一种鸡溶菌酶和鸡β‑防御素7融合基因及其制备方法与流程
本发明涉及一种鸡溶菌酶和鸡β-防御素7融合基因及其制备方法,属于基因工程技术领域。
背景技术:
目前,由于抗生素滥用,导致耐药菌株猛增和食品安全危机等公共安全问题层出不穷,寻找高效、无残留、绿色环保的抗生素替代品已成研究热点。
溶菌酶主要存在于动植物的组织液和某些微生物体内,如鼻粘液、眼泪、唾液、卵蛋白、枯草杆菌培养物和某些蔬菜中。该酶能水解细菌的细胞壁中N-乙酰氨基葡萄糖和N-乙酰胞壁酸之间的β-1,4-糖苷键,故又称胞壁质酶,即N-乙酰胞壁质糖苷聚糖水解酶。其中对鸡蛋清溶菌酶的研究最清楚,它是由129个氨基酸残基构成的一种碱性蛋白,分子量约为14KD,对热稳定,对碱不稳定,对革兰氏阳性细菌有较强的杀菌作用。可药用,具抗菌、清除局部坏死组织、止血、消肿、消炎等作用。在食品工业上可用作防腐剂,还可添加在牙膏中作为防治龋齿的药用牙膏。在发酵工业上是一种重要的溶菌剂,用于分解细胞壁,制备无菌体提取液。
防御素是动物体内广泛存在的一类富含半胱氨酸的抗菌肽,对细菌、真菌、病毒等多种病原微生物均具有抑制作用。防御素抗菌机理独特,不易产生耐受性,是机体抵御外界病原微生物侵袭的重要武器。由于动物防御素具有抗菌谱广、不易产生耐药、安全性高等特点,现已成为抗菌药物研究的热点。研究表明,禽类防御素(Av BD)均属于β-防御素,具备广谱抗菌活性,有开发成抗菌药物的潜力。目前关于禽类防御素的研究较少,仅在鸡、鸭、鹅等常见家禽和少数几种野禽中发现了数十种防御素或防御素基因。
以往对溶菌酶、抗菌肽的研究多是针对单一物质。而冯瑄构建了抗菌肽B-人溶菌酶融合蛋白的表达载体;欧阳萍对人溶菌酶-抗菌肽进行了融合表达,并用于小鼠乳房炎的治疗试验,取得了较好的抗菌疗效,但将鸡溶菌酶-抗菌肽融合表达的研究,还未见报道。
技术实现要素:
本发明旨在为寻找新的抗生素替代品,提供一种鸡溶菌酶和鸡β-防御素7融合基因及其制备方法。
本发明为达到以上目的,是通过这样的技术方案来实现的:提供一种鸡溶菌酶和鸡β-防御素的融合基因,其核苷酸序列是SEQIDNO:1。
该融合基因的核苷酸序列中包含序列SEQIDNO:3、SEQIDNO:4和SEQIDNO:7。基因的5′端为SEQIDNO:3序列,来自于鸡溶菌酶基因,全长393bp。基因的3′端为SEQIDNO:4序列,来自于鸡β-防御素,全长132bp。SEQIDNO:3和SEQIDNO:4之间为连接肽编码序列SEQIDNO:7。
本发明还提供了上述融合基因的制备方法,包括以下步骤:
(1)设计扩增鸡溶菌酶和鸡β-防御素基因片段的PCR引物;
(2)在鸡溶菌酶基因下游引物和鸡β-防御素基因上游引物中加上一段连接肽序列,然后通过PCR反应扩增鸡溶菌酶和鸡β-防御素基因;
(3)根据基因重叠延伸技术,以步骤(2)所得的两个PCR反应产物的混合物为模版,以鸡溶菌酶基因上游引物和鸡β-防御素7基因下游引物为引物,分三步进行基因重叠延伸,获得融合基因。
本发明构建融合基因表达载体时所用的表达载体为pET32a。
本发明构建的融合基因工程菌将融合基因表达载体转入宿主制备而得。
本发明构建的融合基因工程菌为大肠杆菌。
本发明的融合基因表达载体的构建方法:步骤(2)通过设计恰当的引物,通过PCR延伸反应,使鸡溶菌酶基因的下游含有完整的连接子序列,而鸡β-防御素基因的上游含有部分的连接子序列。步骤(3)根据基因重叠延伸技术,以步骤(2)所得的两个PCR产物的混合物为模版,不加引物进行互为引物扩增,此时已获得融合基因。回收扩增产物作为进一步PCR反应的模板,以鸡溶菌酶基因的上游引物和鸡β-防御素基因的下游引物为引物进行扩增反应,获得更大量的融合基因产物。然后依次进行了目的片段割胶回收,并插入克隆载体或表达载体、质粒提取等步骤。
本发明还提供了上述融合基因表达载体的构建方法,包括以下步骤:
(1)以PCR扩增融合基因,得到融合基因扩增片段SEQIDNO:1;
(2)采用双酶切法,双酶切载体与扩增后的片段,并用T4连接酶将双酶切后的产物连接到质粒;
(3)将连接产物转化大肠杆菌获得阳性克隆子,提取质粒,测序验证后,得到所述融合基因的表达载体。
本发明同时提供了上述融合基因编码的融合蛋白,其氨基酸序列是SEQIDNO:2。
该融合蛋白的氨基酸序列SEQIDNO:2同时具有SEQIDNO:5和SEQIDNO:6中的氨基酸序列。
该融合蛋白同时具有鸡溶菌酶和鸡β-防御素的功能,同时该融合蛋白对金黄色葡萄球菌有明显的抑菌活性,其最小抑菌浓度低于4mg/ml。
本发明的设计原理:为了防止鸡溶菌酶或鸡β-防御素在宿主菌中表达成有活性的蛋白形式,而造成的宿主菌的自杀效应,设计连接肽将鸡溶菌酶和鸡β-防御素连接起来导入宿主细胞,将其融合表达,由于通过这种方式表达的产物蛋白没有活性,并且融合蛋白的分子量较大,在宿主细胞中大量表达会形成包涵体,有利于之后的蛋白分离纯化。
鸡溶菌酶和鸡β-防御素7,都是存在于动物体内的高效抗菌(抑菌)多肽,对多种微生物发挥抑杀作用,本身无残留,也不产生赖药性,作为抗菌药物、食品添加剂、防腐剂,发挥作用后还能增加食品的蛋白质含量。
具体实施方式
实施例1、PCR引物设计
在保留鸡溶菌酶基因和鸡β-防御素基因的功能区域基础上,设计PCR引物;本发明是由连接肽过渡连接的融合蛋白,因此在两基因的连接区引入连接肽编码序列(Linker)。
1、鸡溶菌酶基因引物:
提取鸡输卵管总RNA,反转录成cDNA,以其为模板,根据Genbank公布的鸡溶菌酶基因的序列,运用引物设计软件(primer5.0),设计上下游引物,P1和P2,用于扩增删除终止密码子的鸡溶菌酶基因。在上游引物中引入BamH I酶切位点序列,下游引物中加入linker序列。
上游引物序列如下,引物名称为P1:
5′-CGGGATCCGATGAAAGTCTT-3′(划线部分为BamH I酶切位点);
下游引物序列如下,引物名称为P2:
5′-linker-CAGCCGGCAGCCTCTG-3′
(linker为5′-tccacctgaaccacctccacctgaaccacctccacc-3′);
2、鸡β-防御素7基因引物:
提取鸡脊椎总RNA,反转录成cDNA,以其为模板,根据Genbank公布的鸡β-防御素7基因的序列,运用引物设计软件(primer5.0),设计上下游引物,P3和P4,用于扩增删除终止密码子的鸡β-防御素7基因。在下游引物中引入Hind III酶切位点序列,上游引物中加入linker序列。
上游引物序列如下,引物名称为P3:
5′-linker-AGGCCTATTGATACTTGTAGGCTT-3′
(linker为5′-ggtggaggtggttcaggtggaggtggttcaggtgga-3′);
下游引物序列如下,引物名称为P4:
5′-AGAAGCTTAGTTATCAGCTCCTCC-3′(划线部分为Hind III酶切位点)
3、鸡溶菌酶和鸡β-防御素7融合基因的基因引物:
以所述鸡溶菌酶基因和鸡β-防御素7基因的PCR扩增产物作为模板,以引物P1和P4为引物,进行PCR扩增,得到鸡溶菌酶和鸡β-防御素7融合基因。
实施例2、PCR扩增
1、扩增鸡溶菌酶基因片段:
此为PCR1,其反应体系为:含Lyz gene质粒0.5μl,上游引物(P1)0.5μl,下游引物(P2)0.5μl,2×HiFi-PCR Master12.5μl,加ddH2O至总体积25μl;扩增条件为:95℃变性5min后进入循环,95℃变性30s,57℃复性30s,72℃延伸30s,30个循环后在72℃延伸10min。
2、扩增鸡β-防御素7基因片段:
此为PCR2,其反应体系为:含β-Gal7gene质粒0.5μl,上游引物(P1)0.5μl,下游引物(P2)0.5μl,2×HiFi-PCR Master12.5μl,加ddH2O至总体积25μl;扩增条件为:95℃变性5min后进入循环,95℃变性30s,52℃复性30s,72℃延伸30s,30个循环后在72℃延伸10min。
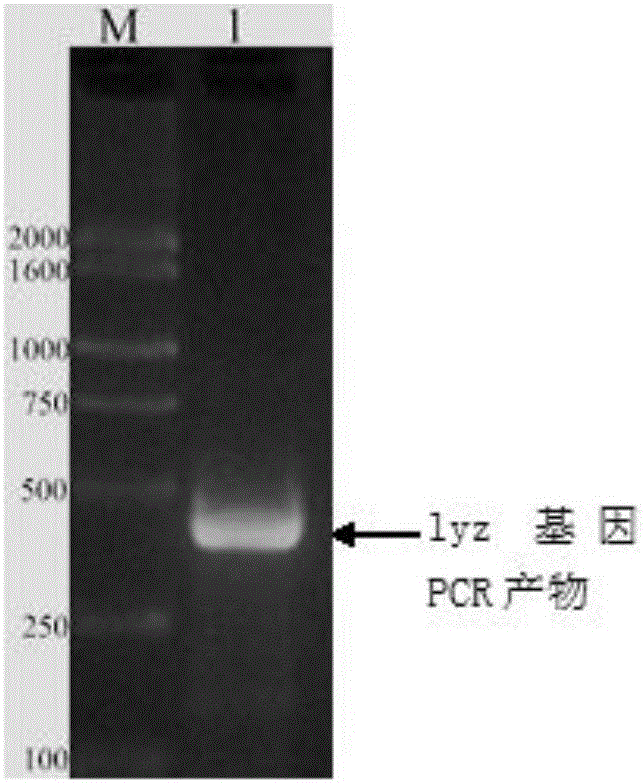
3、鸡溶菌酶基因和鸡β-防御素基因的PCR扩增结果
图1是鸡溶菌酶基因Lyz的PCR扩增结果图,图2是鸡β-防御素7基因β-Gal7的PCR扩增结果图,从图中可以看出鸡溶菌酶基因和鸡β-防御素基因的PCR产物长分别是393bp和132bp,和目的基因的大小一致。
实施例3、融合基因Lyz-β-Gal7的构建(重叠延伸PCR法)
首先,分别进行PCR1反应扩增鸡溶菌酶基因Lyz gene和PCR2反应扩增鸡抗菌肽基因β-Gal7gene,各取PCR产物25ul分别进行1.5%琼脂糖凝胶电泳,并用干净的手术刀切下在100bp至250bp之间、在250bp至500bp之间的目的片段凝胶,并用DNA胶回收试剂盒纯化回收凝胶中的DNA片段,取5~10ul回收溶液用分光光度计测OD,计算其中的基因分子浓度,分别取适当体积的Lyz gene回收液和β-Gal7gene回收液使两基因等量混合,进行PCR3反应构建和扩增Lyz-β-Gal7gene,此处PCR3反应的反应体系和反应条件如表1所示。最后对PCR3反应产物进行1.2%琼脂糖凝胶电泳和目的片段Lyz-β-Gal7gene胶回收纯化。
图3为融合基因Lyz-β-Gal7的PCR扩增结果图,从图中可以看出鸡溶菌酶和鸡β-防御素融合基因Lyz-β-Gal7的PCR产物为573bp,和目的基因的大小一致。
表1 PCR3构建和扩增Lyz-β-Gal7融合基因的体系和操作
实施例4、融合基因Lyz-β-Gal7的T载体克隆与扩增
Taq DNA聚合酶参与PCR反应延伸DNA链时在新生DNA链的3′端添加突出的A碱基,该PCR产物与T Vector 3′端突出的T碱基进行AT配对,再受到连接酶的催化,使目的基因与T载体形成环形质粒,可导入E.coli DH5α扩增。
含目的片段Lyz-β-Gal7gene的纯化PCR3产物10μl,T Vector2.0μl(约20ng),10×连接酶缓冲液1μl,T4DNA连接酶(浓度U/μl)2μl,50%PEG 4000 2μl,10×Ligation Buffer 2μl,ddH2O 2μl,总体积20μl。16℃连接12h,连接产物可导入E.coli DH5α扩增T-Lyz-β-Gal7或于-20℃冰箱保存。
图4为融合基因Lyz-β-Gal7与T Vector连后的质粒图谱。
实施例5、T载体连接与酶切
连接产物转化、感受态的制备、阳性克隆筛选、质粒DNA的提取均按文献所记载的现有技术进行。融合基因片段与和T载体连接并导入宿主菌DH5α后,筛选阳性重组质粒T-Lyz-β-Gal7;然后,提取质粒T-Lyz-β-Gal7,BamH I,Hind III双酶切,重组质粒Lyz-β-Gal7–T双酶切,酶切体系如下:10×MBuffer3.0μl,BamH I限制性内切酶(10U/μl)1.0μl,Hind III限制性内切酶(10U/μl)1.0μl,T–Lyz-β-Gal7重组克隆载体20.0μl,加ddH2O至总体积30μl,反应液混匀,37℃酶切过夜。电泳,割胶回收目的基因Lyz-β-Gal7。
图5为经氨苄青霉素抗性及α-互补筛选得到的阳性重组质粒的PCR扩增结果图。
实施例6、鸡溶菌酶-鸡β-防御素7融合基因表达载体的构建:
提取的Lyz-β-Gal7目的片段与BamH I,Hind III双酶切、纯化的质粒pET32a连接,构建表达载体,导入BL21(DE3),筛选阳性克隆。
目的基因和表达载体pET32a片段连接,连接反应体系如下:酶切载体DNA2.0μl,目的基因10.0μl,T4DNA连接酶2.0μl,50%PEG 40002.0μl,10×Ligation Buffer2.0μl,加ddH2O至总体积20μl,于37℃连接过夜。连接产物大肠杆菌BL21感受态细胞,筛选阳性克隆吧。
实施例7、融合表达产物纯化及活性检测
7.1融合表达产物纯化
取重组工程菌株pET32a/Lyz-β-Gal7/E.coli BL21单菌落或少量菌液接种于2支装有3ml含100ug/ml Amp的LB液体培养基的10ml试管中,37℃振荡培养13~15h活化。按1%取5ml活化菌液接种于500ml含100ug/ml Amp的LB液体培养基(可分装于多个锥形瓶),37℃180r/min培养3h,加终浓度为0.7mmol/L的IPTG诱导4h。
采用超声波破碎仪破碎菌悬液,条件设置为:工作时间5s,间隙时间6s,80个循环,冰浴中超声波破碎菌液20mL左右。对破碎上清液中的目的蛋白用镍柱进行分离纯化,纯化步骤如下:
(1)用50ml灭菌离心管收集菌液,用冰冷10ml PBS清洗菌液1次,离心。
(2)重悬于20ml 1×Bind Buffer,冰浴超声破碎,同实例7,至菌液不在粘稠,分装至10ml离心管。
(3)4℃12000r/min20min,收集上清,沉淀用5ml 1×Bind Buffer重悬,离心取上清。
(4)1ml Ni-NTA预装柱,先用10ml灭菌dH2O洗柱,控制流速0.5ml/min。
(5)10ml 1×Bind Buffer进行平衡,流速:0.5ml/min,检测流出液至A280nm=0为洗干净。
(6)将准备好的样品上样(pET28a-lyz-β-Gal7/BL21表达菌的破碎上清),上清加入柱内(结合时间1h-1.5h)流速控制在0.1-0.5ml/min左右,用一支10ml离心管收集上样后的流出液。
(7)10ml 1×Wash Buffer洗柱子,分管收集洗脱液10-15管,流速在0.5ml/min左右,至A280nm=0为止。
(8)5ml 1×Elution Buffer洗柱子(收集洗脱液5管)。
(9)收集洗脱峰,放入透析袋于4℃,对0.01M pH7.4PBS透析24h,期间更换6次透析液(每次更换一半,最后一次全部更换),透析结束,用PEG8000浓缩,12000rpm 4℃离心10min,收集上清,用于后续试验或-80℃保存。用10ml 1×Refresh Buffer洗柱子,直至A280nm=0为止,再用10ml1×Bind Buffer进行平衡,可接着作下一次纯化。
纯化后的目的蛋白含量测定:收集用镍柱分离纯化的Lyz-β-Gal7融合蛋白溶液,取100ul该蛋白液用镍柱洗脱液进行适当稀释,以镍柱洗脱液作参比,测OD260和OD280,粗略计算蛋白浓度C=1.45*OD280—0.74*OD260(mg/ml)。
图6为镍柱分离纯化前后超声破碎上清液的SDS-PAGE图,图中可见融合蛋白Lyz-β-Gal7条带。图7为超声破碎上清液用镍柱进行分离纯化后的SDS-PAGE图,图中可见杂条带基本消失。
7.2融合表达产物活性检测
蛋白活性检测(Lyz-β-Gal7融合蛋白对金黄色葡萄球菌的抑制):参考混菌琼脂平板打孔法,将固体培养基冷至50℃时,加入终密度为5×106个/ml细菌(乘以混合体积得出要加的菌体数量,菌体数量除以菌体密度得出体积,而菌体密度由比浊法测定),混匀后倒平板,厚度约0.5~1cm,凝固后用打孔器打两个孔,右边孔加入100ul 7mg/ml Lyz-β-Gal7融合蛋白(透明圈直径达到3.9cm);左边孔加入等体积的透析液PBS作对照。图8为Lyz-β-Gal7融合蛋白对金黄色葡萄球菌作用结果图。
探究Lyz-β-Gal7融合蛋白对金黄色葡萄球菌的最小抑制浓度:重复上述步骤,用打孔器打4个孔,孔1、孔2、孔3、孔4,取出圆孔内的琼脂块,分别加入100ul浓度分别为1mg/ml、2mg/ml、3mg/ml、4mg/ml的融合蛋白液。把加样完后的平板正向平放于装有少量水的盒子内,37℃培养18~24h。图9为不同浓度Lyz-β-Gal7融合蛋白对金黄色葡萄球菌作用结果图。
结果表明,本发明的融合蛋白对金黄色葡萄球菌有明显的抑菌活性,最小抑菌浓度低于4mg/ml,纯化后的蛋白电泳得到单一的、大小与理论分子量一致的目的条带。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 奶牛金黄色葡萄球菌乳房炎亚单位的疫苗及其制备方法和应用与流程
- 奶牛金黄色葡萄球菌乳房炎亚单位疫苗的组合物及其制备方法和应用与制造工艺
- 含水流动状组合物及其制造方法、以及溶菌酶加工品和/或其盐、及其制造方法与制造工艺
- 一种纳米材料/群体感应淬灭酶颗粒改性复合膜制备方法与制造工艺
- 一种检测泪液中乳铁蛋白的专用胶体金检测卡的制造方法与工艺
- 一种鸡溶菌酶和鸡β‑防御素7融合基因及其制备方法与制造工艺
- 一种制备治疗敏感菌感染性疾病的药物盐酸头孢他美酯组合物的方法与制造工艺
- 含有复合益生元溶菌酶功能性无糖软糖及其制备方法与制造工艺
- 运用核酸适配体与溶菌酶特异性识别并形成G四链体进行荧光显现潜指纹的方法与制造工艺
- 用于制备结晶DTPMP的方法与制造工艺