一种PEG固载手性双噁唑啉配体的制备方法与流程
本发明属于有机合成和催化技术领域,具体涉及一种peg固载手性双噁唑啉配体的制备方法。
背景技术:
苯乙烯与eda的不对称环丙烷化反应,可以得到用于合成菊酯农药的重要手性中间体。关于催化该类反应的研究已经被成功报道,其中研究最多的是关于手性双噁唑啉。手性双噁唑啉是一类易得、应用性广的化合物。自20世纪80年代以来,双噁唑啉配体催化的反应包括:不对称烯丙基取代反应、烯丙基氧化反应、烯烃及亚胺的环丙烷化反应、diels-alder反应、自由基加成反应、makaiyamaaldol反应、亚胺及醛的亲核加成反应以及硅氢化还原反应等。为了提高该类催化剂的利用率,多种固载方式被报道。固载双噁唑啉时所使用的载体有聚合物和无机载体(如硅胶,分子筛和y型沸石等)。根据载体是否溶于反应体系又可分为不溶性载体和可溶性载体。已见报道的不溶性载体有laponite粘土、nafion-silicananocomposte、cuhy等,可溶性载体有argogel、peg(聚乙烯乙二醇)、meopeg(peg5000的单甲醚)等。可溶性载体peg能与反应溶剂形成均相反应体系,有利于催化剂与底物的作用,有更高的活性和配体的电子效应,还可以提高反应的立体选择性,便于用当前的各种光谱方法进行表征,同时易于分离纯化,因此受到研究者的广泛关注。
2000年,reiser等将可溶性聚合物聚乙二醇单甲醚meopeg-5000接入杂双噁唑啉配体得到固载化的手性配体,再将其与cu(otf)2配位催化了苯乙烯与重氮乙酸甲酯的环丙烷化反应,对映选择性为91%(trans),比非固定化效果要好(87%,trans),产率为69%,而且该催化剂可以进行多达9次的循环利用,催化剂活性和对映选择性均无明显降低;但是合成氮杂双噁唑啉配体产率较低,嫁接到衍生化的peg-5000单甲醚成本较高,取代率较低。2002年,cozzi等通过复杂的合成路线得到了配体并将其嫁接到衍生化的peg-5000单甲醚得到固载化的手性配体,再将其与cuotf配位催化了苯乙烯与重氮乙酸乙酯的环丙烷化反应,对映选择性为91%(trans),产率为63%,但是该催化剂没有被很好地循环利用。如何通过简便方式研发制备高活性、高选择性以及稳定性较好,可重复使用的固载化手性双噁唑啉催化剂仍然是一个有挑战性和重大价值的难题。
技术实现要素:
本发明的目的在于提供一种peg固载手性双噁唑啉配体的制备方法,具体技术方案如下:
一种peg固载手性双噁唑啉配体的制备方法,包括以下步骤:
1)peg与甲基磺酰氯反应,得到oms-peg;
2)步骤1)中oms-peg与碘化钠在丙酮溶液中回流,制得碘代peg;
3)手性双噁唑啉配体和步骤2)中碘代peg在正丁基锂作用下反应,得到peg固载手性双噁唑啉配体。
所述peg为peg-2000、peg-4000、mpeg-10k、4armpeg-10k或4armpeg-20k。
所述手性双噁唑啉配体如结构式i或ii所示:
r1为氢、叔丁基或苯基;r2为甲基、乙基、异丙基、叔丁基、苯基、苄基、2-萘基、3,5-二叔丁基苯基或4-叔丁基苯基。
步骤1)具体包括:将peg溶于甲苯溶液中,升温除水,氮气保护,蒸出甲苯;降至室温加入二氯甲烷和三乙胺,然后加入甲基磺酰氯,室温搅拌反应,结束后加入无水甲醇并过滤;滤液浓缩,加入异丙醇热溶清亮,冰水浴沉淀,过滤,乙醚漂洗,真空干燥,得到oms-peg。
步骤2)具体包括:将步骤1)中oms-peg与碘化钠在丙酮溶液中回流,浓缩,加入质量分数20%的氯化钠,萃取、干燥浓缩、乙醚沉淀、过滤,得到碘代peg。
步骤3)具体包括:氮气保护下,将手性双噁唑啉配体溶于干燥的thf,降至0℃,加入正丁基锂,然后加入步骤2)中的碘代peg,室温搅拌反应,结束后加入质量分数20%的氯化钠,萃取、干燥浓缩、乙醚沉淀、过滤,得到peg固载手性双噁唑啉配体。
步骤1)中peg、甲基磺酰氯和三乙胺的物质的量比为1:3.0:2.5;甲苯为15~20ml/gpeg;甲醇为0.25ml/gpeg;二氯甲烷为1~2ml/gpeg;异丙醇为15~20ml/gpeg;乙醚为2~3ml/gpeg。
步骤1)中室温搅拌反应时间为18-24h,反应结束后采用硅藻土g2沙星漏斗过滤,滤液采用减压浓缩。
步骤2)中oms-peg与碘化钠的物质的量比为1:2;丙酮为10~15ml/gpeg;质量分数20%的氯化钠为10ml/gpeg;二氯甲烷为20ml/gpeg;乙醚为10ml/gpeg。
步骤2)中回流时间为18-24h,回流后采用降温浓缩。
步骤3)中手性双噁唑啉配体、正丁基锂和碘代peg的物质的量比为1:1.1:0.5;thf为10~15ml/gpeg;质量分数20%的氯化钠为10ml/gpeg;二氯甲烷为20ml/gpeg;乙醚为10ml/gpeg。
步骤3)中室温搅拌反应时间为18-24h。
本发明的有益效果为:
1、本发明peg固载手性双噁唑啉配体的催化活性位点与peg载体的连接是利用磺酰化的peg与碘化钠制备得到碘代peg,然后在正丁基锂作用下偶联的,有效提高了活性基团的固载率和稳定性。
2、peg载体可溶于反应溶剂中形成均相反应体系,有利于催化剂与底物的作用,发挥配体的电子效应,其活性和立体选择性更高,反应结束后加入乙醚可简便的从反应体系中分离催化剂,并可重复使用。
3、手性双噁唑啉配体廉价易得、应用性广、活性高。
4、本发明制备过程简单、产率高、活性好、生产成本低、对环境无污染,催化剂与中间产物均可通过常规方法实现固液分离。
附图说明:
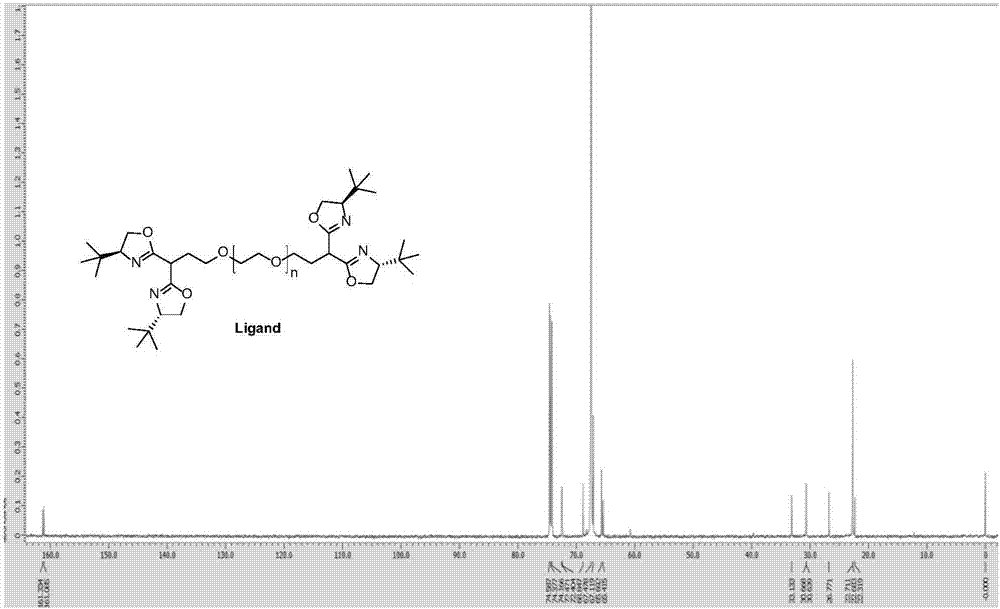
图1是实施例1配体5的1hnmr图谱。
图2是实施例1配体5的13cnmr图谱。
图3是实施例1配体5的红外图谱。
图4是实施例1制备的oms-peg-2000的hplc-elsd图谱。
图5是实施例1制备的i-peg-2000的hplc-elsd图谱。
图6是苯乙烯与eda环丙烷化产物的gc手性拆分图谱。
具体实施方式
本发明为了提高手性合成菊酯产物的产率和立体选择性,降低生产成本,实现绿色化生产,提出了一种peg固载手性双噁唑啉配体的制备方法,下面结合实施例对本发明做进一步介绍。
实施例1
(1)oms-peg-2000的制备
将20gpeg-2000溶于400ml的甲苯溶液中,升温除水,氮气保护,蒸出100ml/g的甲苯。降至室温,加入20ml二氯甲烷和3.46ml三乙胺,搅拌15min后加入2.3ml甲基磺酰氯,室温搅拌18h。反应结束后,加入5ml的甲醇,搅拌30min后,用g2沙星漏斗+硅藻土过滤,滤液减压浓缩后,加入300ml异丙醇热溶清亮,冰水浴沉淀,布氏漏斗过滤,50ml乙醚漂洗,真空干燥,得到18.6goms修饰的2(oms-peg-2000的hplc-elsd图谱如图4),产率为93%。
(2)i-peg-2000的制备
在500ml的单口瓶中加入10g的步骤(1)中2,丙酮100ml,搅拌溶解,加入2.98g碘化钠,回流18h。降温浓缩,用100ml的20%nacl水溶液溶解后,200ml二氯甲烷分三次萃取(100ml+50ml+50ml),无水na2so4干燥,浓缩,100ml乙醚沉淀,过滤得到3为9.2g(i-peg-2000的hplc-elsd图谱如图5),产率为92%。hplc-elsd检测peg端基转化率为100%。
(3)peg固载手性双噁唑啉配体
在氮气保护下,将0.83g手性双噁唑啉配体4溶于20ml干燥的thf,降至0℃,缓慢滴加1.37ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于20ml干燥的thf)3.12g碘代的3,室温搅拌18h。反应结束后,加入30ml的20%nacl水溶液,60ml二氯甲烷分三次萃取(20ml+20ml+20ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体5(配体5的1hnmr图谱如图1,13cnmr图谱如图2,红外图谱如图3)。得到的成品为2.8g,固载率为72%(通过核磁计算所得),产率为90%。
1h-nmr(cdcl3,400mhz)δ4.19-4.15(m,2h),4.08-4.04(m,2h),3.90-3.84(m,2h),3.80-3.74(m,2h),3.66-3.54(m,hofthepeg-signal),3.58-3.52(m,3h),3.26(t,2h,j=4.4),2.26-2.13(m,2h),0.87(s,36h)ppm.13cnmr(100mhz,cdcl3):δ=161.3,161.1,72.5,72.4,68.8,67.5,67.1,65.7,65.4,33.1,30.7,30.6,26.8,22.7,22.3ppm.ir(kbr):ν3525,2669,1967,1664,1573,1468,1359,1281,1243,1111,949,843,732,699,530cm-1;maldi(peg2000):2014da.
实施例2
同实施例1,只是把peg-2000改为peg-4000。
(1)将20gpeg-4000溶于400ml的甲苯溶液中,升温除水,氮气保护,蒸出100ml的甲苯。降至室温,加入20ml二氯甲烷和1.73ml三乙胺,搅拌15min后加入1.15ml甲基磺酰氯室温搅拌18h。反应结束后,加入5ml的甲醇,搅拌30min后,用g2沙星漏斗+硅藻土过滤,滤液减压浓缩后,加入300ml异丙醇热溶清亮,冰水浴沉淀。布氏漏斗过滤,50ml乙醚漂洗,真空干燥,得到19.0goms-peg-4000,产率为95%。
(2)在500ml的单口瓶中加入15g的步骤(1)中oms-peg-4000,丙酮150ml,搅拌溶解,加入2.23g碘化钠,回流18h。降温浓缩,用150ml的20%nacl水溶液溶解后,300ml二氯甲烷分三次萃取(100ml+100ml+100ml),无水na2so4干燥,浓缩,150ml乙醚沉淀,过滤得到产物为13.6g,产率为91%。
(3)在氮气保护下,将1.0g手性双噁唑啉配体4溶于20ml干燥的thf,降至0℃,缓慢滴加1.65ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于60ml干燥的thf)7.52g碘代的peg-4000,室温搅拌18h。反应结束后,加入70ml的20%nacl水溶液,150ml二氯甲烷分三次萃取(50ml+50ml+50ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体,得到的成品为6.99g,产率为93%。
实施例3
同实施例1,只是把peg-2000改为meopeg-10k。
(1)将20gmeopeg-10k溶于400ml的甲苯溶液中,升温除水,氮气保护,蒸出100ml的甲苯。降至室温,加入30ml二氯甲烷和0.35ml三乙胺,搅拌15min后加入0.23ml甲基磺酰氯室温搅拌18h。反应结束后,加入5ml的甲醇,搅拌30min后,用g2沙星漏斗+硅藻土过滤,滤液减压浓缩后,加入350ml异丙醇热溶清亮,冰水浴沉淀。布氏漏斗过滤,50ml乙醚漂洗,真空干燥,得到18.0g,产率为90%。
(2)在500ml的单口瓶中加入18g的步骤(1)中oms-meopeg-10k,丙酮200ml,搅拌溶解,加入0.54g碘化钠,回流18h。降温浓缩,用180ml的20%nacl水溶液溶解后,360ml二氯甲烷分三次萃取(120ml+120ml+120ml),无水na2so4干燥,浓缩,180ml乙醚沉淀,过滤得到产物为16.6g,产率为92%。
(3)在氮气保护下,将0.133g手性双噁唑啉配体4溶于20ml干燥的thf,降至0℃,缓慢滴加0.343ml正丁基锂(浓度为m=1.6的正己烷溶液)搅拌30min后,滴加(溶于45ml干燥的thf)5.0g碘代的meopeg-10k,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体,得到的成品为4.4g,产率为88%。
实施例4
同实施例1,只是把peg-2000改为4armpeg-20k。
(1)将20g4armpeg-20k溶于400ml的甲苯溶液中,升温除水,氮气保护,蒸出100ml的甲苯。降至室温,加入40ml二氯甲烷和0.697ml三乙胺,搅拌15min后加入0.464ml甲基磺酰氯室温搅拌18h。反应结束后,加入5ml的甲醇,搅拌30min后,用g2沙星漏斗+硅藻土过滤,滤液减压浓缩后,加入400ml异丙醇热溶清亮,冰水浴沉淀。布氏漏斗过滤,50ml乙醚漂洗,真空干燥,得到17.0g,产率为85%。
(2)在500ml的单口瓶中加入15g的oms-4armpeg-20k,丙酮225ml/g,搅拌溶解,加入1.19g碘化钠,回流18h。降温浓缩,用150ml的20%nacl水溶液溶解后,300ml二氯甲烷分三次萃取(100ml+100ml+100ml),无水na2so4干燥,浓缩,150ml乙醚沉淀,过滤得到产物为14.0g,产率为93%。
(3)在氮气保护下,将0.266g手性双噁唑啉配体4溶于20ml干燥的thf,降至0℃,缓慢滴加0.687ml正丁基锂(浓度为m=1.6的正己烷溶液)搅拌30min后,滴加(溶于55ml干燥的thf)5g碘代的4armpeg-20k,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.6g,产率为92%。
实施例5
同实施例1,只是把peg-2000改为4armmpeg-10k。
(1)将20g4armpeg-10k溶于400ml的甲苯溶液中,升温除水,氮气保护,蒸出100ml的甲苯。降至室温,加入20ml二氯甲烷和1.4ml三乙胺,搅拌15min后加入0.929ml甲基磺酰氯室温搅拌18h。反应结束后,加入5ml的甲醇,搅拌30min后,用g2沙星漏斗+硅藻土过滤,滤液减压浓缩后,加入350ml异丙醇热溶清亮,冰水浴沉淀。布氏漏斗过滤,50ml乙醚漂洗,真空干燥,得到16.0g,产率为80%。
(2)在500ml的单口瓶中加入15g的oms-4armpeg-10k,丙酮150ml,搅拌溶解,加入2.38g碘化钠,回流18h。降温浓缩,用150ml的20%nacl水溶液溶解后,300ml二氯甲烷分三次萃取(100ml+50ml+50ml),无水na2so4干燥,浓缩,150ml乙醚沉淀,过滤得到产物为12.6g,产率为84%。
(3)在氮气保护下,将0.532g手性双噁唑啉配体4溶于20ml干燥的thf,降至0℃,缓慢滴加0.687ml正丁基锂(浓度为m=1.6的正己烷溶液)搅拌30min后,滴加(溶于40ml干燥的thf)5g碘代的4armpeg-10k,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.1g,产率为82%。
实施例6
同实施例1,只是步骤(3)中把配体改为r2为苯基的手性双噁唑啉。在氮气保护下,将1.53gr2为苯基的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.0g,产率为80%。
实施例7
同实施例1,只是步骤(3)中把配体改为r2为异丙基的手性双噁唑啉。在氮气保护下,将1.19gr2为异丙基的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为3.9g,产率为79%。
实施例8
同实施例1,只是步骤(3)中把配体改为r2为苄基的手性双噁唑啉。在氮气保护下,将1.67gr2为苄基的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.5g,产率为90%。
实施例9
同实施例1,只是步骤(3)中把配体改为r2为2-萘基的手性双噁唑啉。在氮气保护下,将2.03gr2为2-萘基的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.1g,产率为82%。
实施例10
同实施例1,只是步骤(3)中把配体改为r1,r2都为苯基的手性双噁唑啉。在氮气保护下,将2.29gr1,r2都为苯基的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.0g,产率为80%。
实施例11
同实施例1,只是把配体改为如上图结构的手性双噁唑啉。在氮气保护下,将1.65g的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.5g,产率为90%。
实施例12
同实施例1,只是步骤(3)中把配体改为r1为叔丁基,r2为乙基的手性双噁唑啉。在氮气保护下,将1.61g的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.0g,产率为80%。
实施例13
同实施例1,只是步骤(3)中把配体改为r1为苯基,r2为甲基的手性双噁唑啉。在氮气保护下,将1.67g的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.1g,产率为82%。
实施例14
同实施例1,只是步骤(3)中把配体改为r2为3,5-二叔丁基苯基的手性双噁唑啉。在氮气保护下,将2.65g的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.4g,产率为88%。
实施例15
同实施例1,只是步骤(3)中把配体改为r2为4-叔丁基苯基的手性双噁唑啉。在氮气保护下,将2.09g的手性双噁唑啉溶于20ml干燥的thf,降至0℃,缓慢滴加2.2ml正丁基锂(浓度为m=2.5的正己烷溶液)搅拌30min后,滴加(溶于30ml干燥的thf)5g实施例1中碘代的3,室温搅拌18h。反应结束后,加入50ml的20%nacl水溶液,100ml二氯甲烷分三次萃取(50ml+25ml+25ml),无水na2so4干燥,浓缩,乙醚沉淀,过滤得到目标配体为4.6g,产率为92%。
实施例16
为进一步说明本发明,将peg-2000固载的叔丁基双噁唑啉配体与cu(otf)2络合,催化苯乙烯与eda的不对称环丙烷化反应为实例。
peg固载手性金属铜双噁唑啉催化苯乙烯与eda的不对称环丙烷化反应:
在氮气保护下,将3.6mgcu(otf)2和44mg的实施例1中配体5溶于2ml干燥的ch2cl2,搅拌30min后,滴加22μl浓度为5%的苯肼,至反应液完全变色后,加入0.345ml的苯乙烯6后,8h缓慢滴加eda(114mg7溶于8ml干燥的ch2cl2),室温搅拌3h。反应结束后减压浓缩,加入乙醚并降温至0℃左右。通过过滤得到催化剂,滤液过硅胶柱得到目标化合物菊酸乙酯。回收的催化剂可以将其溶于ch2cl2中,下次需要时无需再次苯肼活化,直接即可循环使用。苯乙烯与eda环丙烷化产物的gc手性拆分图谱如图6。目标产物的ee值为92%(trans),产率为83%。
- 还没有人留言评论。精彩留言会获得点赞!