一种胞嘧啶甲基化挖掘的方法与流程
本发明涉及生物信息
技术领域:
,尤其涉及高通量测序序列的甲基化挖掘的方法和系统。
背景技术:
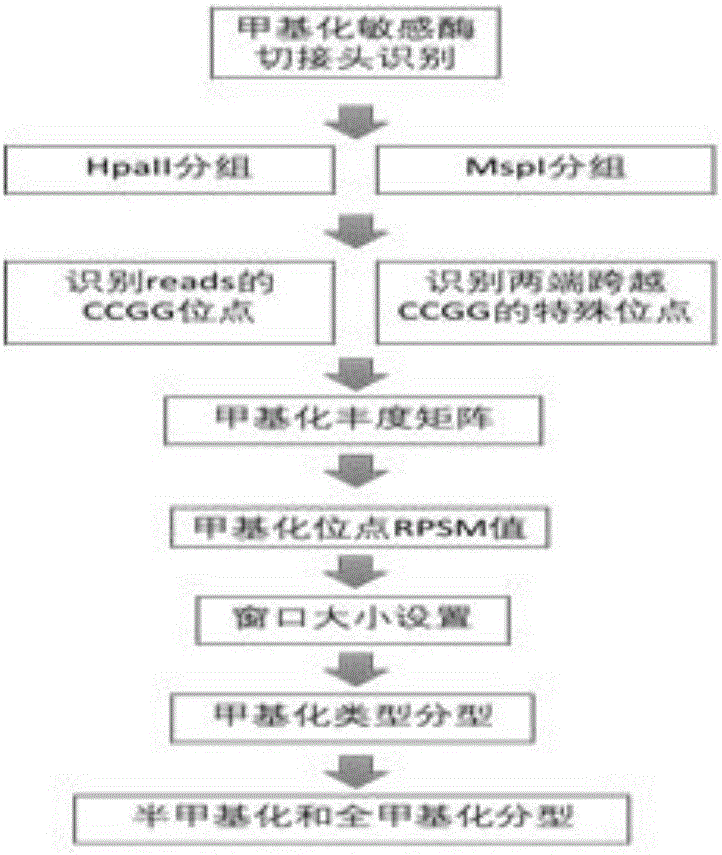
:亚硫酸氢钠测序法(bisulfitegenomicsequencing)是建立在MSP基础上进一步深入研究CpG岛各个位点甲基化情况的方法。重亚硫酸盐使DNA中未发生甲基化的胞嘧啶脱氨基转变成尿嘧啶,而甲基化的胞嘧啶保持不变,进行PCR扩增(引物设计时尽量避免有CpG,以免受甲基化因素的影响)所需片段,则尿嘧啶全部转化成胸腺嘧啶。最后,对PCR产物进行测序,并且与未经处理的序列比较,判断是否CpG位点发生甲基化。此方法虽是一种可靠性及精确度很高的方法,能明确目的片段中每一个CpG位点的甲基化状态。在寻找有意义的关键性CpG位点上,有其他方法无法比拟的优点。测序法以CpG岛两侧不含CpG点的一段序列为引物配对区,所以能够同时扩增出甲基化和非甲基化靶序列。它的不足是耗费时间和耗资过多,至少要测序10个以上的克隆才能获得可靠数据,需要大量的克隆及质粒提取测序,过程较为繁琐、昂贵。甲基化敏感扩增多态性(methylationsensitiveamplificationpolymorphism,MSAP)技术由Reyna-lópez等报道(Reyna-Lópezetal.1997),并被用于检测双相型真菌的DNA甲基化,它是在扩增片段长度多态性(amplifiedfragmentlengthpolymorphism,AFLP)技术的基础上建立起来的。其基本程序是:提取高质量基因组DNA,分别用EcoRI/HpaII,EcoRI/MspI两组酶组合对基因组DNA进行双酶切,并连上相应的限制性内切酶的接头,然后以接头序列设计的预扩增引物,进行PCR扩增。扩增产物稀释后,再加入带有选择性碱基的引物,进行第二次PCR扩增,扩增产物变性后在6%的序列胶上电泳,最后采用银染或同位素放射自显影方法处理序列胶,统计和分析DNA条带。这种方法在研究动植物基因组甲基化上有广泛应用(ShaAHetal.2005)。MSAP技术相对其他测定DNA甲基化程度的技术有如下优点:(1)不需知道被测DNA的序列信息,在不同生物上具有通用性,可用于DNA序列背景知识未知的生物。(2)操作相对简便,在AFLP技术体系的基础无需改进,即可操作。(3)可在全基因组范围检测CCGG位点的胞嘧啶甲基化变化。技术实现要素:本发明就着高通量测序技术的发展,所有开发的基于AFSM测序技术以及同类甲基化敏感位点限制性选择内切酶,高通量的分析甲基化多态性的研究方法进行甲基化数据分型。具体的技术方案为:一种胞嘧啶甲基化挖掘的方法,包括步骤:A)、目标数据获得:对同一来源的基因组由HpaII与MspI甲基化敏感酶切后扩增通过高通量测序;B)通过AFSM技术建库:对同一样品构建HpaII与MpaI酶切DNA文库,分别加上barcode接头序列;C)根据测序原始数据判读酶切位点:通过识别HpaII和MspIbarcode接头序列碱基进行区分HpaII和MspI酶切文库;将识别出的数据标定出酶切编号与样品编号,同时屏蔽标签序列;D)将标记后的原始数据进行图谱分析和组装,产出.bam比对文件;E)对bam文件进行甲基化数据挖掘。进一步的,所述B)步骤barcode接头序列为HpaIIGTCATGCCTCATCTCA,MspIGTCATGCCTCATTAGT。进一步的,所述D)步骤组装为全部数据分成两组进行组装;选择短序列拼接方法,使用Bowtie2软件进行比对到已知参考基因组或者使用Trinity软件进行重头组装再使用Bowtie2软件比对到从头组装的参考基因组;产出.bam比对文件,及样本间变异SNP和Indel。更为具体的,目标数据获得:限制性内切酶对DNA甲基化敏感性不同,因而相同序列就可扩增出不同的带型,以此判断DNA甲基化的程度。同裂酶为HpaⅡ和MspⅠ,这两个酶识别相同的酶切位点CCGG(真核生物中主要的甲基化位点),但对甲基化敏感程度不同,HpaⅡ对于DNA两条链上的该位点内外侧胞嘧啶均甲基化及任一个胞嘧啶甲基化都不能酶切,即不能酶切含mCCGG,CmCGG和mCmCGG的位点,但它可以识别仅一条链上胞嘧啶甲基化的位点。而MSPⅠ可以识别DNA单链或双链上该位点内侧甲基化的胞嘧啶,但不识别外侧甲基化胞嘧啶,即不能酶切mCCGG的位点。所以同一来源的DNA基因组酶切产物测序数据分析后,如果在MspⅠ酶切扩增产物中含有CCGG序列(CCGG位点无法酶切),同时在HpaⅡ酶切扩增产物中没有CCGG序列(CCGG位点被酶切),则说明该位点发生了单链外侧的胞嘧啶甲基化,即mCCGG,如果HpaⅡ酶切扩增产物中含有CCGG序列,同时在MspⅠ酶切扩增产物中没有CCGG序列,则说明该位点发生了双链CCGG位点的内侧胞嘧啶甲基化,即CmCGG。两者扩增产物中都含有CCGG序列,说明该位点发生双链全甲基化。限制性内切酶单核苷酸多态性与甲基化(Amplified-fragmentSinglenucleotidepolymorphismandMethylation,AFSM)是基于测序技术(Xiaetal.,2014)一个简单的,快速和低成本有效的系统,已经用于在非模式生物的测序。,该技术结合RAD原理与MSAP技术,创新性开发一种新的随机扩增序列SNP多态性及基因标记方法AFSM(AmplifiedFragmentSNPandMethylation)。可以同时进行基因组SNP检测和甲基化多态性检测。本发明是第一个将高通量测序的甲基化敏感酶酶切的胞嘧啶甲基化数据进行甲基化分型和识别甲基化位点丰度的技术。该方案也是目前唯一一个对甲基化敏感酶酶切的识别处理方案。对科研人员进行挖掘甲基化信息提供了简单,高效,低成本的方案。极大的加速了表观遗传学研究发展。附图说明图1为甲基化分型软件流程示意图;图2为甲基化分型核心模块示意图;图3针对单端测序和双端测序甲基化数据分型通用方案示意图;图4为针对双端测序甲基化数据分型特殊方案示意图;图5为AFSM技术分析流程示意图。具体实施方式下面结合附图和具体实施例对本发明做进一步详细说明。本发明甲基化分型软件流程主要步骤如图1所示,甲基化分型核心模块如图2所示:Step1、来源的高通量测序原始reads,利用barcode分割模块将reads按照barcode分割为多个样品reads。Step2、利用barcode分割模块将read分为HpaII与MspI两个甲基化库;。Step3、对所有barcode处理好的reads混合,通过基于bowtie2将reads都map到参考基因组,进行组装步骤。Step3、将组装后的数据(.bam格式)进行样品间的变异检测。Step4、对样品进行SNP,Indels,SVs挖掘。Step5、组装后的数据(.bam格式),进行甲基化分型,对甲基化位点识别。Step6、单个样品与单个位点的甲基化识别Step7、设置识别位点的窗口大小,默认值为5,以排除map导致的假阳性结果。Step8、对单个位点的reads数统计,产生丰度矩阵。Step9、计算单位点甲基化RPSM值,作为识别位点与样本的甲基化程度。Step10、识别单个位点的甲基化数据,对两个库的探讨(包括缺失数据讨论)一共可以产生多种数据情况。Step11、对单个位点的甲基化分型,将多种数据情况分类为半甲基化与全甲基化两类。1.针对单端测序和双端测序甲基化数据挖掘通用方案(图3)单个样品所有片段识别“CCGG”位点,同时标记“CCGG”位点中以“CCGG”第二个C为标记在参考基因组中位置。判断该位点的reads数量和酶切信息(来源reads编码的HpaII和MspI)。将单个位点信息分型为HpaII,MspI两个库包含原始reads数的丰度信息。同时标记CCGG在原始reads中位置。通过编写程序定义该位点的甲基化类型和丰度。判断甲基化类型参考表1,如图3中a类序列甲基化类型为mCCGG,b类序列甲基化类型为CmCGG,c类序列甲基化类型为mCCGG。同时计算两个甲基化酶切库map上的的平均reads数。计算该位点的甲基化位点丰度信息。甲基化位点丰度信息定义为RPSM,每1百万个map上的reads中map到甲基化位点的单个碱基上的reads个数(ReadsPeraSinglebaseofDNAmethylationmodelperMillionmappedreads)。RPSM=(totalreads)/(mappedreads(Million)×1bp)2.针对双端测序甲基化数据挖掘特殊方案(图4):对于双端测序两个酶切库(HpaII,MspI)中reads的“序列信息”无法同时含有CCGG位点,但是其双端位置正好越过了此CCGG位点。我们认为此位点为无法被该甲基化酶切。对于这样情况按照图4进行区分甲基化类型,a类序列甲基化类型为CmCGG,b类系列甲基化类型为mCCGG。同时计算两个甲基化酶切库map上的的平均reads数。计算该位点的甲基化位点丰度信息RPSM。程序参数设置:对于CCGG位点中出现的组装错误和CCGG之间距离较短可能出现的识别错误排除。我们设置了识别序列识别的窗口大小。窗口大小为两个CCGG之间的距离,默认值为5bp碱基。使用者可以根据自己物种复杂程度进行设置。对于识别的甲基化位点,下一步可以根据特征进一步分为半甲基化和全甲基化。也可将丰度信息对位点进行可视化展示。最后得到样品间的基因型数据,分型好的样品甲基化矩阵,与单位点的甲基化程度(RPSM)矩阵。用于后续全基因组甲基化分析,甲基化变异,半甲基化全甲基化的深度讨论。本发明分为reads分库模块,甲基化识别模块。reads分库模块将区分甲基化酶切混合库与样本库,判读barcode序列而达区分不同库的目的,与同类bacode软件相比,基于blast的内核,能够判读大于10bp以上序列,同时具备排除测序错误能力,区分双端barcode,以区分甲基化混合库。其他同类软件,无法同时完成双端barcode的数据,无法区分较长的甲基化barcode接头。甲基化识别模块中构架了甲基化识别基础原理和reads各类情况判定处理甲基化识别机制。对在全基因组水平和部分测序水平,大量精确的挖掘甲基化敏感酶酶切的胞嘧啶甲基化提供理论基础和实施方案。如图5所示,AFSM实验技术包括以下步骤:原始数据处理AFSM实验技术为两端测序,两端加上不同的Barcodes,对EcoRI端进行样品区分,对HpaII和MspI端进行甲基化区分。将每个reads进行标注,过滤后保留双端为同时具有HpaII/EcoRI,MspI/EcoRI的reads。数据组装将过滤后的reads进行组装,使用Bowtie2工具map到参考基因组上。生成BAM文件。同时过滤数据。SNP分型使用mPileup进行样品间的SNP挖掘工作,产出样品SNP矩阵。甲基化分型将含有甲基化标记的BAM文件,使用自己编写的甲基化分型软件进行分型(根据本专利甲基化分型原理编译),同时定位到单个样品上。统计甲基化位点。表1为胞嘧啶甲基化类型区分CmCGGmCmCGGmCCGGCmCGGmCmCGGmCCGGH(√)M(√)H(√)M(x)H(√)M(x)H(x)M(√)H(x)M(x)H(x)M(x)表2为AFSM实例中甲基化位点reads统计本发明提供的甲基化敏感酶酶切测序序列分型的实现方法,数据来源于AFSM技术或者由HpaII与MspI甲基化敏感酶切后扩增通过高通量测序得到的原始reads。AFSM技术通过混合选择性双酶切扩增(EcorI分别与MspI、HpaII组合),简化基因组复杂度,同时进行甲基化敏感位点的区分。在接头处设计了96个5碱基序列识别标签,以便使用Hiseq2000高通量测序技术混合测序后对每个样品进行区分。通过AFSM技术建库,对同一样品构建HpaII与MpaI酶切DNA文库,分别加上barcode接头序列HpaIIGTCATGCCTCATCTCA,MspIGTCATGCCTCATTAGT。混合HpaII和MspI文库。程序根据测序原始reads数据判读酶切位点,通过识别HpaII和MspIbarcode接头序列碱基进行区分HpaII和MspI酶切文库。将识别出的reads标定出酶切编号与样品编号。同时屏蔽标签序列。将标记后的原始数据进行map和组装:全部数据分成两组进行组装。选择短序列拼接方法,使用Bowtie2软件进行比对到已知参考基因组或者使用Trinity软件进行重头组装再使用Bowtie2软件比对到从头组装的参考基因组。产出.bam比对文件,及样本间变异SNP和Indel。将map后的bam文件进行甲基化数据挖掘。本发明分为reads分库模块,甲基化识别模块。reads分库模块将区分甲基化酶切混合库与样本库,判读barcode序列而达区分不同库的目的,与同类bacode软件相比,基于blast的内核,能够判读大于10bp以上序列,同时具备排除测序错误能力,区分双端barcode,以区分甲基化混合库。其他同类软件,无法同时完成双端barcode的数据,无法区分较长的甲基化barcode接头。甲基化识别模块中构架了甲基化识别基础原理和reads各类情况判定处理甲基化识别机制。对在全基因组水平和部分测序水平,大量精确的挖掘甲基化敏感酶酶切的胞嘧啶甲基化提供理论基础和实施方案。除非另有说明,否则在这些实施中阐述的部件和步骤的相对布置、数字表达式和数值不构成对发明的限制。对于本领域普通技术人员已知的技术、方法和设备可能不做详细讨论,但在适当情况下,技术、方法和设备应当被视为本说明的一部分。序列表:<110>中国热带农业科学院热带生物技术研究所<120>一种胞嘧啶甲基化挖掘的方法<160>2<210>1<211>16<212>DNA<213>人工序列<220><221><222>(1)...(16)<223><400>1GTCATGCCTCATCTCA<210>2<211>16<212>DNA<213>人工序列<400>2>GTCATGCCTCATTAGT当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1