二芳基硫醚衍生物、其制备方法以及作为五羟色胺转运体靶向显像剂的用途与流程
本发明属于放射性药物化学领域,涉及一类二芳基硫醚衍生物、其制备方法以及作为五羟色胺转运体靶向显像剂的用途。
背景技术:
五羟色胺转运体(sert)是一种选择性五羟色胺(5-ht)跨膜转运蛋白。sert能够重新摄取突触间隙的5-ht,从而调节神经信号的传导。在中枢神经系统中,存在于神经元突触前膜的sert,能够选择性地将神经递质5-ht由突触间隙转回到细胞内胞液中,从而终止该递质对突出后膜5-ht受体的作用,因而在5-ht释放后的再摄取过程中发挥极其重要的作用。研究表明,sert浓度的变化与许多神经精神异常疾病有关,例如抑郁症、忧郁症、焦虑症、精神分裂、药物滥用、酒瘾、饮食失调、阿尔茲海默氏症(alzheimer’sdisease)以及帕金森氏症(parkinson’sdisease)等。sert也是普遍使用的抗忧郁处方药物的主要作用靶点。
正电子发射断层计算机扫描(pet)显像可以无创、动态、定量地研究人体内的化学过程和生理生化过程,在活体水平研究生命物质的代谢,受体的分布和功能,基因调控的变化等,这些使核医学的发展达到真正意义上的分子水平。pet显像离不开正电子核素标记的pet药物,其中18f正电子核素(半衰期为110分钟)比11c(半衰期为20分钟)具有相对较长的半衰期,适合商品化生产和运输,是当前pet显像药物的理想放射性核素,18f药物成为研究的热点。因此,以非侵入性的pet和单光子断层扫描(spect)活体显像技术研究sert在脑部区域的分布,将有助于探讨5-ht系统在神经精神性异常疾病的病理生理学及治疗上所扮演的角色,认识sert密度的变化与精神紊乱疾病及精神退行性疾病病理的关系,也可为治疗药物作用机制研究、个体化治疗策略制定及疗效评估等提供非常有效的信息。
过去十多年中以sert为靶点的显像剂的研究取得了较大的进展,国际上开发出了一系列有潜力的化合物,目前有多个国家在进行该类显像剂的研发或者临床前研究,该领域已经成为当今世界在显像剂研究方面的热点,主要集中于托烷环类、硝基喹啉哌嗪类、二芳基硫醚类先导化合物的修饰方面。近年来,在二芳基醚类化合物上取得了突破性进展,发展了一批体内外性质优良的分子探针。该类化合物是基于抗抑郁药物moxifetin和403u76(其结构式如下)(oyas.etal,5-iodo-2-[[2-2-[(dimethylamino)methyl]phenyl]thio]benzylalcohol,jmedchem,1999;42:333–335)发展而来的,合成简单,没有立体异构体的存在,分离纯化容易,在体内外具有较高的亲和性和选择性。其中用于spect显像的[123i]adam(其结构式如下)和用于pet显像的[11c]dasb(其结构式如下)已经用于人脑sert的临床显像研究。但对于18f标记pet显像剂而言,目前还没有临床广泛应用的显像剂问世,国内外研究者正加紧研发具有sert靶向特异结合的18f显像剂。
sert又是抗抑郁药物的主要作用靶标,二芳基硫醚类配基与sert具有靶向特异结合,其衍生物作为sert抑制剂,已成为治疗抑郁症和精神神经疾病的药物。目前新的sert靶向化合物还在继续研究,其目的是寻找性能更优秀的治疗药物。在sert/pet显像剂的开发中,本发明人研究发现,当在骨架b环4位引入侧链(如下结构式所示),能够调节该系列化合物的药代动力学性质,因此本发明人提供一种含f的二芳基硫醚类衍生物,在骨架b环4位引入了不同侧链,提高了与sert的靶向结合性,从而改善目前此类化合物作为sert显像剂存在的缺陷。同时,当用18f-标记作为放射性靶向药物研究时,使其具有更高的体内靶/非靶比值,更适宜的体内动力学性质,有望满足今后临床sert显像需要。
技术实现要素:
本发明的目的在于提供一种二芳基硫醚衍生物,该类化合物具有较高的脑内sert(五羟色氨转运蛋白)特异性结合效果,同时具有较好的体内生物代谢性质,是较好的脑内摄取的靶向结合显像药物。
本发明的另一目的在于提供一种上述二芳基硫醚衍生物的制备方法。
本发明的还一目的在于提供上述二芳基硫醚衍生物作为五羟色胺转运体靶向显像剂的用途。
根据本发明的一个方面,本发明提供结构式如下的二芳基硫醚衍生物:
其中,r为直接键、
y为氟原子,可以为[18f]或者[19f]。
根据本发明的另一个方面,本发明提供一种上述二芳基硫醚衍生物的制备方法,该方法包括:在碱存在下,2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚与
根据本发明的又一个方面,本发明的二芳基硫醚衍生物,为含有[19f]或[18f]的化合物,[19f]-二芳基硫醚衍生物表现出对中枢sert的高亲合力,同时相对于dat(多巴胺转运体)以及net(去甲肾上腺素转运体)对sert的高选择性。因此,相应的[18f]-二芳基硫醚衍生物,可以作为sert/pet显像剂,这些化合物有望获得较高的脑摄取值,合理的药代动力学性质,有望开发出优良的sert/pet显像剂。
附图说明
图1为化合物a5经过oasishlb柱子初步提纯后hplc谱图中的放射性峰;
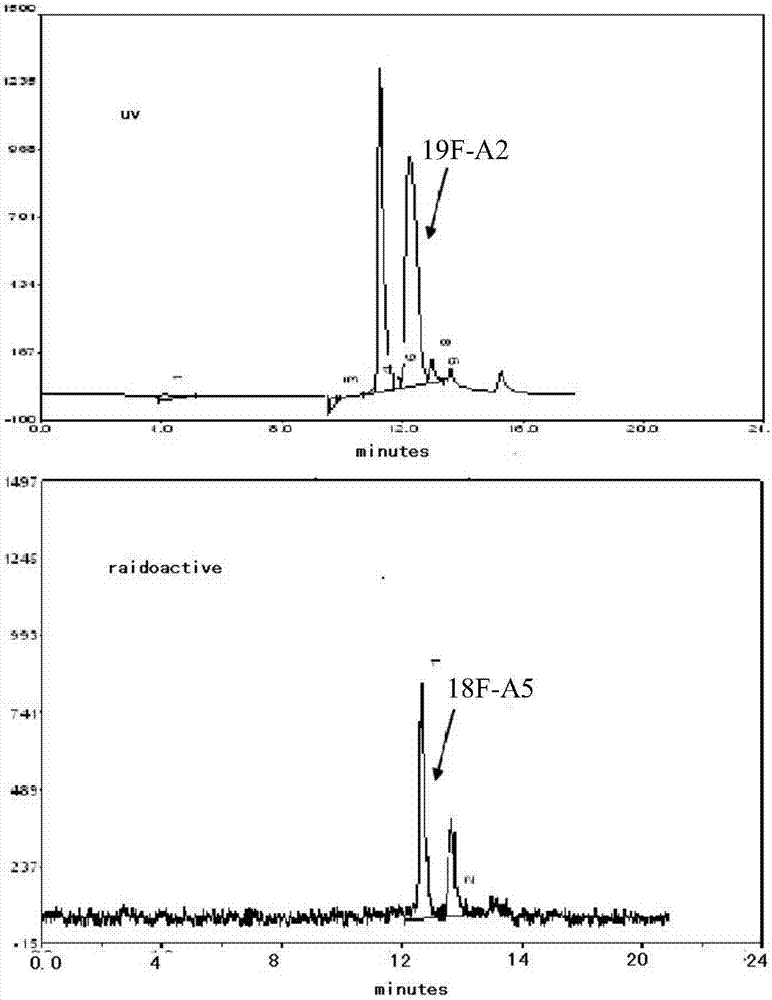
图2为化合物a5即[18f]-a5的hplc确认,即放射性产物[18f]-a5与化合物a2即[19f]-a2共注射谱图;
图3为[18f]-a6的hplc确认,即放射性产物[18f]-a6与[19f]-a3化合物共注射谱图。
具体实施方式
下面更具体地描述本发明。
在有机化学领域,对于氟化合物,如果没有说明,一般是指非放射性的[19f]的化合物。因此,在本发明中对于具体的化合物,如果没有特别指明,则是指非放射性的[19f]的化合物。
在本发明优选的实施方案中,所述二芳基硫醚衍生物,其中,r优选为
通过以下实施例更具体地说明本发明的二芳基硫醚衍生物及其制备,以及对sert的高亲合力,同时相对于dat和net对sert的高选择性,但要理解的是本发明的范围不限于这些实施例。
实施例1
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-氟乙氧基)-乙氧基)-苯)硫醚(化合物a1)的合成
合成反应方程式:
合成方法:
将0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)甲氨基-4-羟基)-苯)硫醚(合成参考文献:(shunichioyaetal,nuclearmedicineandbiology,34(2007),129–139)溶解在4mldmf(hplc纯)中,加入2倍摩尔量的碳酸钾以及1-对甲苯磺酰基-2-氟乙氧基乙烷(1.2倍摩尔量),之后通入氮气3-5min,将反应液在80-90℃条件下油浴加热过夜。反应完成后加入30ml水终止反应,并用乙酸乙酯萃取产物,合并有机相并使用无水硫酸钠干燥,然后通过硅胶柱提纯,所使用的淋洗液为甲醇:二氯甲烷:二乙胺=4:100:1(体积比),得到目标产物化合物a1,产率:24.8%。
化合物a1的确认:
1hnmr(200mhz,cdcl3)δ2.37(s,6h),3.59(s,2h),3.65-3.75(m,1h),3.80-3.92(m,3h),4.05-4.20(m,2h),4.46-4.50(m,1h),4.70-4.74(m,1h),6.68-6.72(m,3h),6.89-7.02(m,2h),7.10-7.17(m,1h),7.35-7.45(m,1h)。
lrms(m++1):理论值365.16,实测值:365.1674
实施例2
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a2)的合成
合成反应方程式:
将0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚溶解在4mldmf中(hplc纯),加入2倍摩尔量的碳酸钾以及1-对甲苯磺酰基-(2-(2-氟乙氧基)-乙氧基)乙烷(1.2倍摩尔量),之后通入氮气3-5min,将反应液在80-90℃条件下油浴加热过夜。反应完后加入30ml水终止反应,并用乙酸乙酯萃取产物,合并有机相混合并使用无水硫酸钠干燥,然后通过硅胶柱提纯,所使用的淋洗液为甲醇:二氯甲烷:二乙胺=4:100:1(体积比),得到目标产物化合物a2,产率:22.6%。
化合物a2的确认:
1hnmr(200mhz,cdcl3)δ2.37(s,6h),3.69-3.82(m,7h),3.83-3.88(m,3h),4.09-4.21(m,2h),4.44-4.48(m,1h),4.68-4.72(m,1h),6.68-6.74(m,3h),6.94-6.98(m,2h),7.12-7.22(m,1h),7.38-7.42(m,1h)。
lrms(m++1):理论值409.19,实测值409.1959。
实施例3
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(顺-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a3)的合成
合成反应方程式:
合成方法:
①1,4-二对甲苯磺酰基-2-丁烯的制备
将0.3ml的顺-2-丁烯-1,4-二醇(3.16mmol)溶解在15ml新蒸四氢呋喃(thf)中,冰浴10min后,加入对甲苯磺酰氯1.76克(9.26mmol),冰浴下搅拌10min后,加入1克叔丁醇钠,得到白色乳状物,继续搅拌过夜。反应完后用旋转蒸发仪除去溶剂,加入20ml水,并用二氯甲烷萃取三次,合并有机相,经无水硫酸钠干燥后通过柱层析法提纯,淋洗液为乙酸乙酯:石油醚=1:4(体积比),得到1,4-二对甲苯磺酰基-2-丁烯。产率:12.2%。
②1-氟代-4-对甲苯磺酰基-2-丁烯的制备
将1mmol1,4-二对甲苯磺酰基-2-丁烯加入两口圆底烧瓶中,并加入3ml乙腈将其溶解,然后加入1ml1mol/l四正丁基氟化铵的四氢呋喃溶液(1mol/l溶液),通氮气3min后将反应液在80℃条件下加热30min,移除溶剂并且将所得紫色油状混合物溶于10ml乙醚中,并加入10ml5%na2co3溶液,使粗产物在两相间平衡10min。之后,使用乙醚进行萃取(10ml×2),合并有机相并用无水硫酸钠干燥,然后使用硅胶柱进行提纯,淋洗液为乙酸乙酯:石油醚=1:4(体积比),得到1-氟代-4-对甲苯磺酰基-2-丁烯。产率:19.8%。
③2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(顺-4-氟代-2-烯丁氧基)-苯)硫醚的制备
将0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚溶解在4mldmf中(hplc纯),加入2倍摩尔量的碳酸钾以及1-氟代-4-对甲苯磺酰基-2-丁烯(1.2倍摩尔量),之后通入氮气3-5min,将反应液在80-90℃条件下油浴加热过夜。反应完后加入30ml水终止反应,并用乙酸乙酯萃取产物,合并有机相并使用无水硫酸钠干燥,然后通过硅胶柱提纯,所使用的淋洗液为甲醇:二氯甲烷:二乙胺=4:100:1(体积比),得到目标产物化合物a3,产率:17%。
结构确认:
①化合物1,4-二对甲苯磺酰基-2-丁烯
1hnmr(400mhz,cdcl3):δ2.39(s,6h),4.47(d,j=5.85hz,4h),5.60-5.62(m,2h),7.28(d,j=8.08hz,4h),7.69(d,j=8.20hz,4h);
②化合物1-氟-4-对甲苯磺酰基-2-丁烯
1hnmr(400mhz,cdcl3):δ2.38(s,3h),4.57(d,j=6.6hz2h),4.83(dd,j=46.6,5.64hz,2h),5.58-5.64(m,1h),5.70-5.76(m,1h),7.28(d,j=8.12hz,2h),7.72(d,j=8.12hz,2h);
③化合物a3的确认:1hnmr(200mhz,cdcl3)δ2.34(s,6h),3.60(s,2h),4.75(dt,jfh=5.2hz,jhh=1.4hz,2h),4.99(dd,jfh=47hz,jhh=1.6hz,2h),5.88-5.91(m,2h)6.72-6.77(m,3h),6.90-6.98(m,2h),7.16-7.19(m,1h),7.42-7.46(m,1h).13cnmr(50mhz,cdcl3)δ43,62,65,81,114,116,116.5,117,126.5,127,129,129.5,130,132,135,148,157.
lrms(m++1):理论值:347.16,实测值:347.1592.
实施例4
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚(化合物a4)的合成
合成反应方程式:
合成方法:
①化合物1,4-二对甲苯磺酰基-2-丁炔的制备
在圆底烧瓶中,将相应的10mmol的1,4-丁炔二醇(860mg)溶解在10ml的二氯甲烷中,冰浴降温至0℃,逐滴加入1.4ml三乙胺(20mmol),以及20mg4-n,n-二甲基吡啶,之后继续搅拌溶液5min;并将3.8gtscl溶解在3ml二氯甲烷中,通过恒压滴液漏斗逐滴加入到圆底烧瓶中,滴完后,反应液继续在冰浴条件下搅拌1h,后于室温反应3-4h;反应完后加入20ml二氯甲烷稀释,并用1mol/lhcl溶液、5%na2co3溶液、饱和食盐水依次洗涤。洗涤后的有机相经过无水硫酸钠干燥后,使用旋转蒸发仪除去溶剂,并用硅胶柱提纯,淋洗液为乙酸乙酯:石油醚=1:4(体积比),得到1,4-二对甲苯磺酰基-2-丁炔。
②化合物1-氟代-4-对甲苯磺酰基-2-丁炔的制备
将1mmol的1,4-二对甲苯磺酰基-2-丁炔加入到两口圆底烧瓶中,并加入3ml乙腈将其溶解,然后加入1ml1mol/l四正丁基氟化铵的四氢呋喃溶液,通氮气3min后,将反应液在80℃条件下加热30min,移除溶剂并且将所得紫色油状混合物溶于10ml乙醚,并加入10ml5%na2co3溶液,让粗产物在两相间平衡10min。之后,使用乙醚进行萃取(10ml×2),合并有机相并用无水硫酸钠干燥,使用硅胶柱提纯产物,淋洗液为乙酸乙酯:石油醚=1:6(体积比),得到1-氟代-4-对甲苯磺酰基-2-丁炔。产率:42%。
③2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚的制备
将0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚溶解在4mldmf中(hplc纯),加入2倍摩尔量的碳酸钾以及1-氟代-4-对甲苯磺酰基-2-丁炔(1.2倍摩尔量),之后通入氮气3-5min,将反应液在80-90℃条件下油浴加热过夜,反应完后加入30ml水终止反应,并用乙酸乙酯萃取产品,将有机相混合后使用无水硫酸钠干燥,并通过硅胶柱提纯。淋洗液为甲醇:二氯甲烷:二乙胺=4:100:1(体积比),得到目标产物,产率:22.1%。
结构确认:
①化合物1,4-二对甲苯磺酰基-2-丁炔
1hnmr(400mhz,cdcl3):δ2.40(s,6h),4.51(s,4h),7.28(d,j=8.12hz,4h),7.69(d,j=8.24hz,4h);
②化合物1-氟代-4-对甲苯磺酰基-2-丁炔
1hnmr(400mhz,cdcl3):δ2.38(s,3h),4.67-4.70(m,2h),4.77(d,j=45.6hz,2h),7.29(d,j=8.04hz,2h),7.74(d,j=8.04hz,2h);
③化合物2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚
1hnmr(200mhz,cdcl3)δ2.34(s,6h),3.60(s,2h),4.72-4.77(m,2h),4.99(dd,jfh=47hz,jhh=1.6hz,2h),6.71-6.77(m,3h),6.91-6.98(m,2h),7.15-7.20(m,1h),7.42-7.46(m,1h);13cnmr(50mhz,cdcl3)δ43,56,62,68,72,85,115,115.5,117,119,128,130,136,138,148,155。
lrms(m++1):理论值345.13,实测值345.1391。
实施例5
[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a5)的合成
(1)[18f]-1-对甲苯磺酰基-2-((2-氟乙氧基)-乙氧基))乙烷的制备
干燥[18f]kf/k222的准备:将水溶液中的[18f-]一次性通过qma小柱(4-(4-甲基哌啶)吡啶阴离子交换树脂小柱)并推干,后使用11mgk222/2mgk2co3溶液(0.8ml乙腈,0.2ml水)淋洗下放射性活度。之后将所得溶液在100℃条件下吹干,并依次加入2ml无水乙腈(每次1ml)吹干,得干燥[18f]kf/k222。
将2mg的1-对甲苯磺酰基-2-((2-对甲苯磺酰基乙氧基)-乙氧基)-乙烷前体溶于1ml无水乙腈中,加入制备好的干燥的[18f]kf/k222中。充分摇荡均匀后置于85℃环境中加热5min,停止加热进行提纯。
提纯方式:反应液无需冷却即加入5ml水,通过oasishlb(反相亲水亲脂小柱)柱子提纯:首先将粗产品一次性以约每秒一滴的速度通过已经处理好的oasishlb柱子,使用5ml水洗柱子,吹干,并且用氮气流吹2min。之后用1.5ml乙腈直接淋洗产品至第二步反应管中(含有7.5mg氢化钠,2mg第二步前体:2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚的10ml试管)。
(2)[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a5)的制备
在氮气流条件下把上述加入了[18f]-1-对甲苯磺酰基-2-((2-氟乙氧基)-乙氧基))乙烷的试管中的溶液在120℃条件下吹干。然后加入1ml无水dmf,摇匀后继续在120℃条件下反应15min,反应结束使用oasishlb柱子初步提纯产品:即先往反应粗产物中加入5ml水,将混合液一次性通过预先活化好的oasishlb柱子。使用5ml水淋洗柱子,推干,并使用1ml乙醇淋洗产物,用hplc检测(参见图1),最终用半制备hplc分离纯化即可得到18f标记目标产物[18f]-a5。
标记产率:标记总体时间约90min,标记中间体产率29.7%,最终产物产率:3.7%(按照投入的[18f-]的活度计算,产率未经时间校正),oasishlb柱子提纯后产品放射化学纯度为64%,主要放射性杂质是放射性中间体。
[18f]-a5的结构确证为将如上所述制得的标记产物[18f]-a5与实施例2制备的化合物a2即[19f]-a2,在相同的条件下共注射进行hplc分析,通过比对保留时间进行确认(参见图2)。
分析用hplc条件如下:溶剂a:acn(乙腈),溶剂b:20mmafb(甲酸铵);梯度:0-3min95%b,3-11min95%-5%b,11-19min5%-95%b,19-21min95%b;柱子:gemini3micron,140x4.6mm,column:gemini3uc18,110a,150×4.6mm,3micron。
实施例6
[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(顺-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a6)的合成
(1)[18f]-1-氟代-4-对甲苯磺酰基-2-丁烯的制备
干燥[18f]kf/k222的准备同实施例5。
将3mg的1,4-二对甲苯磺酰基-2-丁烯前体溶于1ml无水乙腈中,加入制备好的干燥的[18f]kf/k222中。充分摇荡均匀后置于60℃环境中加热10min,停止加热进行提纯。
提纯方式:使用0.5ml乙醚稀释反应液。将反应液合并,并一次性通过硅胶小柱(sep-paksilica)后使用1.5ml乙醚将放射性中间体淋下。
(2)[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(顺-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a6)制备
在氮气流条件下把加入了[18f]-1-氟代-4-对甲苯磺酰基-2-丁烯的试管中的乙醚在60℃条件下吹干。然后加入1ml无水dmf,摇匀后,转移到含有7.5mg碳酸铯,2mg2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羟基)-苯)硫醚的试管中。混合均匀,并在20℃室温下反应10min。反应结束使用oasishlb柱子初步提纯产品:即先往反应粗产物中加入5ml水,将混合液一次性通过预先活化好的oasishlb柱子。使用5ml水淋洗柱子,推干,并使用1ml50%乙醇/水溶液淋洗产物,用hplc检测(图3),[18f]-a6。
标记产率:标记总体时间约2h,以投入的[18f-]活度计算的不经时间校正产率:标记中间体产率约25%,[18f]-a6标记产率约1-2%。经过oasis小柱初步提纯后的产品放射化学纯度大于90%。
[18f]-a6的结构确证为将如上所述制得的标记产物[18f]-a6与实施例3制备的化合物a3即[19f]-a3,在相同的条件下共注射进行hplc分析,通过比对保留时间进行确认(参见图3)。
分析用hplc条件如下:溶剂a:乙腈(acn),溶剂b:20mmafb(甲酸铵);梯度:0-3min95%b,3-11min95%-5%b,11-19min5%-95%b,19-21min95%b;柱子:gemini3micron,140x4.6mm,column:gemini3uc18,110a,150×4.6mm,3micron。
实施例7
体外竞争结合试验(方法参见文献shunichioya,j.med.chem.2002,45,4716-4723)。
体外竞争结合试验中使用的三个测定非特异性结合的化合物如下:citalopram(西酞普兰)、gbr12909(伐诺司林)和nisoxetine(尼索西汀),其中,citalopram是已知的sert抑制剂,gbr12909是已知的dat抑制剂,nisoxetine是已知的net抑制剂。
通过体外竞争实验测得ki常数(数据见下表1),当二芳基硫醚化合物b环4位使用不同小分子链结合后,化合物保有对sert的高亲和能力。由表1中的数据可知,本发明的化合物a1-a4均对sert有非常高的亲和性,尤其是化合物a1(0.09±0.01nm)、a3(0.02±0.003nm)。而且,对dat和net的结合试验显示,这些化合物对dat的亲和性很差,对net的亲和性也一般,从而表现出了高选择性。例如,化合物a3的dat/sert选择性高达34300,net/sert选择性也高达4010,利用18f标记后可有望作为sert显像剂。
表1:二芳基硫醚系列衍生化合物与sert、dat、net受体的竞争性结合常数kia
a.每个ki值都是三次实验所得。
b.sert结合性能测定实验中,llc-sert细胞膜匀浆与0.1nm[125i]idam(kd=0.2nm);dat结合性能测定实验中,llc-dat细胞膜匀浆与0.08nm[125i]ipt(kd=1.2);net结合性能测定实验中,llc-net细胞膜匀浆与0.06nm[125i]2-inxt(kd=0.06nm)。
c.n=1。
上述用于放射性受体结合实验的标准化合物化学结构如下:
- 还没有人留言评论。精彩留言会获得点赞!