磺胺类兽药亲水性磁性分子印迹材料的制备方法及应用与流程
本发明属于有机物检测领域,具体涉及一种磺胺类药物分子印迹材料的制备方法,还涉及上述的磺胺类药物分子印迹材料聚合物在磺胺类兽药检测中的应用。
背景技术:
磺胺类药物是最早应用于全身神经系统的抗生素类药物,它的诞生为抗生素药物的在医药领域的发展铺垫了道路。因为磺胺类药物具有廉价和高效的优点,进而被广泛的应用于医药、畜牧业、食品等领域。然而,随着磺胺类药物在很多领域的广泛应用,其滥用现象也随之出现,它的潜在致癌性和抗药性也逐渐显现出来,人们越来越担心其自身的健康会因为磺胺类药物的滥用而受到危害。与此同时,一旦磺胺类药物流入到环境中,它们就会进入饮用水系统,并被土壤吸收,引发更严重的问题。因此,为了保证食品的安全和人们的健康,许多国家和组织对磺胺类药物的最大残留限量做了相关规定。比如,我国和欧盟等国家规定,动物性食品和组织中磺胺总量或者单种磺胺类药物的最大残留限量为100μg/kg。
目前,样品前处理及检测药物残留的简单、快速、经济的方法有很多种,比如固相萃取、液相色谱与低分辨率的串联质谱分析、高分辨率质谱检测、气相色谱法、毛细管电泳法等。在理化法中,最常用的为高效液相色谱法,其主要适用于分析挥发性、半挥发性及热不稳定性物质,目前已经成为磺胺类药物定性、定量分析的有效方法。
分子印迹技术是一种有效合成具有高效性、亲和性、特异性聚合物的方法,合成的聚合物可以模仿自然识别实体,高效的吸附目标产物。分子印迹技术与固相萃取技术联用,使样品的前处理过程变得简单、劳动力损耗降低,溶剂利用率提高,并且装置可循环利用。但是,普通的分子印迹聚合物材料,表面活性低,很难与样品进行分离。
磁性颗粒被应用于基质分散固相萃取技术中,它们因为具有了磁性,所以对于合成材料在检测样品中的分离具有快速、高效的特点。随着磁性颗粒与分子印迹聚合物的结合,一项新的技术“磁性分子印迹”技术,得到了高度的重视和利用。然而,由于材料缺乏亲水性,限制了其在含水量高的牛奶,蜂蜜等样品检测中的应用。所以,研究一种可以在水相中快速、方便富集多种磺胺类兽药的磁性分子印迹材料具有较深远的意义。
CN 105085843 A披露了一种分子印迹材料的制备方法及由其制备的分子印迹材料,在制备方法中,首先采用共沉淀法在类石墨相氮化碳表面沉积Fe3O4得到Fe3O4-g-C3N4纳米材料,进而得到表面修饰有丙烯基的磁性二氧化硅类石墨相氮化碳纳米材料,然后以其为载体,通过优化功能单体,采用表面印迹技术制备得到能同时快速识别七种环境内分泌干扰物的分子印迹材料。该分子印迹材料吸附容量大、选择性好、响应快、化学稳定性好、便于操作、可重复利用率高,可同时实现多种痕量类固醇类和酚类环境内分泌干扰物的识别和分离富集,扩展了现有磁性纳米材料、类石墨相氮化碳材料、分子印迹材料的应用范围。
上述方法制备的分子印迹材料虽然具有附容量大、选择性好、响应快和化学稳定性好的有点,但是由于分子印迹材料具备生物抗体的特点,所以只能对特定的目标物(多种痕量类固醇类和酚类环境内分泌干扰物)具有选择性的吸附性能,而对其他物质(如磺胺类兽药)的吸附性差。同时,该方法是以甲基丙烯酸和2-乙烯基吡啶采用有机聚合的方法制备分子印迹聚合物,限制了其在含水相样品基质(如动物源性食品)中的应用。
技术实现要素:
为解决上述的技术问题,本发明提供了一种可以在水相中快速、方便富集多种磺胺类兽药的磁性分子印迹材料的制备方法。
另外,本发明针对食品中农兽药残留的问题,将上述的制备所得的印迹材料应用于磺胺类兽药检测中,建立了快速便捷且准确率较高的的检测方法。
采用本发明的方法制备得到的印迹材料,在动物源性食品中对磺胺二甲基嘧啶、磺胺嘧啶、磺胺甲基嘧啶、磺胺噻唑等其他磺胺类兽药检测的应用,也是本发明所要保护的范围。
磺胺类兽药亲水性磁性分子印迹聚合物材料的制备方法,其步骤如下:
(1)采用共沉淀法制备Fe3O4粒子;
(2)采用stober法制备硅烷化的Fe3O4,得表面包覆有SiO2的Fe3O4磁性微球;
(3)制备磺胺类分子印迹聚合物聚合物:
a.将模板分子溶解在乙腈中,在氮气保护下,混合均匀;
b.在a中加入功能单体,搅拌均匀;
c.在b中加入交联剂,搅拌均匀;
d在c中加入磁性微球,搅拌均匀;
e.在d中加入引发剂,搅拌,热引发,得聚合物。
具体的,步骤(1)为:
将FeSO4.7H2O与FeCl3.6H2O混合,加入去离子水,搅拌至其溶解,随后在氮气保护下升温,加入氨水,继续加热搅拌反应;
反应结束后,利用外加磁场将所得的黑色产物与上清液分离,并用去离子水与无水乙醇反复洗涤黑色产物直至洗液呈中性,将黑色产物真空干燥至恒重,所得黑色产物Fe3O4粒子研磨后备用;
步骤(2)为:
在步骤(1)中合成的Fe3O4粒子中加入正硅酸乙酯,搅拌混匀,然后在氮气的保护下加入氨水,继续搅拌反应,紧接着水浴,对Fe3O4粒子进行表面改性;
用磁铁收集反应产物,并用去离子水与无水乙醇反复冲洗反应产物直至洗液呈中性,将反应产物真空干燥,制得表面包覆有SiO2的Fe3O4磁性微球。
更具体的,步骤(1)为:
将2.5g FeSO4.7H2O与4.0g FeCl3.6H2O置于锥形瓶中,加入200mL的去离子水,搅拌至其溶解,随后在氮气保护下升温至60℃,加入5mL氨水,继续加热搅拌反应1h;
反应结束后,利用超强磁铁的外加磁场将黑色产物与上清液分离,并用去离子水与无水乙醇反复洗涤黑色产物直至洗液呈中性,将黑色产物置于真空干燥箱中于60℃下干燥至恒重,所得黑色产物Fe3O4粒子经研磨后备用;
步骤(2)为:
取步骤(1)中合成的Fe3O4粒子倒入盛有水和无水乙醇的圆底烧瓶中,加入正硅酸乙酯,搅拌混匀4min,然后在氮气的保护下加入5mL氨水,继续搅拌反应3h,紧接着在50℃水浴1h,对Fe3O4粒子进行表面改性;
用磁铁收集反应产物,并用去离子水与无水乙醇反复冲洗反应产物直至洗液呈中性,将反应产物置于真空干燥箱中于60℃下干燥,制得表面包覆有SiO2的磁性微球;
Fe3O4粒子的质量与正硅酸乙酯的体积比为1g:4mL,水与无水乙醇的体积比为1:4。
步骤(3)为:
a.以磺胺二甲基嘧啶作为模板分子,将其溶解在乙腈中,用氮气将体系中的氧气置换出来,并超声波处理,混合均匀;
b.在a中加入功能单体,搅拌4小时;
c.在b中加入交联剂;
其中模板分子与功能单体、交联剂的比例为:0.1mmol:6mmol:6mmol;
乙腈:模板分子+功能单体+交联剂组成的溶剂=6ml:12.1mmol;
d.在c中加入表面包覆有SiO2的磁性微球,搅拌均匀;
表面包覆有SiO2的磁性微球与乙腈的质量体积比为:1g:20ml;
e.在d中加入引发剂,引发剂与表面包覆有SiO2的磁性微球的质量比为1:12,机械搅拌,热引发,得磁性磺胺分子印迹聚合物。
更具体的,步骤(3)为:
a.以磺胺二甲基嘧啶作为模板分子,将其溶解在乙腈中,用氮气将体系中的氧气置换出来,并超声波处理15min,混合均匀;
b.在a中加入摩尔比为1:1的功能单体甲基丙烯酸和甲基丙烯酸羟乙酯,搅拌4小时;
c.在b中加入摩尔比为1:1的交联剂乙二醇二甲基丙烯酸酯和3-(三甲氧基甲硅烷基)丙基丙烯酸酯;
其中模板分子与功能单体、交联剂的比例为:0.1mmol:6mmol:6mmol;
乙腈:模板分子+功能单体+交联剂组成的溶剂=6ml:12.1mmol;
d.在c中加入表面包覆有SiO2的磁性微球,搅拌均匀;
表面包覆有SiO2的磁性微球与乙腈的质量体积比为:1g:20ml;
e.在d中加入引发剂偶氮二异丁腈,偶氮二异丁腈与表面包覆有SiO2的磁性微球的质量比为1:12,机械搅拌,并于50℃水浴5h,60℃水浴24h,85℃水浴2h;得磁性磺胺分子印迹聚合物。
更进一步的,可以将磁性磺胺分子印迹聚合物在真空烘箱中100℃老化8h,研磨,定性滤纸进行包裹,用索氏提取器萃取50h-110h,萃取溶剂为甲醇,80℃烘干至恒重,得到磁性印迹聚合物颗粒。
磺胺类兽药亲水性磁性分子印迹材料的制备方法,包括下述的步骤:
(1)将2.5g FeSO4.7H2O与4.0g FeCl3.6H2O置于锥形瓶中,加入200mL的去离子水,搅拌至其溶解,随后在氮气保护下升温至60℃,加入5mL氨水,继续加热搅拌反应1h;
反应结束后,利用超强磁铁的外加磁场将黑色产物与上清液分离,并用去离子水与无水乙醇反复洗涤黑色产物直至洗液呈中性,将黑色产物置于真空干燥箱中于60℃下干燥至恒重,所得黑色产物Fe3O4粒子经研磨后备用;
(2)取步骤(1)中合成的Fe3O4粒子倒入盛有水和无水乙醇的圆底烧瓶中,加入正硅酸乙酯,搅拌混匀4min,然后在氮气的保护下加入5mL氨水,继续搅拌反应3h,紧接着在50℃水浴1h,对Fe3O4粒子进行表面改性;
用磁铁收集反应产物,并用去离子水与无水乙醇反复冲洗反应产物直至洗液呈中性,将反应产物置于真空干燥箱中于60℃下干燥,制得表面包覆有SiO2的磁性微球;
Fe3O4粒子的质量与正硅酸乙酯的体积比为1g:4mL,水与无水乙醇的体积比为1:4;
(3)a.以磺胺二甲基嘧啶作为模板分子,将其溶解在乙腈中,用氮气将体系中的氧气置换出来,并超声波处理15min,混合均匀;
b.在a中加入摩尔比为1:1的功能单体甲基丙烯酸和甲基丙烯酸羟乙酯,搅拌4小时;
c.在b中加入摩尔比为1:1的交联剂乙二醇二甲基丙烯酸酯和3-(三甲氧基甲硅烷基)丙基丙烯酸酯;
其中模板分子与功能单体、交联剂的比例为:0.1mmol:6mmol:6mmol;
乙腈:模板分子+功能单体+交联剂组成的溶剂=6ml:12.1mmol;
d.在c中加入表面包覆有SiO2的磁性微球,搅拌均匀;
表面包覆有SiO2的磁性微球与乙腈的质量体积比为:1g:20ml;
e.在d中加入引发剂偶氮二异丁腈,偶氮二异丁腈与表面包覆有SiO2的磁性微球的质量比为1:12,机械搅拌,并于50℃水浴5h,60℃水浴24h,85℃水浴2h;得磁性磺胺分子印迹聚合物。
本发明的实验证明,采用本发明制备所得的磁性分子印迹聚合物材料可以直接用于动物源性食品中磺胺二甲基嘧啶、磺胺嘧啶、磺胺甲基嘧啶、磺胺噻唑、磺胺多辛、磺胺对甲氧嘧啶、磺胺甲基恶唑、磺胺甲氧嗪磺胺异噁唑和磺胺氯哒嗪十种磺胺类兽药的检测分析,其对磺胺类药物具有高选择性吸附和高效富集。
按以上的方法印迹材料在检测磺胺类兽药中的应用,包括下述的步骤:
(1)活化:取按权利要求1的方法制备所得的磺胺磁性分子印迹聚合物颗粒加入到锥形瓶中,依次采用甲醇与去离子水进行活化处理,利用外加磁场分离两相,倒掉废液,得到活化好的磁性分子印迹聚合物;
其中磺胺磁性分子印迹聚合物的质量与甲醇的质量体积与比为1g:40mL,甲醇与去离子水的体积比为1:1;
(2)萃取:将待检测的样品溶液或样品萃取溶液加入到锥形瓶中,与磺胺磁性分子印迹聚合物颗粒一同置于振荡器上振荡混合,此时存在样品中的痕量目标物先从样品相转移至溶液相,再被吸附到磁性分子印迹聚合物表面,其中样品的质量与溶液的体积比为1g:15mL;
样品萃取溶液,以甲醇或乙腈作为萃取溶剂;
(3)分离:萃取完成后,在锥形瓶外壁放置磁铁,利用外加磁场的作用将磺胺磁性分子印迹聚合物与溶液分离,倒掉废液;
(4)淋洗:向装有磁性分子印迹聚合物的锥形瓶中加入淋洗液,振荡混合,洗下杂质,再次利用磁场分离两相,倒掉废液,除去杂质,其中磁性分子印迹聚合物的质量与淋洗液的体积比为1g:50mL;
淋洗液为去离子水或甲醇;
(5)洗脱:向锥形瓶中加入2mL洗脱剂洗脱磺胺磁性分子印迹聚合物,充分振荡混合,将吸附在磁性分子印迹聚合物表面的待测目标物洗脱下来,然后利用外加磁场分离两相,收集洗脱液;将分离的洗脱液于50℃条件下用氮气吹干,并用1.0mL甲醇重新溶解,经0.45μm有机系滤膜过滤后注入高效液相色谱检测分析。
本发明的有益效果在于,采用本发明的方法所制备的亲水性的磁性分子印迹材料具有可在含水相食品样品中对10种磺胺类兽药(动物源性食品中磺胺二甲基嘧啶、磺胺嘧啶、磺胺甲基嘧啶、磺胺噻唑、磺胺多辛、磺胺对甲氧嘧啶、磺胺甲基恶唑、磺胺甲氧嗪磺胺异噁唑和磺胺氯哒嗪)快速的吸附能力、强大的吸附功能、较强的特异性、回收率高和较低的制备成本,同时改善了传统固相萃取样品前处理技术的所需溶剂量大和耗时的缺点。通过比较亲水性和非亲水性磁性材料在不同含水比例的溶剂中对模板分子的吸附性能发现,非亲水性聚合物在含水量较高的溶剂(60%以上)中对模板分子水溶液的吸附量为0,在乙腈溶液中的吸附量为0.22mg/g。亲水性聚合物在水相环境中的吸附量为0.98mg/g,在乙腈中的吸附量为0.10。说明该亲水性聚合物在水相环境中的吸附量大于非亲水性聚合物在乙腈溶液中的吸附量。
此方法准确,灵敏度高,检测速度快,效率高,成本低,磁性材料可以多次循环使用,解决了样品前处理复杂的问题以及商品化固相萃取柱的高售价和一次性使用的难题等,同时此方法还具有一定的经济效益和社会效益。
附图说明
图1:5mL 30mg/L的磺胺二甲基嘧啶的甲醇溶液在50mg磁性分子印迹材料上的吸附动力学曲线图;
图2:磁性分子印迹聚合物的准二级吸附动力学方程拟合图;
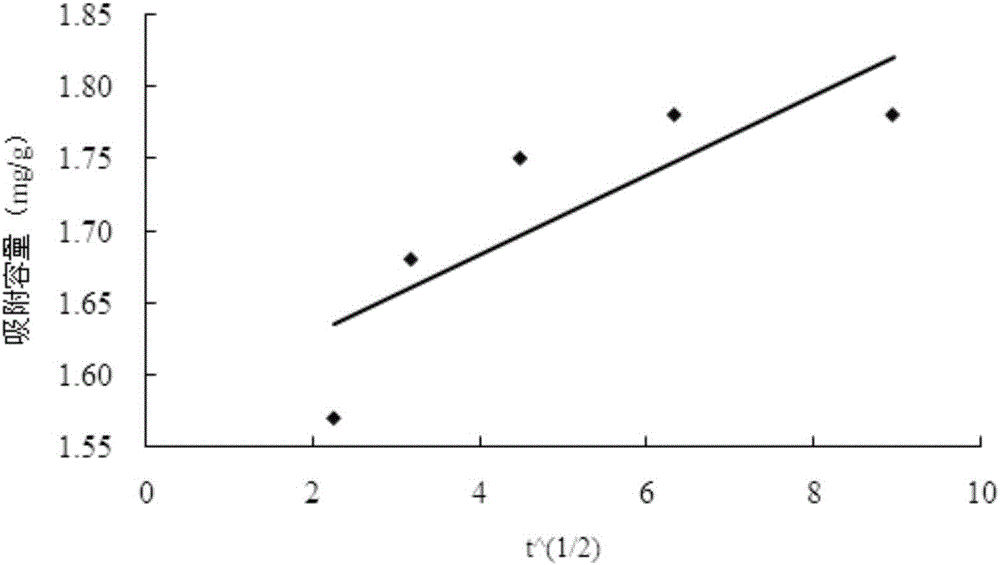
图3:磁性分子印迹聚合物的粒子扩散方程拟合图;
图4:磁性分子印迹聚合物的Elovich方程的拟合图;
图5:磁性印迹聚合物和磁性非印迹聚合物对磺胺二甲基嘧啶的等温吸附曲线;
图6:磁性印迹聚合物和磁性非印迹聚合物的Scatchard分析;
图7:磁性分子印迹聚合物合成图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步的说明,但并不以此限制本发明。
实施例1
亲水性磁性磺胺分子印迹聚合物的制备
1)采用共沉淀法合成Fe3O4粒子
将2.5g FeSO4.7H2O与4.0g FeCl3.6H2O置于锥形瓶中,加入200mL的去离子,机械搅拌使之溶解,随后在氮气保护下升温至60℃,一次性加入5mL氨水,继续加热搅拌反应1h;反应结束后利用超强磁铁的外加磁场将黑色产物与上清液分离,并用去离子水与无水乙醇反复洗涤至洗液呈中性;将合成物置于真空干燥箱中于60℃下干燥至恒重,所得黑色产物研磨后备用;
2)采用stober法制备硅烷化的Fe3O4
准确称取步骤1)制得的Fe3O4粒子0.625g倒入装有2.5mL水和8mL无水乙醇的圆底烧瓶中,加入2mL TEOS,机械搅拌5min,混合;然后加入1.5mL氨水,继续搅拌反应2h,紧接着60℃水浴1h,对Fe3O4粒子进行表面改性。用磁铁收集反应产物,并用去离子水与无水乙醇反复冲洗沉淀物直至洗液呈中性,置于真空干燥箱中于60℃下干燥,制得表面包覆有SiO2的磁性微球;
3)以磺胺二甲基嘧啶作为模板分子,将其溶解在乙腈中,用氮气将体系中的氧气置换出来,并超声波处理15min,混合均匀;
b.在a中加入摩尔比为1:1的功能单体甲基丙烯酸和甲基丙烯酸羟乙酯,搅拌4小时;
c.在b中加入摩尔比为1:1的交联剂乙二醇二甲基丙烯酸酯和3-(三甲氧基甲硅烷基)丙基丙烯酸酯;
其中模板分子与功能单体、交联剂的比例为:0.1mmol:6mmol:6mmol;
乙腈:模板分子+功能单体+交联剂组成的溶剂=6ml:12.1mmol;
d.在c中加入表面包覆有SiO2的磁性微球,搅拌均匀;
表面包覆有SiO2的磁性微球与乙腈的质量体积比为:1g:20ml;
e.在d中加入引发剂偶氮二异丁腈,偶氮二异丁腈与表面包覆有SiO2的磁性微球的质量比为1:12,机械搅拌,并于50℃水浴5h,60℃水浴24h,85℃水浴2h;得磁性磺胺分子印迹聚合物。
将磁性磺胺分子印迹聚合物在真空烘箱中100℃老化8h,研磨,定性滤纸进行包裹,用索氏提取器萃取50h-110h,萃取溶剂为甲醇,80℃烘干至恒重,得到磁性印迹聚合物颗粒。
非印迹聚合物除不加模板外,其他步骤同印迹聚合物的制备过程。为了比较亲水性聚合物材料的亲水性,实验的过程制备了非亲水性的磺胺类兽药的磁性分子印迹聚合物,其制备过程除不加亲水性功能单体外,其他步骤相同。
实施例2
对实施例1中所得的印迹聚合物进行表征,确定印迹结合位点的存在;
所述的表征方法包括动态吸附、静态吸附和选择性实验。
磁性分子印迹聚合物的表征
(1)吸附动力学实验
准确称取50mg磁性分子印迹聚合物,置于25mL容量瓶中,用移液管准确移取5mL 30mg/L的SMZ-水溶液中,分别在室温下振荡5min,10min,20min,40min,80min,120min后,在外加磁体的吸附下将材料进行分离;
在波长270nm条件下采用紫外-可见分光光度计测定上清液中磺胺二甲基嘧啶的浓度;吸附动力学实验是为了确定合成的磁性分子印迹聚合物的吸附效率,本专利测定了不同吸附时间,吸附材料对目标物的吸附能力。结果图1。由图可知,吸附5分钟后,吸附容量基本达到最大吸附容量的88.2%,25min即可达到吸附平衡。
经实验测定,可知,初始磺胺二甲基嘧啶的浓度越低,达到吸附平衡的时间会越短。这可能与合成材料的结构性质有关,由于聚合物合成后,要经过研磨处理,可能导致聚合物表面产生的空穴深浅不一,较浅的空穴有利于目标物的快速吸附,所以实验的初始阶段的吸附效率会很高。当较浅的空穴完全吸附后,深处的空穴在吸附目标物时会存在一定的空间位阻,会使吸附的速率变慢,最终达到吸附平衡。
以往用本体聚合法制备的聚合物,达到吸附平衡的时间较长,有的甚至要吸附24h,而采用该制备的表面分子印迹聚合物对模板分子的吸附动力学较快,聚合物的传质速度很快,所以可以将此材料用做固相萃取-高效液相色谱联用对磺胺类药物的检测。
为了确定吸附过程中材料的吸附特性和动力学参数,对实验结果进行准一级吸附动力学、准二级吸附动力学、粒子扩散和Elovich方程的拟合。拟合结果如图1和如表1所示。R2作为拟合模型与实验数据相符度的重要参数,R2越接近1,实验数据与拟合模型的相符度越高。由表2可知,准二级吸附动力学的的线性相关系数R2为0.999,线性关系良好,呈现显著相关水平。准二级吸附动力学可以反映出吸附过程中发生的两个反应,一个是控制整个反应时间的慢反应,一个是快速达到平衡的反应。本实验中限制吸附速率的步骤主要为化学吸附
表1四种方程的吸附动力学拟合结果对比
2)平衡结合实验
为了考察所制备的亲水性磁性分子印迹聚合物对磺胺二甲基嘧啶的吸附能力,实验中准确称取50mg的吸附材料于25mL容量瓶中,分别加入5mL不同浓度的SMZ-水溶液(20mg/L,30mg/L,40mg/L,70mg/L,80mg/L),摇床充分振荡30min。在波长270nm条件下,采用紫外-可见分光光度计测定上清液的吸光度值,并计算吸附容量。在相同条件下做非印迹聚合物对磺胺二甲基嘧啶的平衡结合实验。
吸附容量是评价磁性分子印迹聚合物对模板分子结合能力强弱的重要参数。由图5可知,磁性印迹聚合物和磁性非印迹聚合物对磺胺二甲基嘧啶的吸附量均有不同程度的增加。当磺胺二甲基嘧啶的初始浓度为70mg/L时,印迹聚合物和非印迹聚合物对模板分子磺胺二甲基嘧啶的吸附容量分别为4.1mg/g和0.27mg/g,印迹聚合物对磺胺二甲基嘧啶的吸附量大约是非印迹聚合物吸附量的15倍,说明相较于非印迹聚合物,印迹聚合物对模板分子的吸附性较好。
制备的磁性分子印迹聚合物对磺胺二甲基嘧啶具有选择性吸附,原因在被二氧化硅包裹的磁性小球表明有化学键生成与聚合物相连,聚合反应过程中,以磺胺二甲基嘧啶作为模板分子,磺胺二甲基嘧啶与甲基丙烯酸形成配体,聚合物内部具有与磺胺二甲基嘧啶相匹配的活性基团,当磺胺二甲基嘧啶被洗脱后,原来的部位会有特异性空穴存在,这种特异性空穴使得磁性分子印迹聚合物对磺胺二甲基嘧啶具有良好的结合能力。
Scatchard分析:
将所获得的数据用于印迹聚合物和非印迹聚合物的Scatchard分析。分析结果见图6
Scatchard方程:Q/C=Q/b+Qmax/b
方程式中C指溶液的初始浓度,Q指达到吸附平衡时的吸附容量,Qmax指最大饱和吸附荣量,以Q/C对Q作图即得Scatchard方程。由Scatchard曲线的斜率和截距可得聚合物的Qmax和Kd。
由图可以看出,磺胺二甲基嘧啶磁性分子印迹聚合物的Scatchard方程是一条线性良好的直线,其R2可达到0.99,回归方程为y=-0.0014x+0.0624。说明该方法合成的磁性分子印迹聚合物对目标物磺胺二甲基嘧啶是一种结合方式。而磁性非印迹聚合物不能模拟出线性方程,呈散点状态,从侧面显示出非印迹聚合物没有特异性结合位点。
3)选择性实验
本实验选择与模板分子结构相似的塑化剂(BBP、DCHP)和速灭威,同时与十种磺胺类药物进行竞争,测定磁性分子印迹聚合物对这十三种溶液的选择性情况。结果见表2。
从表2中可以看出,磁性印迹聚合物对磺胺类药物的吸附量要远远大于非印迹聚合物对磺胺类药物的吸附容量。磁性非印迹聚合物对塑化剂及速灭威的吸附量可以达到其对磺胺类药物吸附量的10倍以上,而磁性印迹聚合物,对BBP、DCHP及速灭威的吸附量仅为20.58μg/g、0μg/g、0μg/g。从结果看来,磁性印迹分子聚合物对磺胺类药物具有特异的吸附性,而磁性非印迹分子聚合物则不具有特异性。
磁性分子印迹聚合物具有对磺胺类药物有吸附特异性,是因为在聚合物合成的过程中,模板分子磺胺二甲基嘧啶与功能单体键合,形成了一定的内部立体化学结构。当模板分子洗脱下来后,其存在的区域形成了特定的空穴,该空穴可以特异性吸附目标物,而非印迹聚合物不存在这种特异性的空穴,只是通过物理或者是化学性吸附,所以其吸附的特异性很低。
表2磁性分子印迹聚合物对13种溶液的选择性情况
从表2中可以看出,磁性印迹聚合物对磺胺类药物的吸附量要远远大于非印迹聚合物对磺胺类药物的吸附容量。磁性非印迹聚合物对塑化剂及速灭威的吸附量可以达到其对磺胺类药物吸附量的10倍以上,而磁性印迹聚合物,对BBP、DCHP及速灭威的吸附量仅为20.58μg/g、0μg/g、0μg/g。从结果看来,磁性印迹分子聚合物对磺胺类药物具有特异的吸附性,而磁性非印迹分子聚合物则不具有特异性。
磁性分子印迹聚合物具有对磺胺类药物有吸附特异性,是因为在聚合物合成的过程中,模板分子磺胺二甲基嘧啶与功能单体键合,形成了一定的内部立体化学结构。当模板分子洗脱下来后,其存在的区域形成了特定的空穴,该空穴可以特异性吸附目标物,而非印迹聚合物不存在这种特异性的空穴,只是通过物理或者是化学性吸附,所以其吸附的特异性很低。
实施例3
亲水性磁性磺胺类分子印迹聚合物的应用
为了评价方法的适用性,以鸡肉,羊奶和牛奶样品为原料,以制备的亲水性磺胺类兽药磁性分子印迹聚合物为基质分散固相萃取材料,在优化的萃取条件下,对样品进行加标回收实验,添加后样品的磺胺类药物的浓度为5ug/kg、10ug/kg和20ug/kg三个水平,样品经吸附、富集和净化后后,测定样品的回收率和相对标准偏差,结果分别见下表(a),(b)和(c)。由表可知,三种样品的添加回收率除牛奶样品的添加回收率相对较低以外,其它两种样品的10种磺胺类药物的回收率范围为65%以上。
(a)鸡肉样品加标回收率(Mean±RSD)
(b)羊奶样品加标回收率(Mean±RSD)
(c)牛奶样品加标回收率(Mean±RSD)
- 还没有人留言评论。精彩留言会获得点赞!