二氢双苯并恶庚衍生物及其组合物与应用的制作方法
本发明涉及二氢双苯并恶庚衍生物及其组合物与应用,属于抗肿瘤药学技术领域。
背景技术:
恶性肿瘤严重威胁人类的生命健康,每年全世界约有700万人死于癌症,约占总死亡人数的四分之一。目前我国现有癌症患者死亡率逾30%,已成为我国居民死亡的第二大因素。药物治疗已成为对恶性肿瘤有效而又普遍使用的一种治疗方法。尽管自1942年耶鲁大学的Gilman等首次证明盐酸氮芥对小鼠Gardner淋巴瘤有治疗作用以来,肿瘤的药物治疗取得了长足的进展,并成为当前临床治疗不可或缺的主要措施。但高毒副作用、耐药等问题仍然是临床肿瘤药物治疗遇到的主要障碍。临床上应用的抗肿瘤药物种类繁多,其中化疗药物主要有烷化剂钼络合物抗肿瘤药物、蒽环类抗肿瘤药物、破坏DNA的抗生素等。此外,天然抗肿瘤药物的研究也占有相当大的比例,如目前临床上常用一些药物有喜树碱、长春新碱、紫杉醇等。
多聚(ADP-核糖)聚合酶(poly(ADP-ribose)polymerase-1,PARP1)是一个高度保守的酶,主要集中在DNA损伤的维护和维修,参与多种生物学过程,包括细胞凋亡,染色体的稳定性、基因扩增、转录调控和细胞分裂。当DNA损伤时,PARP1结合到单链断裂(SSBs)位点,成为催化活性形式。它利用还原型烟酰胺腺嘌呤二核苷酸(NAD)为底物,合成直链和支链的聚(ADP-核糖)(PAR)到PARP1本身以及核靶蛋白如组蛋白拓扑异构酶,DNA聚合酶和DNA连接酶。合成的PAR链介导的碱基切除修复(BER)通过招募BER蛋白如XRCC1,DNA连接酶和DNA聚合酶III,β(POLβ)受损的DNA位点。抑制PARP1会导致SSBs的积累和阻碍DNA修复机制,最终导致双链断裂(DSBs)。有趣的是,在许多类型的癌症中PARP1发生过表达,如黑色素瘤显示,胶质母细胞瘤和乳腺癌。此外,parp1高表达与三阴性乳腺癌(TNBC)密切相关。因此,针对PARP1抑制其相关的生物学功能可以为我们提供了潜在的癌症治疗的新策略,尤其是在TNBC。
以前的研究已经报道,抑制PARP1导致合成致死性在BRCA1/2突变的卵巢癌和乳腺癌中,这使BRCA1/2基因缺失肿瘤细胞被parp1抑制剂选择性地靶向。目前,各种各样的PARP抑制剂,如olaparib,rucaparib,bmn-673以及Niraparib,处于不同的临床实验阶段。从化学角度来看,PARP抑制剂的大多数化学支架含有酰胺结构,更多的新的化学结构可以在将来被发现;从生物学的角度来看,虽然这些PARP抑制剂有高PARP1/2抑制和抗肿瘤活性;但是,长期用药会伴随药物的耐药性,导致肿瘤复发和转移。因此,除了深入的探讨抑制剂耐药机制与PARP依赖途径和肿瘤特异性的缺陷,开发一种新型的具有结构多样性表现出更好的疗效和更好的安全性的PARP抑制剂是治疗TNBC很有前途的策略。
技术实现要素:
本发明解决的技术问题是提供一种作为PARP抑制剂的新化合物。
本发明提供可如式Ⅰ所示的化合物或其药学上可接受的盐:
其中,R1为-H或C1~C4烷基;R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
进一步的,优选R1为-H;R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
作为优选方案,R1为-H;R2为-H、-CH3、-NH2。
进一步的,优选R1为-H;R2为-NH2。
本发明还提供上述化合物或其药学上可接受的盐在制备抗肿瘤药物中的用途。
进一步的,所述抗肿瘤药物优选为PARP抑制剂类药物。
所述抗肿瘤药物优选为治疗三阴性乳腺癌的药物。
本发明还提供一种药物组合物,它是包含有效剂量的上述化合物或其药学上可接受的盐的制剂。
本发明制备的化合物或其药学上可接受的盐,可以作为PARP抑制剂,具有一定的抗肿瘤活性,能有效的抑制某些癌细胞的生长。本发明化合物对多种肿瘤细胞,特别是乳腺癌细胞具有明显的抑制作用。
附图说明
图1A为用不同浓度的化合物2处理MCF-7,MDA-MB-231,MDA-MB-436,MDA-MB-468细胞系,通过MTT测定的细胞的存活率。
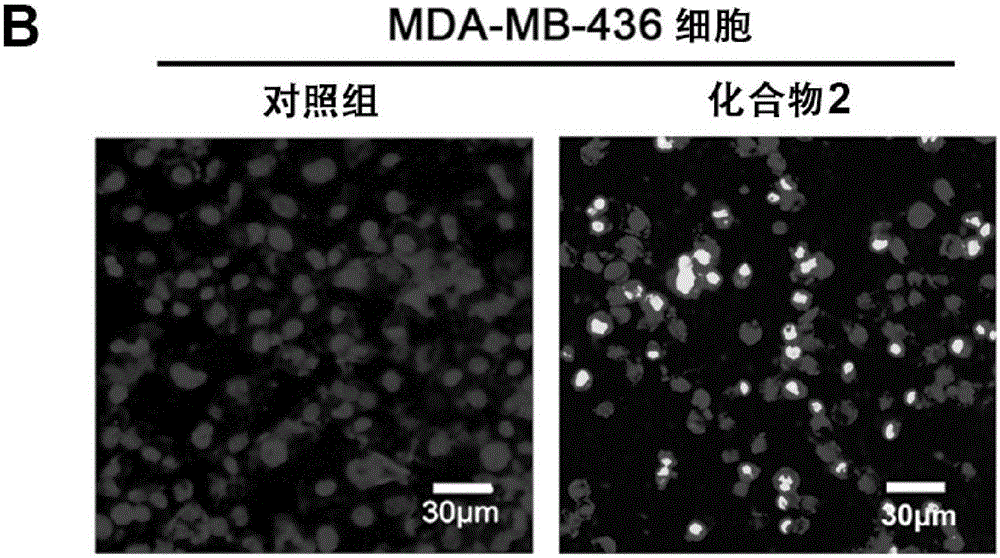
图1B为Hoechst 33258荧光染色检测细胞DNA损伤(Scale bar=200μm)。
图1C为用不同浓度的化合物2处理MDA-MB-436细胞系24小时,通过流式分析测定的凋亡率。
图1D为化合物2处理MDA-MB-436细胞,利用蛋白质印记法测定了Bax,Bcl-2,Caspase-3,PARP1和PAR的表达量。
图1E为用化合物2处理MDA-MB-436细胞,测定其抑制细胞转移的能力(Scale bar=100μm)。
图2A为不同组小鼠肿瘤相对体积,**,P<0.01,与对照组相比。
图2B为不同组小鼠相对瘤重,*,P<0.05;***,P<0.001,与对照组相比。
图2C为.治疗期间不同组的小鼠体重,**,P<0.01,与对照组相比。
图2D为不同组小鼠的肝、脾、肾,*,P<0.05;**,P<0.01,与对照组相比。
图2E为Ki-67和PAR的免疫组化分析(Scale bar=200μm,化合物2为高剂量组)。
图2F为肿瘤离体组织的PARP,PAR和Caspase-3的蛋白印记分析。
具体实施方式
本发明提供可如式Ⅰ所示的化合物或其药学上可接受的盐:
其中,R1为-H或C1~C4烷基;R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
其中,优选R1为-H;R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。作为优选方案,R1为-H;R2为-H、-CH3、-NH2。进一步的,优选R1为-H;R2为-NH2。
其中,优选R1为-CH3,R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基;更优选R1为-CH3,R2为-CH3或-CH2CH3。
其中,优选R1为-CH2CH3,R2为-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基;更优选R1为--CH2CH3,R2为-CH2CH3。
下面是本发明的化合物的一些优选结构。
本发明还提供上述化合物或其药学上可接受的盐在制备抗肿瘤药物中的用途。
进一步的,所述抗肿瘤药物优选为PARP抑制剂类药物。
所述抗肿瘤药物优选为治疗三阴性乳腺癌的药物。
本发明还提供一种药物组合物,它是包含有效剂量的上述化合物或其药学上可接受的盐的制剂。
可以通过本领域已知的方法可将本发明化合物制成以下形式:片剂、胶囊剂、水性或油性溶液剂、混悬剂、乳剂、乳膏剂、软膏剂、凝胶剂、喷鼻剂、栓剂、用于吸入的细小分散的粉剂或气雾剂或喷雾剂、用于胃肠道外(包括静脉内、肌内或输注)的无菌水性或油性溶液或混悬剂或无菌乳剂。可采用无菌水或水-丙二醇溶液作为溶剂来制备液体制剂,还可将活性组分配制在聚乙二醇水溶液中。用于口服给予的水性溶液可通过将活性组分溶解在水中并按需要加入合适的着色剂、矫味剂、稳定剂和增稠剂来制备。口服使用的水性混悬剂可通过将细小分散的活性组分与粘性物质一道分散在水中,所述粘性物质如为天然合成胶、树脂、甲基纤维素、羧甲基纤维素和其他药剂领域已知的悬浮剂。
药物组合物可为单位剂量形式。在这些形式中,将所述组合物分成含适量活性组分的单位剂量。该单位剂量形式可为包装制剂,包装中包括分隔量的制剂,例如盒装片剂、胶囊剂和在管形瓶或安瓿中的粉剂。单位剂量形式还可为胶囊剂、扁囊剂或片剂或其可为适当数量的任何这些包装形式。
本发明的药物组合物,其活性成分可仅为本发明的化合物,也可与其他抗肿瘤化合物组合作为活性成分。
在治疗肿瘤的过程中,可采用本发明的药物组合物与其他抗肿瘤药进行联合治疗。例如,与用于医学肿瘤学的抗增殖/抗肿瘤药、细胞生长抑制剂、抗入侵药物、生长因子功能抑制剂、抗血管生成剂、血管损伤剂等联用。
在治疗肿瘤时,可通过同时、序贯或单独给予各种治疗成分可实现这种联合治疗。此类组合产品应用有效剂量范围内的本发明化合物和准许剂量范围内的其他药学活性剂。
下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实施例范围之中。
实施例1化合物2的合成
取isobenzofuran-1(3H)-one 10.0g,对羟基苯乙酸11.3g溶于60ml DMF中,加热至120℃。再缓慢加入30ml含有8.0g甲醇钠的甲醇溶液,完毕后减压除去甲醇,回流过夜。加入200ml冰水,浓盐酸调节pH到12,过滤得到粗品,70%的乙醇重结晶,得到白色固体A,收率60%。1H-NMR(DMSO-d6,400MHz),δH 12.65(2H,s),7.92(1H,dd,J=7.6,1.2Hz),7.63(1H,dd,J=7.1,1.1Hz),7.56(1H,td,J=7.1,1.1Hz),7.44(1H,td,J=7.6,1.2Hz),7.17(2H,d,J=8.6Hz),6.90(2H,d,J=8.6Hz),5.43(2H,s),3.48(2H,s);13C-NMR(DMSO-d6,100MHz)δC173.4,168.6,157.6,138.9,132.6,132.6,130.9,130.9,129.9,128.3,128.1,127.8,114.9,114.9,68.0,40.0。
取白色固体A(即2-(11-oxo-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetic acid)5.0g,85%的磷酸1.8mmol,乙酰氯1.2eq,溶于50ml甲苯中,加热至100℃反应10小时。再加入活性炭500mg,在100℃搅拌1小时,过滤冷却至室温,得到粗品,乙酸乙酯-正己烷重结晶得浅黄色固体B,收率70%。1H-NMR(CDCl3,400MHz),δH8.12(1H,d,J=2.3Hz),7.88(1H,dd,J=7.6,1.1Hz),7.55(1H,td,J=7.4,1.3Hz),7.46(1H,td,J=7.6,1.1Hz),7.42(1H,dd,J=8.4,2.3Hz),7.36(1H,d,J=7.4Hz),7.03(1H,d,J=8.4Hz),5.18(2H,s),3.67(2H,s);13C-NMR(CDCl3,100MHz),δC 190.9,177.2,160.6,140.4,136.4,135.5,132.8,132.6,129.5,129.3,127.8,127.1,125.2,121.2,73.6,39.9。
取8.0g 1,3-二溴丙烷,三苯基膦10.40g溶于50ml甲苯中,混合物加热至130℃反应2小时,冷却至室温,过滤得到白色粉末三苯基膦溴丙烷,产率90%。
取三苯基膦溴丙烷10.0g,二甲胺水溶液20ml,溶于100ml乙醇中,70℃反应6小时,加压除去溶剂,乙醇重结晶得到白色晶体C,收率99%。
取白色晶体C 10.0g溶于20ml THF中,在-10℃下缓慢加入95mmol(eq)的正丁基锂,搅拌1小时后缓慢加入含有4.0g浅黄色固体B的THF溶液,反应液加热回流10小时,冷却至室温,减压除去溶剂,加入20ml水,乙醚萃取3次,水层酸化至pH=2,乙酸乙酯萃取。水层调pH=7,除去溶剂得到产物D,收率30%。1H-NMR(DMSO-d6,400MHz),δH12.3(1H,br s),7.25-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.1,2.2Hz),6.78(1H,d,J=8.1Hz),5.65(1H,t,J=7.2Hz),5.12(2H,br s),3.56(2H,s),3.25(2H,t,J=7.7Hz),2.79(2H,q,J=7.4Hz),2.72(6H,br s);13C-NMR(CDCl3,100MHz),δC 173.4,154.3,145.0,141.4,133.9,132.3,131.1,129.6,128.2,128.2,127.7,127.3,126.4,123.3,119.7,69.9,55.9,42.2,42.2,40.0,24.9
取上步产物D 10.0g,溶于30ml甲醇中,加入3滴浓硫酸,室温反应过夜,加压蒸干溶剂,硅胶柱纯化,乙酸乙酯-石油醚系统纯化的油状产物E,收率72%。1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.67(1H,s),3.45(3H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 172.2,154.6,145.6,139.7,133.7,132.0,130.7,130.0,129.1,127.5,127.5,126.3,125.7,123.9,119.8,70.4,59.5,53.4,52.0,45.4,40.2,28.1.MS(ESI),m/z352.2[M+H]+.
取上步产物E 5.0g,溶于20ml水合肼中,室温搅拌10小时,乙酸乙酯萃取,饱和食盐水洗涤3次,无水硫酸钠干燥,过滤,除去溶剂,硅胶柱层析纯化,得终产物(即化合物2),收率77%。1H-NMR(CDCl3,400MHz),δH 7.51-7.16(4H,m),7.08(1H,d,J=2.2Hz),7.02(1H,dd,J=8.3,2.2Hz,1H),6.88(1H,d,J=8.3Hz),5.96(1H,t,J=7.2Hz),5.43(2H,brs,),3.63(2H,s),3.41(2H,m),2.34(2H,t,J=7.2Hz),2.23(6H,br s);13C-NMR(CDCl3,100MHz),δC 172.4,152.1,144.8,139.7,133.7,130.9,130.6,129.9,129.6,128.7,128.7,127.9,127.2,126.7,117.8,71.2,58.2,45.6,39.9,28.8.MS(ESI),m/z352.7[M+H]+。
实施例2化合物1的合成
化合物1的合成方法同实施例2,唯一的不同为将水合肼改为氨水,即得化合物1。收率:64%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.58(2H,br s),3.45(2H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz),δC173.6,154.8,145.5,139.6,133.6,132.2,130.9,130.0,129.2,127.6,127.5,126.6,126.3,124.2,120.3,70.4,59.4,45.4,45.4,42.5,28.3.MS(ESI),m/z337.8[M+H]+。
实施例3化合物8的合成
取中间体酸(即实施例1的产物D)5.0g,DIEA(5eq)溶于30ml DMF中,室温反应20min,再加入环丙胺1.0eq,HBTU 1.0eq,室温反应8小时。加入纯净水50ml,乙酸乙酯萃取两次,水洗3次,饱和食盐水洗3次,无水硫酸钠干燥,加压除去溶剂得粗品,石油醚-乙酸乙酯硅胶柱层析的纯品。收率,66%。1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.71(1H,t,J=7.2Hz),5.60(2H,br s),3.44(2H,s),2.64(1H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),0.71(2H,q,J=7.0Hz),0.39(2H,q,J=7.0Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.8,28.2,22.7,6.6,6.6.MS(ESI),m/z377.2[M+H]+。
实施例4化合物3的合成
化合物3的合成方法同实施例3,唯一的不同为将环丙胺改为间甲胺,即得化合物3。收率:65%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.49(2H,br s),3.48(2H,s),3.18(2H,q,J=6.9Hz),2.73(3H,d,J=4.8Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.8,28.3,26.5.MS(ESI),m/z351.2[M+H]+。
实施例5化合物4的合成
化合物4的合成方法同实施例3,唯一的不同为将环丙胺改为间乙胺,即得化合物4。收率:70%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.22(2H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.04(3H,t,J=7.2Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.4,45.3,42.9,34.5,28.2,14.8.MS(ESI),m/z365.1[M+H]+。
实施例6化合物5的合成
化合物5的合成方法同实施例3,唯一的不同为将环丙胺改为间二甲胺,即得化合物5。收率:55%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.18(2H,q,J=6.9Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.38(2H,m),1.24(2H,m),0.86(3H,t,J=7.2Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.9,39.4,31.6,28.2,20.0,13.8.MS(ESI),m/z393.4[M+H]+。
实施例7化合物6的合成
化合物6的合成方法同实施例3,唯一的不同为将环丙胺改为丙胺,即得化合物6。收率:85%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.15(2H,q,J=6.6Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.43(2H,m),0.83(3H,t,J=7.4Hz).13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.3,43.0,41.5,38.6,28.1,22.6,11.2.MS(ESI),m/z379.4[M+H]+。
实施例8化合物7的合成
化合物7的合成方法同实施例3,唯一的不同为将环丙胺改为异丙胺,即得化合物7。收率:70%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.37(2H,q,J=7.1Hz),3.29(2H,q,J=7.1Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.11(6H,t,J=7.1Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.3,43.0,41.5,38.6,28.1,22.6,22.6.MS(ESI),m/z379.2[M+H]+。
实施例9化合物9的合成
化合物9的合成方法同实施例3,唯一的不同为将环丙胺改为羟乙胺,即得化合物9。收率:67%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.58(2H,m),3.45(2H,s),3.30(2H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,60.9,59.6,45.4,45.4,43.0,41.4,28.5.MS(ESI),m/z381.3[M+H]+。
实施例10化合物10的合成
化合物10的合成方法同实施例3,唯一的不同为将环丙胺改为3-甲氧基丙胺,即得化合物10。收率:82%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.30(4H,m),3.02(3H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.66(2H,m).13C-NMR(CDCl3,100MHz)δC 171.3,154.7,145.7,139.6,133.5,132.4,130.9,130.2,129.1,127.5,127.5,126.6,126.3,124.2,120.1,71.8,70.4,59.4,58.5,45.4,45.4,43.0,38.5,28.8,28.1.MS(ESI),m/z409.3[M+H]+。
实施例11化合物11的合成
化合物11的合成方法同实施例3,唯一的不同为将环丙胺改为2-氨基噻唑,即得化合物11。收率:80%,1H-NMR(DMSO-d6,400MHz),δH 12.28(1H,s),7.46(1H,d,J=3.6Hz),7.20-7.40(4H,m),7.19(1H,d,J=3.6Hz),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.78(1H,d,J=8.3Hz),5.68(1H,t,J=7.2Hz),5.15(2H,br s),3.68(2H,s),2.46(2H,m),2.35(2H,t,J=7.2Hz),2.07(6H,br s);13C-NMR(DMSO-d6,100MHz)δC 169.8,158.5,154.4,145.7,139.1,138.1,134.0,132.2,131.8,130.5,129.6,128.2,127.9,127.3,126.3,123.8,119.7,113.9,69.9,59.1,45.3,45.3,41.1,27.9.MS(ESI),m/z422.1[M+H]+。
实施例12~15
化合物12~15的合成方法同实施例3,唯一不同为将环丙胺改为异丁胺、正丁胺、甲基乙基胺或二乙胺,得到化合物12~15。
试验例1化合物1~11的激酶抑制活性及体外细胞增殖抑制测试
本实验的目的是检测本发明的化合物对体外PARP酶抑制活性以及MDA-MB-436细胞增殖抑制活性,采用的方法分别为商业试剂盒及MTT法。
parp1酶活实验是利用Trevigen’s PARP1试剂盒(Trevigen,cat.no.4676-096-K)测得,通过GraphPad Prism5software(San Diego,CA,USA)计算得到IC50,结果见表1。
MDA-MB-436细胞增殖抑制实验结果见表1。
表1
表中,a:PARP-1的IC50是由4点剂量响应曲线计算的估算值。
b:MDA-MB-436的IC50是由4点剂量响应曲线计算的估算值。
c:n.d即not determinded,表示无法确定。
实验结果表明,化合物1~7、9、12、13对parp1具有较强的抑制活性以及在体外对MDA-MB-436细胞具有较强的增殖抑制活性。其中,Iniparib、Olaparib为现有的PARP抑制剂。
试验例2化合物2对多种肿瘤细胞的抗增值试验
采用化合物2做多种肿瘤细胞(包括MDA-MB-231,MDA-MB-468and MDA-MB-436细胞系)的抗增值活性试验,其结果见图1。其中,图1A为用不同浓度的化合物2处理MCF-7,MDA-MB-231,MDA-MB-436,MDA-MB-468细胞系,通过MTT测定了细胞的存活率。图1B为Hoechst 33258荧光染色检测细胞DNA损伤(Scale bar=200μm)。图1C为用不同浓度的化合物2处理MDA-MB-436细胞系24小时,通过流式分析测定的凋亡率。图1D为化合物2处理MDA-MB-436细胞,利用蛋白质印记法测定了Bax,Bcl-2,Caspase-3,PARP1和PAR的表达量。图1E为用化合物2处理MDA-MB-436细胞,测定其抑制细胞转移的能力(Scale bar=100μm)。
从图1中可以明显看出,化合物2对多种肿瘤细胞具有显著的抗增殖活性,包括MDA-MB-231,MDA-MB-468and MDA-MB-436等细胞系。特别是在BRCA1突变的MDA-MB-436细胞系中,其IC50为5.14μM.(Figure 4A)。通过Hoechst 33258染色在荧光显微镜下细胞发生凋亡的形态学特征(Figure 4B)。并且细胞凋亡率呈现显著的剂量依赖(Figure 4C)。
BRCA1功能缺失会导致基因组不稳定,从而导致通过同源重组完成的DNA修复缺陷。因此,BRCA1缺失的细胞DNA损伤率高,并且对parp依赖的DNA修复更加敏感。因此,我们检测了化合物2对parp1的抑制活性以及下游底物蛋白,如PAR。我们发现,化合物2可显著抑制PARP1的活性,并且PAR的表达量也显著降低(Figure 4D)。随后,我们检测了化合物2诱导细胞死亡的凋亡标志物。发现,化合物2上调Bax的表达以及下调Bcl-2的表达,caspase-3的活性形式是显著增加。此外,我们还发现,化合物2可抑制乳腺癌细胞迁移(Figure 4E)。这些结果表明,化合物2可以诱导细胞凋亡,通过抑制PARP1抑制BRCA1基因突变的乳腺癌细胞的迁移。
试验例3化合物2的体内抗肿瘤实验
本实验的目的是检测发明化合物的体内抗肿瘤效果。本实验采用模型,测试发明化合物2的体内抗肿瘤活性。所用细胞株为MDA-MB-436。
1.实验方法
将培养好的MDA-MB-436细胞经消化胰酶消化后,再用PBS液清洗2次,然后用1%台盼兰染色,于细胞记数板上进行细胞记数,并调节活细胞浓度至1×107/ml,在无菌条件下施行裸鼠右侧胸壁第二乳垫,脂肪层下接种0.2ml/只,共接种3只,术后荷瘤裸鼠继续饲养于SPF环境中,待肿瘤长至0.8cm3时,行裸鼠间原位移植。在无菌条件下取出乳腺癌组织,剪切成1mm3左右的小块,分别移植于24只裸鼠右侧胸壁的第二乳垫脂肪层下,4d后即可见肿瘤生长,成瘤率100%。裸鼠分组(n=6)并注射给药。对照组注射PBS,药物对照组注射Iniparib,用量为100mg/kg/d,实验组分别注射低剂量的化合物2,用量为12.5mg/kg/d),高剂量的化合物2,用量为25mg/kg/d,
肿瘤生长曲线:从第一次注射开始每隔2天,观察一次荷瘤裸鼠的全身情况及各组肿瘤生长情况,在无菌的条件下测量各组荷瘤裸鼠移植瘤的直径,并连续观察纪录。以时间为横坐标,肿瘤的体积为纵坐标,肿瘤体积计算公式为:V=1/2*ab2,a为肿瘤长径,b为短径,并据此绘制移植瘤生长曲线。
抑瘤率:注射药物28天后,脱颈处死各组裸鼠,用动物天平称取裸鼠体重,在分离各组瘤体,称取瘤重,计算抑瘤率。
观察指标:每3天观测一次裸鼠,观察有无腹泻,抽搐,皮疹,体重明显减轻等反应。2实验数据
实验测定的生长曲线如图2A~图2D,实验完毕后解剖所得肿瘤照片如图2E和图2F。其中,图2A为小鼠肿瘤相对体积(n=6)注射PBS,Iniparib(100mg/kg/d),化合物2,低剂量(12.5mg/kg/d),高剂量(25mg/kg/d),**,P<0.01。图2B为小鼠相对瘤重,注射PBS,Iniparib(100mg/kg/d),化合物2,低剂量(12.5mg/kg/d)高剂量(25mg/kg/d)*,P<0.05;***,P<0.001与对照组相比。图2C为.治疗期间的小鼠体重,**,P<0.01;与对照组相比。图2D为不同组小鼠的肝、脾、肾*,P<0.05;**,P<0.01;与对照组相比。图2E为Ki-67和PAR的免疫组化分析(Scalebar=200μm)。图2F为肿瘤离体组织的PARP,PAR和Caspase-3的蛋白印记分析。
实验结果表明,化合物2对MDA-MB-436细胞具有明显的体内生长抑制活性,在每天12.5mg/kg及以上剂量下,可以明显抑制肿瘤生长或完全消退肿瘤。给药过程中未发现裸鼠出现体重降低、皮疹、腹泻等不良反应,表明在测试剂量下,化合物2在给药剂量范围内毒性低。
- 还没有人留言评论。精彩留言会获得点赞!
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 一种杂环衍生物及使用该杂环衍生物的有机发光器件的制造方法与工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法