硫酸沃拉帕沙中间体的制备方法与流程
本发明涉及药物合成技术领域,特别是涉及硫酸沃拉帕沙中间体的制备方法。
背景技术:
2014年5月8日,默沙东抗凝血剂硫酸沃拉帕沙(vorapaxar)获FDA批准,用于遭受心脏病发作的患者或腿部动脉有堵塞的患者,以降低进一步的心脏病发作、中风、心血管死亡和需要手术的风险。Vorapaxar是一种首创的蛋白酶激活受体1(PAR-1)拮抗剂,旨在减少血小板聚集倾向,抑制血凝凝块的形成,有着广阔的市场需求和社会价值。至今默沙东已投入80亿美元研发vorapaxar,现在vorapaxar也已经获得用于防止初次心脏病患者复发的许可。
化合物I是合成硫酸沃拉帕沙的重要中间体,在Journal of Medicinal Chemistry,1988,vol.31,#8p.1558–1566以及专利WO2006076415A2中,其合成均以消旋的丁炔醇为原料,经过拆分得手性化合物R-丁炔醇,再经过羟基保护等反应制得目标化合物。由于消旋的丁炔醇原料成本较高,导致化合物I的制备成本较高。
硫酸沃拉帕沙在心血管药物的市场需求较大,因此,有效控制硫酸沃拉帕沙的合成成本具有重大的社会意义。
技术实现要素:
本发明的目的是提供硫酸沃拉帕沙中间体化合物I的制备方法,该制备方法具有生产成本低、选择性高、收率高、易于工业化生产等优点。
本发明提供了硫酸沃拉帕沙中间体化合物I的制备方法,化合物I的结构如下式所示,
以D-乳酸酯为起始原料,经羟基保护、还原取代和羟基脱保护,得到所述化合物I。
可选地,包括以下步骤:
S101:在碱的存在下,用R2L对D-乳酸酯中的羟基进行保护,得到化合物III,
S102:对化合物III进行还原,得到化合物IV,
S103:在第一强碱的存在下,用对甲苯磺酰氯对化合物IV进行取代反应,得到化合物V,
S104:在第二强碱的存在下,化合物V与化合物VII进行反应,得到化合物VI,
S105:用酸脱除化合物VI中的R2保护基,得到目标产物化合物I,
其中,R1选自烷基或杂环基,R2为保护基,L为第一离去基,R3为第二离去基。优选地,R1选自C1~C6的直链或支链的烷基、四氢吡咯基,R2选自TBS、TMS、THP,R3选自卤素或咪唑基。
可选地,S101中,碱选自咪唑、三乙胺、DIPEA、吡啶、DBU;D-乳酸酯、R2L、碱的摩尔比为1:(1.0~1.5):(1.2~1.8)。
可选地,S102中,对化合物III进行还原的方法选自:使用第一还原剂将化合物III直接还原为化合物IV;或者使用第二还原剂将化合物III还原为化合物IIIA,再使用第一氧化剂将化合物IIIA氧化为化合物IV,
其中,第一还原剂选自DIBAL-H、氢化二乙氧基铝锂、氢化三乙氧基铝锂、二氢双(2-甲氧乙氧基)铝酸钠;化合物III、第一还原剂的摩尔比为1:(1.05~1.3);
第二还原剂选自LiBH4、LiAlH4、NaBH4、I2/NaBH4、LiCl/NaBH4、ZnCl2/NaBH4、AlCl3/NaBH4;化合物III、第二还原剂的摩尔比为1:(1.0~2.0);
第一氧化剂选自DMSO/草酰氯/三乙胺、次氯酸钠/TEMPO、IBX。
可选地,S103中,所述第一强碱选自LDA、NaH、LiHMDS、NHMDS;化合物IV、二氯甲烷、对甲苯磺酰氯、第一强碱的摩尔比为1:(2.5~3.5):(1.0~1.5):(1.2~1.8);
S104中,所述第二强碱选自正丁基锂、正己基锂、仲丁基锂;化合物V、化合物VII、第二强碱的摩尔比为1:(1.0~1.5):(0.002~0.004)。
可选地,S105中,所述酸选自盐酸、硫酸、磷酸、硝酸、氢溴酸,化合物VI、酸的摩尔比为1:(1.5~3)。
可选地,化合物II为D-乳酸甲酯。
可选地,所述D-乳酸甲酯通过发酵制得。
本发明采用廉价易得的D-乳酸酯为起始原料,经过五步反应制备得到硫酸沃拉帕沙的关键中间体化合物I,从而大大降低了硫酸沃拉帕沙的合成成本,且根据本发明的方法适合工业化生产并具有高收率和高选择性。
附图说明
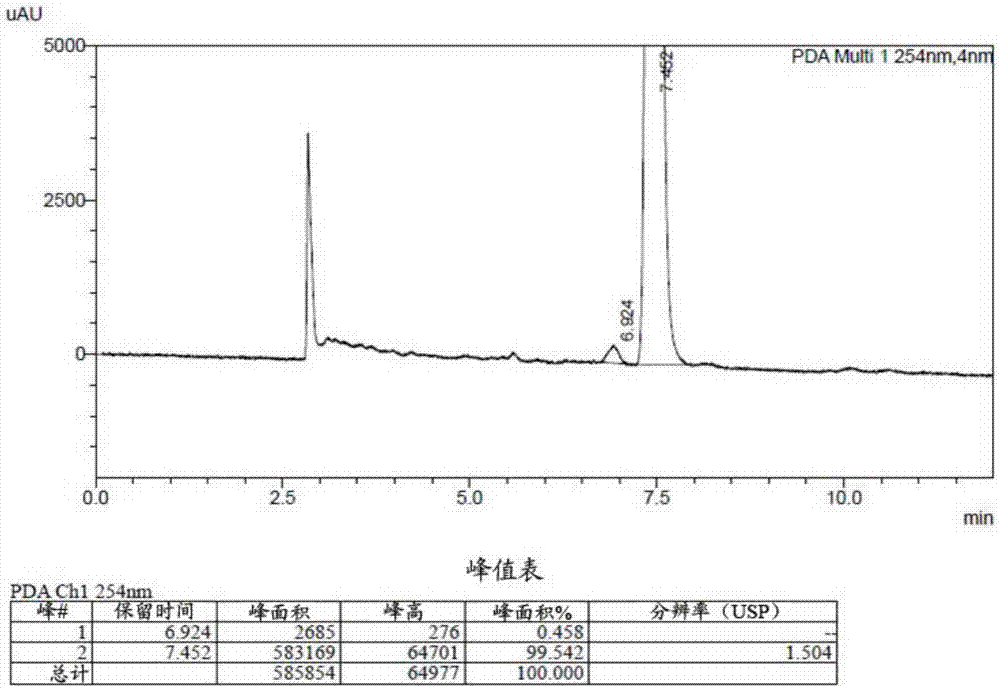
图1为化合物I的消旋体的手性HPLC检测结果图;
图2为本发明制备的化合物I的手性HPLC检测结果图;
图3为本发明制备的化合物I的化学纯度HPLC检测结果图。
具体实施方式
具体实施方式仅为对本发明的说明,而不构成对本发明内容的限制,下面将结合具体的实施方式对本发明进行进一步说明和描述。
本发明提供了硫酸沃拉帕沙中间体化合物I的制备方法,化合物I的结构如下式所示,
以D-乳酸酯为起始原料,经羟基保护、还原取代和羟基脱保护,得到所述化合物I。
本发明采用廉价易得的D-乳酸酯为起始原料,经过五步反应制备得到硫酸沃拉帕沙的关键中间体化合物I,从而大大降低了硫酸沃拉帕沙的合成成本,且根据本发明的方法适合工业化生产并具有高收率和高选择性。
可选地,包括以下步骤:
S101:在碱的存在下,用R2L对D-乳酸酯中的羟基进行保护,得到化合物III,
S102:对化合物III进行还原,得到化合物IV,
S103:在第一强碱的存在下,用对甲苯磺酰氯对化合物IV进行取代反应,得到化合物V,
S104:在第二强碱的存在下,化合物V与化合物VII进行反应,得到化合物VI,
S105:用酸脱除化合物VI中的R2保护基,得到目标产物化合物I,
其中,R1选自烷基或杂环基,R2为保护基,L为第一离去基,R3为第二离去基。优选地,R1选自C1~C6的直链或支链的烷基、四氢吡咯基,R1例如可以为甲基、乙基、异丙基等;R2选自叔丁基二甲基硅基(TBS)、三甲基硅基(TMS)、四氢吡喃基(THP),R3选自卤素(F、Cl、Br)或咪唑基。
S101中:
反应在惰性气体(如N2、Ar等)保护下进行;碱选自咪唑、三乙胺、N,N-二异丙基乙胺(DIPEA)、吡啶、1,8-二氮杂二环[5.4.0]十一碳-7-烯(DBU);D-乳酸酯、R2L、碱的摩尔比为1:(1.0~1.5):(1.2~1.8),优选为1:1:1.5;溶剂选自二氯甲烷、DMF;R2L优选以滴加方式加入反应液中,以减少副产物的产生;反应时间为10~24h,滴加R2L期间及加完后1~2h内,反应液的温度为0~5℃,之后升温至20~30℃进行反应。
优选地,羟基保护反应结束后,进行后处理。所述后处理的方法和条件可为本领域后处理常规的方法和条件,本发明优选下列方法和条件:将羟基保护反应的反应液萃取,合并有机相,干燥,过滤,减压浓缩得粗品,再经减压蒸馏得化合物III。
S102中:
对化合物III进行还原的方法选自:
a.使用第一还原剂将化合物III直接还原为化合物IV;其中,反应在惰性气体(如N2、Ar等)保护下进行;反应温度为-78℃,反应时间为1~3h;第一还原剂选自二异丁基氢化铝(DIBAL-H)、氢化二乙氧基铝锂、氢化三乙氧基铝锂、二氢双(2-甲氧乙氧基)铝酸钠(红铝);化合物III、第一还原剂的摩尔比为1:(1.05~1.3),优选1:1.1;第一还原剂优选以滴加方式加入反应液中,以减少副产物的产生;反应结束后用甲醇淬灭反应。
优选地,反应结束后,进行后处理。所述后处理的方法和条件可为本领域后处理常规的方法和条件,本发明优选下列方法和条件:将羟基保护反应的反应液萃取,合并有机相,干燥,过滤,减压浓缩得粗品,再经减压蒸馏得化合物IV。
或者b.使用第二还原剂将化合物III还原为化合物IIIA,再使用第一氧化剂将化合物IIIA氧化为化合物IV,
其中,还原反应在惰性气体(如N2、Ar等)保护下进行;第二还原剂选自LiBH4、LiAlH4、NaBH4、I2/NaBH4、LiCl/NaBH4、ZnCl2/NaBH4、AlCl3/NaBH4;化合物III、第二还原剂的摩尔比随第二还原剂种类的变化而变化,本领域技术人员可以根据反应程度、产物情况等在一定范围内适当调整,例如为1:(1.0~2.0);第一氧化剂选自DMSO/草酰氯/三乙胺、次氯酸钠/TEMPO、IBX;化合物IIIA、第一氧化剂的摩尔比随第一氧化剂种类的变化而变化,本领域技术人员可以根据反应程度、产物情况等在一定范围内适当调整。
当第二还原剂选自I2/NaBH4、LiCl/NaBH4、ZnCl2/NaBH4、AlCl3/NaBH4,I2、LiCl、ZnCl2、AlCl3与NaBH4的摩尔比分别选自1:(1.5~2.0)、1:(1.0~1.5)、1:(1.0~1.5)、1:(1.0~1.5),且分别优选为1:1.5、1:1、1:1、1:1。当第一氧化剂选自DMSO/草酰氯/三乙胺时,DMSO/草酰氯/三乙胺的摩尔比为1:(1.0~1.5):(1.0~3.0),优选1:1.2:2.0;当第一氧化剂选自次氯酸钠/TEMPO时,次氯酸钠/TEMPO的摩尔比为1:(0.1~0.5),优选1:0.2。
S103中:
反应在惰性气体(如N2、Ar等)保护下进行;所用溶剂均为无水处理后的无水溶剂;所述第一强碱选自二异丙基氨基锂(LDA)、NaH、双(三甲基硅基)氨基锂(LiHMDS)、双(三甲基硅基)氨基钠(NHMDS);化合物IV、二氯甲烷、对甲苯磺酰氯、第一强碱的摩尔比为1:(2.5~3.5):(1.0~1.5):(1.2~1.8),优选为1:3:1:1.5;溶剂选自2-甲基四氢呋喃、四氢呋喃;第一强碱优选以滴加方式加入反应液中,以减少副产物的产生;滴加第一强碱期间及加完后0.5~1h内,反应液的温度为-78℃,之后升温至0℃进行反应,加入对甲苯磺酰氯后10~15min,再次升温至20~30℃,反应1~3h,结束后用水淬灭反应。
优选地,反应结束后,进行后处理。所述后处理的方法和条件可为本领域后处理常规的方法和条件,本发明优选下列方法和条件:将反应液分别用10%HCl和1N NaOH洗涤,收集有机相,干燥,过滤,减压浓缩得化合物V。
S104中:
反应在惰性气体(如N2、Ar等)保护下进行;所用溶剂均为无水处理后的无水溶剂;所述第二强碱选自正丁基锂、正己基锂、仲丁基锂;化合物V、化合物VII、第二强碱的摩尔比为1:(1.0~1.5):(0.002~0.004),优选1:1:0.003;溶剂选自四氢呋喃、2-甲基四氢呋喃;正丁基锂优选以滴加方式加入反应液中,以减少副产物的产生;滴加正丁基锂期间及加完后0.5~1h内,反应液的温度为-78℃,之后升温至0℃反应1~2h后再降温至-78℃,加入化合物VII后10~15min,再次升温至20~30℃,反应3~8h,结束后用饱和氯化铵溶液淬灭反应。
优选地,反应结束后,进行后处理。所述后处理的方法和条件可为本领域后处理常规的方法和条件,本发明优选下列方法和条件:将反应液分别用1N NaOH溶液和饱和氯化钠溶液洗涤,收集有机相,干燥,过滤,减压浓缩得化合物VI。
S105中:
所述酸选自盐酸、硫酸、磷酸、硝酸、氢溴酸,优选浓度为2M的酸;化合物VI、酸的摩尔比为1:(1.5~3),优选1~2;反应温度为20~30℃,时间为1~2h。
优选地,反应结束后,进行后处理。所述后处理的方法和条件可为本领域后处理常规的方法和条件,本发明优选下列方法和条件:将反应液依次用1N NaOH溶液和饱和氯化钠溶液洗涤,收集有机相,干燥,过滤,减压浓缩得粗品,之后重新溶解析晶,过滤,减压干燥,得到目标化合物I。
可选地,化合物II为D-乳酸甲酯。
可选地,所述D-乳酸甲酯通过发酵制得,可以进一步降低了成本。
为了更加详细地说明本发明的制备方法,下面将列举上述化合物的具体合成方法对本发明进行进一步的描述。
实施例1化合物III的制备方法
在2L的三口烧瓶中,加入D-乳酸甲酯(104g,1mol)、二氯甲烷(208ml)和咪唑(102g,1.5mol),冷却反应液冷却至0-5℃,氮气保护,滴加TBSCl(150g,1mol)和二氯甲烷(300ml)的溶液,保持温度0-5℃,约1h滴加完毕,0-5℃搅拌反应1h,升温至20-30℃,搅拌反应18h,反应液加入水(500ml),搅拌10min,水层用二氯甲烷(300ml)萃取,合并有机相,用饱和氯化钠溶液(50mlx3)洗涤,收集有机相,用无水硫酸镁干燥1h,过滤,收集滤液,减压浓缩得粗品,粗品经减压蒸馏得产品197g,收率90%。
实施例2化合物IV的第一种制备方法
在2L的三口瓶中加入化合物III(65.4g,0.3mol)和正己烷(600ml),冷却至-78℃,氮气保护;另取DIBAL-H(220ml,1.5M甲苯溶液,0.33mol),预冷至-78℃,将DIBAL-H滴加到化合物III的溶液中,控温-78℃,用时约30min,滴加完毕,-78℃搅拌反应1h,然后向反应液中加入甲醇(30ml),淬灭反应,加毕,-78℃搅拌15min。另取2L三口瓶,换用机械搅拌,加入饱和酒石酸钾钠溶液(600ml),将上述反应液加入到酒石酸钾钠溶液中,升温至20-30℃,搅拌2-3h,静置,分出有机相,水相用正己烷(200ml)萃取,合并有机相,用饱和氯化钠溶液(100ml)洗涤,无水硫酸镁干燥1h,减压浓缩除去溶剂,得化合物IV粗品,减压蒸馏得产品47g,收率83%。
实施例3化合物IV的第二种制备方法
在500ml三口烧瓶中加入氯化锂(7.8g,183mmol)和四氢呋喃(200ml),加入硼氢化钠(6.9g,183mmol),氮气保护,20-25℃下搅拌3h,加入化合物III(20g,91.7mmol),氮气保护下,20-25℃下搅拌反应48h,反应完毕后用1N HCl(50ml)淬灭,加入乙酸乙酯(200ml)萃取,收集有机相,用饱和食盐水洗涤,无水硫酸镁干燥,浓缩干得粗品,40-45℃下减压蒸馏得产品15.2g,收率87.2%。
在500ml三口烧瓶中加入DCM(50ml)和草酰氯(27.8g,276mmol),氮气保护,冷却至-78℃,滴加DMSO(27.9g,276mmol)的DCM(50ml)的溶液,控温-78℃搅拌2h,滴加上一步产品(15.0g,78.9mmol),保温反应2h,滴加三乙胺(88.3g,789mmol),搅拌升温至室温,加水100ml,分层,收集有机相,分别用1N盐酸、饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩得粗产品,40-45℃减压蒸馏得产品13.2g,收率:88%。
实施例4化合物V的制备方法
在2L的三口瓶中加入化合物IV(45.1g,0.24mol)、干燥二氯甲烷(45.8ml,0.72mol)和无水四氢呋喃(500ml),冷却至-78℃,氮气保护,滴加LDA溶液(2.0M THF溶液,180ml,0.36mol),控温-78℃,用时约1h,滴加完毕,-78℃搅拌反应30min,升温至0℃,控温搅拌30min,将p-TsCl(45.6g,0.24mol)加入反应体系,搅拌反应10min,升温至20-30℃,搅拌反应1.5h,反应液用水(10ml)淬灭,搅拌30min,反应液分别用10%HCl(250ml)和1N NaOH(150ml)洗涤,收集有机相,水相分别用乙酸乙酯(500ml)萃取,合并乙酸乙酯相和四氢呋喃相,有机相用饱和食盐水(100ml)洗涤,无水硫酸镁干燥1h,减压浓缩得产品82.2g,收率80%。直接进行下一步反应。
实施例5化合物VI的制备方法
在1L的三口瓶中加入上述制得的化合物V(80g,187mmol)和无水四氢呋喃(500ml),冷却至-78℃,氮气保护,滴加正丁基锂(232ml,2.5M正己烷溶液,0.58mol),控温-78℃,用时约1h滴完,滴加完毕,控温-78℃搅拌反应30min,然后升温至0℃搅拌反应1h,反应液颜色变深,将反应液重新冷却至-78℃,加入化合物VII(43g,187mmol),搅拌15min,升温至20-30℃,搅拌反应4h,向反应液中加入饱和氯化铵溶液(200ml)淬灭反应,分出有机相,水相用乙酸乙酯萃取(200mlx3),合并有机相分别用1NNaOH溶液(100ml)和饱和氯化钠溶液(50ml)洗涤,无水硫酸镁干燥1h,过滤,收集滤液,减压浓缩至干,得产品54.5g,收率76.8%。直接进行下一步反应。
实施例6化合物I的制备方法
在1L的三口瓶中加入化合物VI(50g,132mmol)和甲醇(500ml),加入2M HCl(132ml,264mmol),20-30℃下搅拌反应1h,反应完毕后反应液中加入乙酸乙酯(500ml)和水(500ml),搅拌10min,分出水相,有机相用1NNaOH(100ml)洗涤,收集有机相,用饱和氯化钠(100ml)洗涤,无水硫酸镁干燥1h,过滤,收集滤液,减压浓缩至干,得油状物粗品,将粗品加入甲苯(25ml),搅拌溶解,冷却至0℃,滴加正庚烷(125ml),搅拌,析晶2h,过滤,正庚烷淋洗,50℃减压干燥2h,得产品25g,收率71%,ee大于99%。Mp 105℃。1H NMR(400MHz,DMSO-d6)δ1.04(d,J=6.4Hz,3H),δ4.27(dq,J=5.6Hz,6.4Hz,1H),δ5.49(d,J=5.6Hz,1H),δ7.2-7.5(m,10H);13C NMR(DMSO-d6)δ23.7,56.3,76.9,96.4,126.8,127.0,128.5,129.2,129.4,129.6,141.5,142.2,152.9。
从图1和图2的比对中可以看出,本发明方法具有很高的选择性,化合物I的含量高达99.5%以上,其对映异构体的含量低于0.5%。从图3中可以看出,本发明制备得到的化合物I的纯度高达98.8%。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!