM(7mL)中,随后在二乙醚(150mL)中沉淀。过滤沉淀物,并且在真空中干燥过夜,得到0.13gm的白色粉末(10)。化合物10的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.28ppm(大s)和2.33ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。POZ末端Boc基团的1.37ppm峰大大降低,这表明去除了末端Boc基团。末端酰胺NH出现在7.82ppm(t,未溶解,1H,-CONH-)处。侧基生物素相关峰中的一些包括3.79ppm(t,未溶解,2H/三唑-CH2CH2O-)、4.12ppm(m,在生物素上1H/-CONHCH(CH2S-)-)和4.30ppm(m,在生物素上1H/-CONHCH)、4.46ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)(2H/-NH-CO-NH-)和6.40ppm(s)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-VC-PABC-脱乙基秋水仙碱峰中的一些(下面的H的数量是指每个侧基PEG4-VC-PABC-脱乙酰基秋水仙碱)包括:0.827ppm,d,3H;0.860ppm,d,3H;1.37ppm,m,1H;1.43ppm,m,1H;1.59ppm,m,1H;1.69ppm,m,1H;1.83ppm,m,1H;2.58ppm,m,1H;2.95ppm,m,1H;3.02ppm,m,1H;3.53ppm,m,-CH2CH2O-;3.59ppm,s,3H;3.79ppm,s,3H;3.83ppm,s,3H;3.88ppm,s,3H;4.15ppm,m,1H;4.23ppm,m,1H;4.38ppm,m,1H;4.45ppm,t,2H;4.90ppm,q,2H;5.41ppm,s,NH2-;5.98ppm,t,未溶解,1H;6.77ppm,s,1H;7.03ppm,d,1H;7.12ppm,d,1H;7.23ppm,d,3H;7.58ppm,d,2H;7.82ppm,t,1H;7.87ppm,d,1H;8.11ppm,m,2H;9.98ppm,s,1H。
将POZ侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]a-EDA 20K(10,0.11gm,0.0042mmol,1当量)溶解于ACN(5mL)中,然后通过旋转蒸发将其蒸发至干。将残余物溶解于DCM(5mL)中。加入琥珀酰亚胺碘乙酸酯(2.5mg,0.0083mmol,2当量),随后加入TEA(4.6μL,0.033mmol,8当量)。将溶液在氩气气氛下在黑暗中搅拌。反应过夜后,将溶液沉淀到80mL的二乙醚中。过滤沉淀物,用二乙醚(5mL)冲洗,并且在真空中干燥,得到99mg的白色粉末(11)。如在与3-巯基丙酸反应之后通过离子交换色谱法测定的,11的碘乙酰胺取代度为81%。
实例8.POZ 10p-EDA-Fmoc 20K(13)的合成
将POZ 10p-EDA-BOC 20K(7,1.0gm)溶解于DCM(6mL)中,随后加入TFA(6mL)。将溶液在室温下在氩气气氛下搅拌2小时。然后将混合物在28℃下蒸发至干。将残余物溶解于去离子水(10mL)中,并且通过1N NaOH将溶液pH调节至11。向溶液中加入NaCl以制备15%盐水,将其用DCM(4×30mL)萃取。相分离后,收集DCM相,用无水MgSO4(1gm)和Na2SO4(90gm)干燥。滤除固体后,将滤液蒸发至干。将残余物溶解于DCM(10mL)中,将其加入道二乙醚(200mL)中以沉淀聚合物。过滤沉淀物,并且在真空中干燥过夜,得到0.86gm的白色粉末(12)。化合物12的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.28ppm(大s)和2.33ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。侧基峰出现在1.64ppm(m,2H/-CH2CH2CH2CC≡H),以及2.77ppm(m,1H/CH2CH2CH2CC≡H)处。POZ末端Boc基团(1.37ppm)消失,这表明完全去除了末端Boc基团。
通过旋转蒸发将POZ 10p-EDA 20K(12,0.42gm,0.022mmol,1当量)在10mL的ACN(10mL)中的溶液蒸发至干。将残余物溶解于DCM(6mL)中。加入DIPEA(15μL,0.087mmol,4当量)和Fmoc氯化物(12mg,0.044mmol,2当量)。在氩气下搅拌过夜后,将溶液蒸发至干。将残余物溶解于去离子水(10mL)中,随后溶解NaCl(1gm)。将水溶液用二乙醚(15mL)洗涤,随后用DCM(3×15mL)萃取剩余的水溶液。将DCM溶液用Na2SO4(20gm)干燥,并且然后过滤以去除固体。通过旋转蒸发将滤液浓缩至约4mL,并且然后在二乙醚(80mL)中沉淀。通过过滤收集沉淀物,并且在真空中干燥过夜,得到0.36gm的白色粉末(13)。在CDCl3中的NMR显示转化为Fmoc。化合物13的1H NMR(Varian,10mg/mL CDCl3,δ)在1.12ppm(s,3H,CH3CH2CO-)、2.31ppm(小s)和2.41ppm(大s)(总面积2H,CH3CH2CO-),以及3.45ppm(s,4H,-NHCH2CH2NH-)处显示常见的主链峰。侧基峰出现在1.84ppm(m,2H,-CH2CH2CH2CC≡H),以及2.03ppm(m,1H,CH2CH2CH2CC≡H)处。Fmoc基团峰出现在4.20ppm(t,1H)、4.37ppm(m,2H)、7.31ppm(t,2H)、7.40ppm(t,2H)、7.59ppm(d,2H),以及7.76ppm(d,2H)处。
实例9.POZ 20K侧基[(生物素)2(PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱)5]a-碘乙酰胺(17)的合成
POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-EDA-Fmoc,14
POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-EDA,15
POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-碘乙酰胺,16
POZ 20K侧基[(生物素)2(PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱)5]a-碘乙酰胺,17
向100mL圆底烧瓶中的N3-PEG4-Phe-Lys(Trt)-PABC-碳酸酯-脱乙酰基秋水仙碱(5,193mg,0.145mmol,6当量)和POZ 10p-EDA-Fmoc 20K(13,470mg,0.0242mmol,1当量)中加入预先溶解于1mL的DMF中的生物素-PEG3-叠氮化物(22mg,0.048mmol,2.0当量)。使用DMF(1mL)冲洗并将残余的生物素-PEG3-叠氮化物转移到反应混合物中。然后加入无水TF1F(15mL)。将溶液在氩气下搅拌15分钟。然后加入CuI(29mg,0.15mmol,6.4当量),随后立即加入TEA(22μL,0.15mmol,6.4当量)。将绿色溶液在50℃下在氩气下搅拌过夜。将反应混合物通过0.2μm过滤器过滤,并且通过旋转蒸发将滤液蒸发至干。在真空下蒸发DMF。加入甲醇(10mL)以溶解残余物。将澄清的绿色溶液通过填充有M4195介质(9gm)的柱以去除铜。用另外的甲醇(90mL)洗脱所述柱。将洗脱液浓缩至干。将残余物溶解于DCM(12mL)中。通过缓慢加入到二乙醚(200mL)中来沉淀DCM溶液。过滤沉淀物,并且然后在真空中干燥,得到0.64gm的琥珀色粉末(14)。化合物14的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCB2CH2NH-)处显示常见的POZ主链峰。Fmoc基团峰出现在4.12ppm(m,1H)、4.30ppm(m,2H)处。侧基生物素相关峰中的一些包括3.79ppm(t,未溶解,2H/三唑-CH2CH2O-)、4.12ppm(m,在生物素上1H/-CONHCH(CH2S-)-)和4.30ppm(m,在生物素上1H/-CONHCH)、4.44ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)(2H/-NH-CO-NH-)和6.40ppm(s)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱相关峰中的一些(下面的H的数量是指每个侧基PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)为3.52ppm,s,3H;3.78ppm,s,3H;3.82ppm,s,3H;3.87ppm,s,3H;4.12ppm,m,1H;4.44ppm(t,未溶解,2H/三唑-CH2CH2O-);4.53ppm,t,1H,未溶解;4.91ppm,s,2H;6.76ppm,s,1H;7.02ppm,d,1H;7.11-7.21ppm,m,4H;7.37ppm(d,来自Trt基团);7.56ppm,d,2H;7.80ppm(m,1H/三唑环=CH-N);8.03ppm,d,1H;8.11p pm,m,2H;9.99ppm,s,1H。
在Ar下将POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-Fmoc(14,0.44gm)溶解于无水DMF(4mL)中。向溶液中加入哌啶(0.20mL)。将溶液在氩气气氛下搅拌1小时。然后将溶液在40℃下在真空下蒸发至干。将残余物溶解于DCM(4mL)中,随后在二乙醚(80mL)中沉淀。将沉淀物用二乙醚(5mL)洗涤,并且然后在真空中干燥,得到0.42gm的粉末。将干燥的粉末溶解于DCM(15mL)中,并且然后在分液漏斗中用7%盐水(30mL)洗涤。相分离后,将DCM溶液用Na2SO4干燥。过滤以去除Na2SO4后,将滤液浓缩至约7mL,将其沉淀到二乙醚(150mL)中。通过过滤去除沉淀物,用二乙醚(10mL)冲洗,并且然后在真空中干燥过夜,得到0.37gm的粉末(15)。化合物15的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。侧基生物素相关峰中的一些包括4.44ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)和6.40ppm(s)(2H/-NH-CO-NH-)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱相关峰中的一些(下面的H的数量是指每个侧基PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)是3.44ppm,s,PEG4-CH2CH2O-;3.52ppm,s,3H;3.78ppm,s,3H;3.82ppm,s,3H;3.86ppm,s,3H;4.13ppm,m,1H;4.44ppm(t,未溶解,2H/三唑-CH2CH2O-);4.56ppm,t,1H,未溶解;4.91ppm,m,2H;6.76ppm,s,1H;7.03ppm,d,1H;7.14-7.26ppm,m,Ar;7.37ppm(d,Trt基团);7.56ppm,d,2H;7.80ppm(m,1H/三唑环=CH-N);8.03ppm,d,1H;8.11ppm,m,2H;9.99ppm,s,1H。
将POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-EDA(15,0.36gm,MW 26685Da,0.013mmol,1当量)溶解于ACN(5mL)中,然后通过旋转蒸发将其蒸发至干。将残余物溶解于无水DCM(5mL)中并且在氩气气氛下保护。加入琥珀酰亚胺基碘乙酸酯(12mg,0.040mmol,3.0当量),随后加入TEA(11μL,0.080mmol,6当量)。在黑暗中搅拌过夜后,将溶液沉淀到二乙醚(150mL)中。将溶液过滤,用二乙醚(10mL)冲洗,并且在真空中干燥,得到0.36gm的白色粉末(16)。
将POZ 20K侧基[(生物素)2(PEG4-Phe-Lys(Trt)-PABC-脱乙酰基秋水仙碱)5]a-碘乙酰胺(16,0.16gm)溶解于DCM(2mL)中,随后加入TFA(0.10mL)。将溶液在室温下在黑暗中在氩气下搅拌过夜。然后通过在28℃下旋转蒸发将混合物蒸发至干。将溶液溶解于DCM(4mL)中,并且然后在二乙醚(80mL)中沉淀。过滤沉淀物,用大量乙醚洗涤,并且在真空中干燥。将干燥的粉末(0.15gm)溶解在去离子水(20mL)中。使用具有PLBC膜(44.5mm,NMWL 3000)的艾美康恩(amicon)超滤单元通过超滤将溶液浓缩至5mL。将浓缩的溶液用去离子水稀释至50mL,再次浓缩至5mL。将浓缩的溶液稀释至30mL,随后浓缩至5mL。将浓缩的溶液转移至50mL小瓶中。将超滤单元用去离子水(10mL)冲洗并转移到50mL小瓶中。将溶液在冻干机中冷冻干燥,得到0.15gm的白色粉末(17)。化合物17的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.44(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。POZ末端碘乙酰胺(-COCH2I)峰出现在3.62ppm(s,2H)处。侧基生物素相关峰包括4.44ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)和6.40ppm(s)(2H/-NH-CO-NH-)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱相关峰中的一些(下面的H的数量是指每个侧基PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱)包括:3.46ppm,s,PEG4-CH2CH2O-;3.52ppm,s,3H;3.79ppm,s,3H;3.83ppm,s,3H;3.87ppm,s,3H;4.13ppm,m,1H;4.44ppm(t,未溶解,2H/三唑-CH2CH2O-);4.56ppm,t,1H,未溶解;4.92ppm,m,2H;6.76ppm,s,1H;7.04ppm,d,1H;7.13ppm,m,Ar;7.23ppm,m,Ar;7.57ppm,d,2H;7.64ppm,m,Ar;7.80ppm(m,1H/三唑环=CH-N);8.05ppm,d,1H;8.11ppm,m,1H;10.0ppm,s,1H。
实例10.POZ 20K侧基[(PEG4-VC-PABC-MMAE)9]a-马来酰亚胺(19)
向玻璃小瓶中的N3-PEG4-VC-PABC-MMAE(72mg,0.049mmol,9当量)中加入THF(4mL)。然后将聚[(EOZ)n-co-(PtynOZ)10]-酸20K(106mg,0.00544mmol,1当量)加入到溶液中。将溶液在氩气下搅拌10分钟。然后向混合物中加入CuI(9mg,0.05mmol,9当量),随后立即加入TEA(7μL,0.05mmol,9当量)。将溶液在氩气下在45℃下搅拌过夜。通过反相HPLC分析绿色混合物,所述绿色混合物表明反应完成,因为反应混合物中没有残余的N3-PEG4-VC-PABC-MMAE。通过旋转蒸发将溶液蒸发至干。加入2mM HCl(4mL)和ACN(4mL)以溶解残余物,将其通过填充有Dowex M4195介质(2mL)的柱进行提纯以去除铜。通过1:1v/v的2mM HCl和ACN(10mL)洗脱所述柱。通过旋转蒸发蒸发ACN。向剩余的水溶液中加入NaCl以制备15%盐水。将溶液用DCM(3×30mL)萃取。相分离后,将DCM溶液浓缩至约20mL,并且用无水硫酸钠(10gm)干燥。将混合物通过玻璃料过滤,并且将滤液浓缩至约4mL。然后将溶液沉淀到二乙醚(80mL)中。通过过滤收集沉淀物并且在真空中干燥,得到108mg的白色粉末18。化合物18的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.47(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。侧基PEG4-VC-PABC-MMAE相关峰中的一些(下面H的数量是指每个侧基PEG4-VC-PABC-MMAE)包括:0.70-0.95ppm,m,24H,-CH3;3.50ppm,s,-(CH2CH2O)3-;3.79ppm(t,未溶解,2H/三唑-CH2CH2O-;3.99ppm,m,2H;4.23ppm,m,2H;4.45ppm(主峰,t,未溶解,2H/三唑-CH2CH2O-);5.04ppm,m,2H;5.41ppm,s,2H;6.04ppm,s,1H;7.17ppm,m,1H,在MMAE上的Ar;7.27ppm,d,2H,PAB的Ar;7.31ppm,m,4H,在MMAE上的Ar;7.59ppm,d,2H,PAB的Ar;7.82ppm(m,1H/三唑环=CH-N);8.13ppm,d,1H;10.03ppm,s,1H。
将POZ 20K侧基-(PEG4-VC-PABC-MMAE)9-酸(18,93mg,0.0029mmol,1.0当量)溶解于无水ACN(2mL)中,随后加入HOBT(0.4mg,0.002mmol,0.8当量)和N-(2-氨基乙基)马来酰亚胺TFA盐(1mg,0.004mmol,1.3当量)。通过旋转蒸发将溶液蒸发至干。将残余物溶解于无水DCM(3mL)中,随后加入TEA(0.6μL,0.004mmol,1.5当量)和DCC(0.7mg,3.5101×10-6mol,1.2当量)。将溶液在氩气气氛下搅拌过夜。将混合物过滤,并且加载到小硅胶柱(10mL)上,随后用DCM洗脱。通过旋转蒸发将洗脱液(30mL)浓缩至1mL。将溶液加入到乙醚(15mL)中以沉淀。通过过滤收集沉淀物并且在真空中干燥,得到30mg的白色粉末(19)。化合物19的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.47(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。POZ末端马来酰亚胺基团峰出现在7.00ppm(s,2H)处。侧基PEG4-VC-PABC-MMAE相关峰中的一些(下面的H的数量是指每个侧基PEG4-VC-PABC-MMAE)包括:0.70-0.95ppm,m,24H,-CH3;3.50ppm,s,-(CH2CH2O)3-;3.79ppm,t,未溶解,2H/三唑-CH2CH2O-);3.99ppm,m,2H;4.23ppm,m,2H;4.45ppm(主峰,t,未溶解,2H/三唑-CH2CH2O-);5.04ppm,m,2H;5.41ppm,s,2H;6.04ppm,s,1H;7.17ppm,m,1H,在MMAE上的Ar;7.27ppm,d,2H,PAB的Ar;7.31ppm,m,4H,在MMAE上的Ar;7.59ppm,d,2H,PAB的Ar;7.82ppm(m,1H/三唑环=CH-N);8.13ppm,d,1H;10.03ppm,s,1H。
实例11.POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]a-碘乙酰胺(22)
反应方案:
20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]a-EDA-Boc(20)的结构
POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]a-EDA(21)的结构
POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]a-碘乙酰胺(22)的结构
向50mL圆底烧瓶中的N3-PEG4-VC-PABC-MMAE(132mg,0.0944mmol,6.0当量)和POZ 10p-EDA-Boc 20K(7,305mg,0.0157mmol,1当量)中计入生物素-PEG3-叠氮化物(14mg,0.032mmol,2当量,溶解于0.6mL的DMF中),随后加入9mL的无水THF(9mL)。将溶液在氩气下保护并且在室温下搅拌15分钟。然后加入CuI(19mg,0.10mmol,6.8当量),随后立即加入TEA(52μL,0.37mmol,24当量)。将绿色溶液在氩气下在50℃下搅拌过夜。将溶液通过0.2μm注射器式过滤器过滤,并且将滤液蒸发至干。在真空下蒸发DMF。将甲醇(10mL)加入到残余固体中。通过使甲醇溶液通过填充有M4195介质(5gm)的柱来去除铜离子。将洗脱液浓缩至干并溶解于DCM(6mL)中。在搅拌下,将澄清溶液在二乙醚(150mL)中沉淀。通过过滤收集沉淀物并且在真空中干燥,得到400mg的琥珀色粉末(20)。化合物20的1H NMR(Varian,10mg/mL DMSO-d6,δ)在0.95ppm(s)和0.97(s)(总面积3H/CH3CH2CO-)、2.27ppm(大s)和2.32ppm(小s)(总面积2H/CH3CH2CO-),以及3.35ppm(大s)和3.47(小s)(总面积4H/-NHCH2CH2NH-)处显示常见的POZ主链峰。POZ末端Boc基团峰出现在1.37ppm(s,9H)处。侧基生物素相关峰中的一些包括3.79ppm(t,未溶解,2H/三唑-CH2CH2O-)、4.12ppm(m,在生物素上1H/-CONHCH(CH2S-)-)和4.30ppm(m,在生物素上1H/-CONHCH)、4.45ppm(t,未溶解,2H/三唑-CH2CH2O-)、6.34ppm(s)(2H/-NH-CO-NH-)和6.40ppm(s)、7.81ppm(m,1H/三唑环=CH-N,1H/-CONH-)。侧基PEG4-VC-PABC-MMAE相关峰中的一些(下面的H的数量是指每个侧基PEG4-VC-PABC-MMAE)包括:0.70-0.95ppm,m,-CH3;3.50ppm,s,-(CH2CH2O)3-;3.79ppm(t,未溶解,2H/三唑-CH2CH2O-);3.99ppm,m,2H;4.23ppm,m,2H;4.45ppm(主峰,t,未溶解,2H/三唑-CH2CH2O-);5.04ppm,m,2H;5.40ppm,s,2H;5.90ppm,s,1H;7.16ppm,m,1H,在MMAE上的Ar;7.27ppm,d,2H,PAB的Ar;7.31ppm,m,4H,在MMAE上的Ar;7.58ppm,d,2H,PAB的Ar;7.82ppm(m,1H/三唑环=CH-N);8.13ppm,d,1H;9.98ppm,s,1H。
将化合物20(390gm)溶解于无水DCM(2mL)中,随后加入TFA(2mL)。将溶液在室温下在氩气下搅拌3小时。然后将混合物蒸发至干。将残余固体溶解于ACN(5mL)中,随后与去离子水(15mL)混合。然后通过旋转蒸发蒸发ACN。用1N NaOH将剩余溶液的pH调节至11,随后将氯化钠(1.5gm)溶解于溶液中。将水溶液用DCM(3×30mL)萃取。收集DCM相,用无水MgSO4(1.5gm)和Na2SO4(20gm)干燥。将混合物过滤以去除固体,并且通过旋转蒸发将滤液蒸发至干。将残余物在真空中干燥过夜,得到289mg的固体(21)。化合物21的1H NMR(Varian,10mg/mL DMSO-d6,δ)表明末端Boc基团完全裂解,如在1.37ppm处的单峰的消失所示。化合物21的1H NMR(Varian,10mg/mL DMSO-d6,δ)表明末端Boc基团完全裂解,如在1.37ppm处的单峰的消失所示。
将化合物21(150mg,5.86×10-3mmol,1当量)溶解于氯仿(6mL)中,然后通过旋转蒸发将其蒸发至干。将残余物溶解于DCM(6mL)中并且在氩气气氛下保护。加入琥珀酰亚胺基碘乙酸酯(3.4mg,1.2×10-2mmol,2.0当量),随后加入TEA(6.5μL,4.7×10-2mmol,8当量)。将溶液在黑暗中搅拌过夜。然后在搅拌下将溶液缓慢加入到80mL的二乙醚中以沉淀产物。过滤沉淀物,用二乙醚(5mL)冲洗,并且在真空中干燥,得到140mg的白色粉末。将固体溶解于50mM NaH2PO4(7mL,pH 4.6)中。然后将溶液在Slide-A-Lyzer(3-12mL,2,000MWCO)透析盒中针对DI水(3×1L)透析过夜。通过旋转蒸发将溶液蒸发至干。将残余物溶解于氯仿(5mL)中并且蒸发。将残余物再溶解于氯仿(5mL)中,并且蒸发至干。然后将残余物溶解于DCM(6mL)中,并且然后在搅拌下沉淀在二乙醚(80mL)中。过滤沉淀物,并且用二乙醚(5mL)冲洗,并且在真空中干燥,得到118mg的白色粉末(22)。22的反相HPLC分析显示纯度>99%。22的碘乙酰胺取代度为67%,如通过与3-巯基丙酸反应后的离子交换色谱法测定的。
实例12.POZ(生物素)10马来酰亚胺与抗体的结合
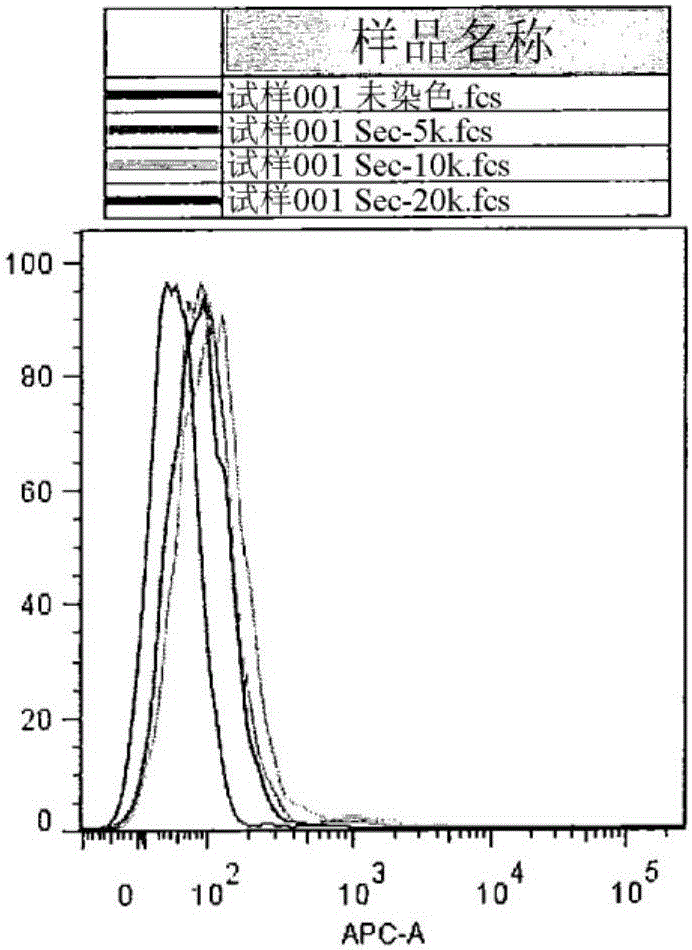
将5、10和20kDa的POZ(生物素)10-马来酰亚胺试剂附着到R11sc-Fv-FC特异性ROR1抗体上。典型的结合条件是溶解于100μM乙酸钠缓冲液(pH 5.2)中并且在室温下混合1小时的抗体4μM、POZ-生物素100μM,DTT 0.1mM。然后使用蛋白质-A分离柱,利用100mM乙酸钠(pH 5.2)的洗涤和洗脱缓冲液将未结合的POZ聚合物与抗体和POZ-抗体结合物分离。然后借助于单体抗生物素蛋白流通试剂盒将结合的抗体与未结合的抗体分离,其中与聚合物主链连接的生物素对抗生物素蛋白具有强亲和力。然后将不同分子量的POZ结合物与HBL-2(ROR+)和MEC-1(ROR-)细胞温育。图1显示当连接到抗体上的单个位点时,POZ聚合物的大小似乎不改变其与表达抗原的细胞(HBL-2)的选择性结合的能力;聚合物-结合物显示很少与不表达抗原的细胞(MEC-1)结合。
POZ-ADC结合活性也可以通过ELISA来验证。用hROR1ECD抗原包覆96孔板的每个孔,并且在37℃下在TBS中温育1小时。用150μL 3%(w/v)BSA/TBS在37℃封闭1小时后,加入连接有或没有连接有POZ聚合物的R11sc-Fv-FC特异性ROR1抗体,并且在37℃下温育2小时。在该实验中使用的POZ分子是上面所描述的POZ 10p马来酰亚胺聚合物。在洗涤每个孔后,加入片段特异性第二抗体、具有稳定的HRP活性的抗人驴IgG Feγ的稀释液。在用水洗涤后,使用2,2'-连氮基-双(3-乙基苯并噻唑啉)-6-磺酸进行比色检测。使用酶标仪在405nm处测量吸光度。绘制吸光度对ROR1抗体浓度(图2)的图,以描绘天然抗体与聚合物-ADC结合的抗体的结合效率。该图显示天然抗体和聚合物-ADC结合的抗体以类似的方式结合hROR1ECD抗原,导致POZ聚合物不干扰抗原结合的结论。
实例13.POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)1]r-碘乙酰胺和POZ20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]r-碘乙酰胺与抗体的结合
将POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)1]a-碘乙酰胺和POZ 20K侧基[(生物素)2(PEG4-VC-PABC-MMAE)5]a-碘乙酰胺与R11sc-Fv-Fc特异性ROR1抗体、R12sc-Fv-Fc特异性ROR2抗体和CD79b抗体附着。上文对结合和提纯条件进行了说明。使用蛋白质-A分离柱,利用100mM乙酸钠(pH 5.2)的洗涤和洗脱缓冲液将未结合的POZ聚合物与天然抗体和POZ-抗体结合物分离。然后借助于单体亲和素流通试剂盒将POZ结合的抗体与未结合的抗体分离,其中聚合物主链上的生物素对抗生物素蛋白具有强亲和力。图3中的CD-79b POZ val-cit-PABC-MMAE结合物的SDS PAGE图谱(考马斯染色)显示了结合和提纯的每个步骤。然后将提纯的POZ抗体结合物与HBL-2(ROR+)、MEC-1(ROR-)和拉莫斯细胞温育。流式细胞术显示所有POZ结合物与拉莫斯细胞而不与MEC-1细胞进行高细胞表面结合(数据未显示)。下面示出了通道分配。
通道样品
1CD79b-终止
2蛋白MW标志物
3在结合前CD79b-Sec
4蛋白A分离后CD79b-POZ-(MMAE)1
5单-抗生物素蛋白流过CD79b-POZ-(MMAE)1
6单-抗生物素蛋白分离后CD79b-POZ-(MMAE)1
7蛋白A分离后CD79b-POZ-(MMAE)5
8单-抗生物素蛋白流过CD79b-POZ-(MMAE)5
9单-抗生物素蛋白分离后CD79b-POZ-(MMAE)5
10CD79b-vc-MMAE
实例14.POZ 20K侧基[(生物素)2(PEG4-VC-PABC-脱乙酰基秋水仙碱)6]α-碘乙酰胺和POZ 20K侧基[(生物素)2(PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱)5]α-碘乙酰胺与抗体的结合
将POI(生物素)2(Val-Cit-PAB-DC)6碘乙酰胺和POZ(生物素)2(Phe-Lys-PAB-DC)5碘乙酰胺与R11sc-Fv-FC特异性ROR1抗体、R12sc-Fv-FC特异性ROR2抗体和CD79b抗体附着。上文对典型的结合和提纯条件进行了说明。在结合和提纯的每个步骤期间,CD-79b POZ(phe-lys-PAB-DC)5结合物的SDS PAGE图谱(考马斯染色)显示发生结合和提纯。然后将纯的POZ抗体结合物与HBL-2(ROR+)、MEC-1(ROR-)和拉莫斯细胞温育。流式细胞术数据显示所有POZ结合物与HBL-2和拉莫斯细胞而不与MEC-1细胞进行高细胞表面结合。
实例15.POZ 20K侧基(PEG4-VC-PABC-MMAE)1、POZ 20K侧基(PEG4-VC-PABC-MMAE)5、POZ(PEG4-VC-PABC-脱乙酰基秋水仙碱)6和POZ 20K侧基(PEG4-Phe-Lys-PABC-脱乙酰基秋水仙碱)5的CD79b结合物的效力
在细胞毒性测定中,将拉莫斯和MEC-1细胞与不同浓度的CD79b结合的POZ药物结合物温育96小时。图4显示增加浓度的CD79b-POZ结合物对体外拉莫斯细胞生存力(%)的影响。数据显示具有一个附着的MMAE分子的POZ Val-Cit-PAB结合物具有约20nM的IC50。当与具有平均5个MMAE分子的结合物相比时,IC50降低约一个log至约2nM。该数据显示,增加每个POZ结合物的MMAE分子的数量使IC50增加10倍;即较高的药物抗体比例(DAR)改善聚合物-ADC的活性和效力。DC的POZ val-cit-PAB结合物具有约200nM的IC50,并且DC的POZ Phe-Lys-PAB结合物具有降低的活性,即使两种CD-79b结合物与拉莫斯细胞进行高细胞表面结合。这表明在拉莫斯细胞研究中,脱乙酰基秋水仙碱(DC)不如MMAE有效。另外,Val-Cit连接基团似乎在细胞内释放时更容易,这可能对于其中细胞内释放是一个因素的一些应用具有吸引力。图5显示CD79b-POZ结合物的浓度对MEC-1细胞的生存力(%)的影响。数据显示MMAE的POZ val-cit-PAB结合物具有大于300nM的IC50,并且利用DC作为细胞毒素剂的结合物没有活性。由于CD79b抗体的低浓度的抗原,如所预期的,所有这些结合物与MEC-1细胞进行较差的细胞表面结合。
实例16.POZ 20K侧基(PEG4-VC-PABC-MMAE)5的R11结合物的效力
在细胞毒性测定中将HBL-2和MEC-1细胞与不同浓度的与POZ-MMAE结合的R11温育96小时。图6显示了R11-POZ-(PEG4-VC-PABC-MMAE)5结合物的浓度对体外HBL-2(ROR+)和MEC-1(ROR-)细胞的生存力(%)的影响。数据显示,该化合物对HBL-2细胞的IC50为约200nM,以及对MEC-1细胞没有活性。
- 还没有人留言评论。精彩留言会获得点赞!