一种丁二酸酐改性纤维素的制膜方法与流程
本发明涉及膜材料制备领域,具体涉及一种丁二酸酐改性纤维素的制膜方法。
背景技术:
近年来,生物质衍生的碳水化合物是一种很有前途的以碳为基础的替代能源和可持续的化学原料,纤维素作为一种天然的可再生碳水化合物,在制备和加工样品中受到了化学工作者的青睐。由于其具有资源丰富、价格低廉、对环境友好、可再生、可生物降解、机械性能优良等优良性能,拓宽了它的应用范围。纤维素(cellulose)是由葡萄糖组成的大分子多糖。不溶于水及一般有机溶剂。是植物细胞壁的主要成分。纤维素是自然界中分布最广、含量最多的一种多糖,占植物界碳含量的50%以上。棉花的纤维素含量接近100%,为天然的最纯纤维素来源。一般木材中,纤维素占40~50%,还有10~30%的半纤维素和20~30%的木质素。
丁二酸酐是无色针状或粒状结晶,溶于乙醇、三氯甲烷和四氯化碳,与热水可水解为丁二酸。分子式:C4H4O3。有机工业用作合成有机化合物的中间体。
采用丁二酸酐对纤维素进行改性,使纤维素的羟基基团转化为羧基基团,从而增加了纤维素的水溶性。同时由于纤维素的羧基基团也是介质改性和应用的功能性基团,因而其可应用于食品加工行业中新型乳化剂开发、色素和有害重金属物的去除剂等领域。
虽然以纤维素为原料制备膜材料的技术很成熟,但是丁二酸酐改性纤维素的物理化学性质发生改变,从而使得以纤维素为原料制备膜材料的方法难以应用于丁二酸酐改性纤维素。
技术实现要素:
为了解决现有技术的不足,本发明的目的之一是提供一种丁二酸酐改性纤维素的制膜方法,该方法能够以丁二酸酐改性纤维素制备膜材料。
为了实现上述目的,本发明的技术方案为:
一种丁二酸酐改性纤维素的制膜方法,将丁二酸酐改性纤维素溶解在1-烯丙基-3-甲基咪唑氯盐中混合均匀形成改性纤维素溶液,将改性纤维素溶液转移至模具中,在室温下放置一段时间,再向模具中加入冰水,静置一段时间后去除上层液体,然后向模具中加入丙三醇溶液,静置一段时间后去除上层液体即可获得膜材料。
首先,本发明选用1-烯丙基-3-甲基咪唑氯盐离子液体作为溶剂,能够完全溶解丁二酸酐改性纤维素。其次,由于丁二酸酐的改性使得不同的改性纤维素链缠绕速度降低,从而阻碍了丁二酸酐改性纤维素的成膜,本发明将改性纤维素溶液静置,使得改性纤维素链在离子液体中充分舒展,同时使不同的改性纤维素的链充分缠绕,从而大大提高改性纤维素的成膜性。第三,加入冰水,使得改性纤维素与离子液体分离,从而使改性纤维素沉降成膜。第四,加入丙三醇溶液能够去除膜中杂质,进一步提高改性纤维素的成膜性。
本发明的目的之二是提供一种上述制膜方法制备的膜材料。本发明制备的膜材料具有较高的吸湿性、较好的机械性能及优良的阻光性能。
本发明的目的之三是提供一种上述膜材料在电泳、离子交换、渗透、过滤或包装材料中的应用。
本发明的有益效果为:
1.本发明以丁二酸酐改性纤维素为制膜原料制备出了膜材料,制备方法简单、操作方便、成膜性能良好。
2.本发明制备的膜材料具有优良的吸湿性能,180h时最大吸水值可达226.72%;本发明制备的膜材料具备优良的机械性能,与纯纤维素膜相比,本发明制备的膜材料抗张强度较低、断裂伸长率较高及较低的杨氏模量;本发明制备的膜材料具备较好的阻光性能。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。
图1为实施例5制备的改性纤维素及实施例15制备的负载dom的改性纤维素的傅里叶红外(FTIR)光谱图;
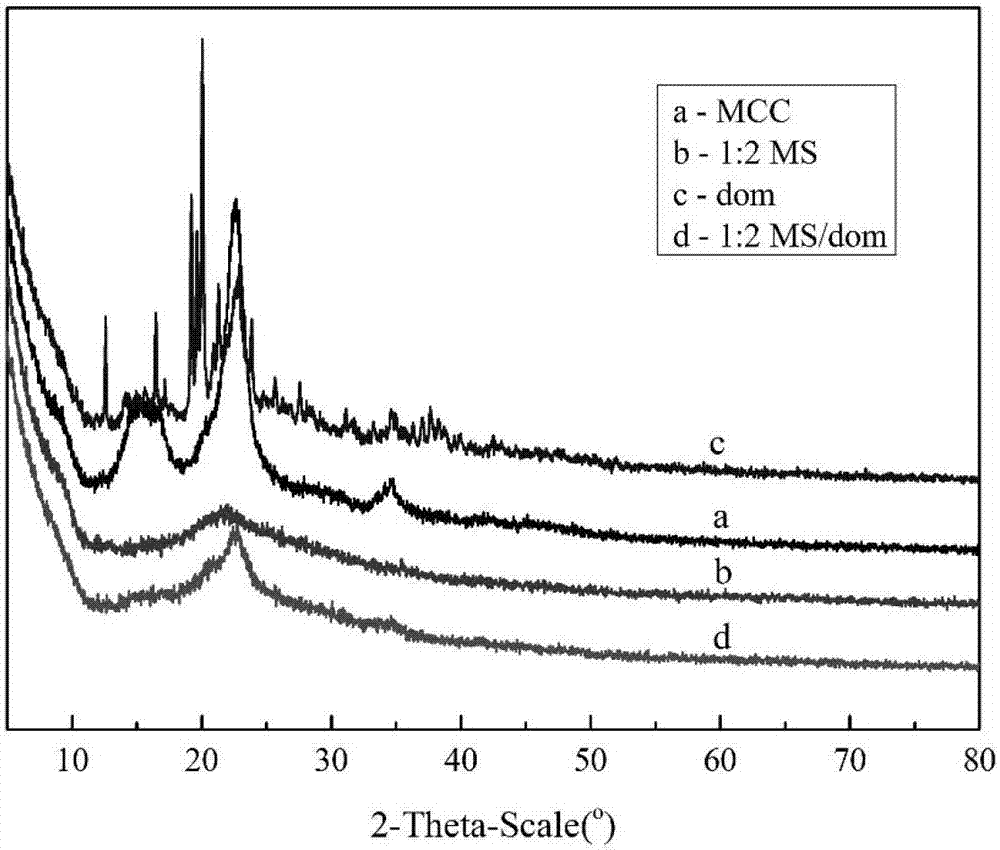
图2为为实施例5制备的改性纤维素及实施例15制备的负载dom的改性纤维素的X射线衍射(XRD)光谱图;
图3为实施例5制备的改性纤维素及实施例15制备的负载dom的改性纤维素的热力学分析图;
图4为实施例4~8制备的的改性纤维素的吸水性曲线;
图5为实施例4~8制备的的改性纤维素的降解性能曲线;
图6为不同膜的相同倍率下表面和断面的扫描电镜(SEM)图片,其中,a1,a2均为纤维素制备的膜,b1,b2均为改性纤维素制备的膜,c1,c2均为负载药物的改性纤维素制备的膜;
图7为不同膜的不同倍率下表面的扫描电镜(SEM)图片,其中,A1,A2均为改性纤维素制备的膜,B1,B2均为负载药物的改性纤维素制备的膜;
图8为实施例14~18制备的膜的药物释放曲线。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本申请提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本申请所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
本发明中所述的丁二酸酐又称琥珀酸酐,均以SAD作为其简写。
本发明中所述的1-烯丙基-3-甲基咪唑氯盐是一种离子液体,简写为AMIMCl。
本发明中所述的丙三醇溶液为丙三醇的水溶液。
正如背景技术所介绍的,现有技术中存在以纤维素为原料制备膜材料的方法难以应用于丁二酸酐改性纤维素不足,为了解决如上的技术问题,本申请提出了一种丁二酸酐改性纤维素的制膜方法。
本发明的一种典型的实施方式中,提供了一种丁二酸酐改性纤维素的制膜方法,将丁二酸酐改性纤维素溶解在1-烯丙基-3-甲基咪唑氯盐中混合均匀形成改性纤维素溶液,将改性纤维素溶液转移至模具中,在室温下放置一段时间,再向模具中加入冰水,静置一段时间后去除上层液体,然后向模具中加入丙三醇溶液,静置一段时间后去除上层液体即可获得膜材料。
首先,本实施方式选用1-烯丙基-3-甲基咪唑氯盐离子液体作为溶剂,能够完全溶解丁二酸酐改性纤维素。其次,由于丁二酸酐的改性使得不同的改性纤维素链缠绕速度降低,从而阻碍了丁二酸酐改性纤维素的成膜,本实施方式将改性纤维素溶液静置,使得改性纤维素链在离子液体中充分舒展,同时使不同的改性纤维素的链充分缠绕,从而大大提高改性纤维素的成膜性。第三,加入冰水,使得改性纤维素与离子液体分离,从而使改性纤维素沉降成膜。第四,加入丙三醇溶液能够去除膜中杂质,进一步提高改性纤维素的成膜性。
为了更好的成膜,本实施方式优选的,将改性纤维素溶液转移至模具中在室温下放置2h。
优选的,加入冰水后静置的时间为30min。能够充分的将丁二酸酐改性纤维素沉积成膜。
优选的,加入丙三醇溶液前将模具中沉积的膜采用冰水进行洗涤。去除膜中的离子液体等其他溶于水的杂质。
进一步优选的,采用冰水洗涤3~5次。充分去除溶于水的杂质。
优选的,加入丙三醇溶液后静置的时间为30min。能够充分去除膜中的有机物杂质。
优选的,所述丙三醇溶液中丙三醇的浓度为5%(质量)。
为了降低制膜成本,本发明的另一种典型实施方式提供了一种改性纤维素溶液的制备方法,将纤维素溶于1-烯丙基-3-甲基咪唑氯盐中混合均匀形成纤维素溶液,向所述纤维素溶液中加入丁二酸酐和二甲基亚砜,搅拌一段时间后即得改性纤维素溶液。加入二甲基亚砜能够降低纤维素溶液的粘度,使反应更加充分。
优选的,所述纤维素溶液的制备步骤是,将纤维素放入1-烯丙基-3-甲基咪唑氯盐中90℃下搅拌至纤维素完全溶解。
进一步优选的,所述纤维素与1-烯丙基-3-甲基咪唑氯盐的质量比为0.3:4。
优选的,纤维素中葡萄糖单元与丁二酸酐的摩尔比为1:1~5。保证纤维素改性完全。
本发明中采用的丁二酸酐可以从上海阿拉丁生化科技股份有限公司、邯郸市华骏化工有限公司等企业购得,也可以自行制备。
本发明的另一种典型实施方式提供了一种丁二酸酐的制备方法,4g丁二酸和6.4mL乙酸酐加入圆底烧瓶中,混合物在100℃下搅拌直至变为澄清溶液,继续搅拌1h,冷却至室温出现沉淀物,此混合物于室温下放置2h,再在8℃下保持30min确保反应充分。反应后产物经过抽滤、乙醚洗涤、干燥处理,得到透明的白色固体,即为丁二酸酐。
本发明中采用的1-烯丙基-3-甲基咪唑氯盐可以从上海麦克林生化科技有限公司、上海阿拉丁生化科技股份有限公司等企业购得,也可以自行制备。
本发明的另一种典型实施方式提供了一种1-烯丙基-3-甲基咪唑氯盐的制备方法,将105.84mL烯丙基氯与79.61mL 1-甲基咪唑加入500mL烧瓶(摩尔比1.3:1,烯丙基氯过量),在N2氛围下,55℃回流,并磁力搅拌(8-9h)。蒸出剩余烯丙基氯,再旋蒸,然后用乙醚洗涤。产物为浅黄色透明液体,即为1-烯丙基-3-甲基咪唑氯盐(AMIMCl)。.
本发明还提供了一种上述制膜方法制备的膜材料。本发明制备的膜材料具有较高的吸湿性、较好的机械性能及优良的阻光性能。
本发明还提供了一种上述膜材料在电泳、离子交换、渗透、过滤或包装材料中的应用。
为了使得本领域技术人员能够更加清楚地了解本申请的技术方案,以下将结合具体的实施例与对比例详细说明本申请的技术方案。
实施例1丁二酸酐的合成
4g丁二酸和6.4mL乙酸酐加入圆底烧瓶中,混合物在100℃下搅拌直至变为澄清溶液,继续搅拌1h,冷却至室温出现沉淀物,此混合物于室温下放置2h,再在8℃下保持30min确保反应充分。反应后产物经过抽滤、乙醚洗涤、干燥处理,得到透明的白色固体。反应产物的结构用1H NMR证明,1H NMR谱图中,δ2.9ppm(s,4H),δ2.5和3.3ppm(m,DMSO)。
实施例2离子液体AMIMCl的合成
1-甲基咪唑在使用前应进行旋蒸处理。将105.84mL烯丙基氯与79.61mL1-甲基咪唑加入500mL烧瓶(摩尔比1.3:1,烯丙基氯过量),在N2氛围下,55℃回流,并磁力搅拌(8-9h)。蒸出剩余烯丙基氯,再旋蒸,然后用乙醚洗涤。产物为浅黄色透明液体。.1H NMR(400MHz,D2O):δ3.94ppm(s,3H),δ4.85ppm(d,2H),δ5.43ppm(m,1H),δ6.09ppm(m,2H),δ7.48ppm(s,1H),δ7.50ppm(s,1H),δ8.79ppm(s,1H).ESI-MS:m/z(+)123.8,m/z(-)35.5.
实施例3纤维素溶液的制备
将0.3g微晶纤维素放于盛有4g AMIMCl离子液体中,在90℃下反应,直至纤维素全部溶解,得到纤维素溶液。
实施例4
SAD按一定比例加入到纤维素离子液体溶液中(纤维素葡萄糖单元与SAD之间的摩尔比为1:1),然后,二甲基亚砜(DMSO)(五滴)加入到纤维素离子液体溶液中以降低反应溶液的粘度使该溶液混合均匀。1小时后,得到改性纤维素溶液;加入蒸馏水而再生,过滤后,再与蒸馏水混合再次强力搅拌3次以去除丁二酸酐和离子液体残留物,然后进行冷冻干燥即得改性纤维素。
实施例5
SAD按一定比例加入到纤维素离子液体溶液中(纤维素葡萄糖单元与SAD之间的摩尔比为1:2),然后,二甲基亚砜(DMSO)(五滴)加入到纤维素离子液体溶液中以降低反应溶液的粘度使该溶液混合均匀。1小时后,得到改性纤维素溶液;加入蒸馏水而再生,过滤后,再与蒸馏水混合再次强力搅拌3次以去除丁二酸酐和离子液体残留物,然后进行冷冻干燥即得改性纤维素。
实施例6
SAD按一定比例加入到纤维素离子液体溶液中(纤维素葡萄糖单元与SAD之间的摩尔比为1:3),然后,二甲基亚砜(DMSO)(五滴)加入到纤维素离子液体溶液中以降低反应溶液的粘度使该溶液混合均匀。1小时后,得到改性纤维素溶液;加入蒸馏水而再生,过滤后,再与蒸馏水混合再次强力搅拌3次以去除丁二酸酐和离子液体残留物,然后进行冷冻干燥即得改性纤维素。
实施例7
SAD按一定比例加入到纤维素离子液体溶液中(纤维素葡萄糖单元与SAD之间的摩尔比为1:4),然后,二甲基亚砜(DMSO)(五滴)加入到纤维素离子液体溶液中以降低反应溶液的粘度使该溶液混合均匀。1小时后,得到改性纤维素溶液;加入蒸馏水而再生,过滤后,再与蒸馏水混合再次强力搅拌3次以去除丁二酸酐和离子液体残留物,然后进行冷冻干燥即得改性纤维素。
实施例8
SAD按一定比例加入到纤维素离子液体溶液中(纤维素葡萄糖单元与SAD之间的摩尔比为1:5),然后,二甲基亚砜(DMSO)(五滴)加入到纤维素离子液体溶液中以降低反应溶液的粘度使该溶液混合均匀。1小时后,得到改性纤维素溶液;加入蒸馏水而再生,过滤后,再与蒸馏水混合再次强力搅拌3次以去除丁二酸酐和离子液体残留物,然后进行冷冻干燥即得改性纤维素。
实施例9
将实施例4制备的3.5g改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中。
实施例10
将实施例5制备的3.5g改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中。
实施例11
将实施例6制备的3.5g改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中。
实施例12
将实施例7制备的3.5g改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中。
实施例13
将实施例8制备的3.5g改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中。
实施例14
向实施例4制备的改性纤维素溶液中加入多潘立酮(多潘立酮记为dom)在45℃下搅拌24小时以保证足够的药物负载,然后取3.5g负载多潘立酮的改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中,使得膜中多潘立酮的负载浓度为2.5mg/g(每克改性纤维素膜中负载2.5mg多潘立酮)。
实施例15
向实施例5制备的改性纤维素溶液中加入多潘立酮(多潘立酮记为dom)在45℃下搅拌24小时以保证足够的药物负载,然后取3.5g负载多潘立酮的改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中,使得膜中多潘立酮的负载浓度为2.5mg/g(每克改性纤维素膜中负载2.5mg多潘立酮)。
实施例16
向实施例6制备的改性纤维素溶液中加入多潘立酮(多潘立酮记为dom)在45℃下搅拌24小时以保证足够的药物负载,然后取3.5g负载多潘立酮的改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中,使得膜中多潘立酮的负载浓度为2.5mg/g(每克改性纤维素膜中负载2.5mg多潘立酮)。
实施例17
向实施例7制备的改性纤维素溶液中加入多潘立酮(多潘立酮记为dom)在45℃下搅拌24小时以保证足够的药物负载,然后取3.5g负载多潘立酮的改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中,使得膜中多潘立酮的负载浓度为2.5mg/g(每克改性纤维素膜中负载2.5mg多潘立酮)。
实施例18
向实施例8制备的改性纤维素溶液中加入多潘立酮(多潘立酮记为dom)在45℃下搅拌24小时以保证足够的药物负载,然后取3.5g负载多潘立酮的改性纤维素溶液转入聚四氟乙烯模具(Φ=8cm),并在室温下放置2h。将其浸入冰水中30min,然后用冰水洗涤至少三次,再浸入5%的丙三醇溶液中30min,室温下成膜。将干燥后的膜剥离并储存在相对湿度≤20%的干燥器中,使得膜中多潘立酮的负载浓度为2.5mg/g(每克改性纤维素膜中负载2.5mg多潘立酮)。
将纤维素记为MCC,将改性纤维素记为MS,其中,η代表MS中纤维素葡萄糖单元与SAD之间的摩尔比,将改性纤维素负载多潘立酮记为MS/dom,进行表征如下:
用FTIR、TG/DSC、和XRD来分析再生的载药和未载药的SAD改性纤维素样品的结构和结晶度的变化。
FTIR光谱(图1)充分地描述了载药和未载药的MCC,MS的特征峰,对于原纤维素(曲线a),在2900.81cm-1出现的是羟基的伸缩振动吸收峰,在1647.40cm-1处出现的峰是与水的吸收相关。此外,在1112.48和1164.39cm-1处的特征峰是C-O的伸缩振动,它分别属于C-OH和C-O-C。对于改性纤维素MS(曲线c)红外光谱显示了MCC的所有特征峰,此外,在1727.90cm-1处出现了新的吸收峰,这是羰基的伸缩振动吸收峰,这属于MCC与SAD交联后新形成的酯羰基。在2888.03cm-1处的吸收峰强度明显增加,这是改性后CH2和CH相互作用的结果。所有的证据都证明MCC与SAD通过形成酯羰基而改性成功。与此同时,曲线d,载药样品MS/dom,显示了改性纤维素MS的主要的吸收峰而没有新的峰出现,这表明药物知识包封在MS中而没有新化学键的形成。此外,MS/dom的曲线在1727.90cm-1处表现出明显的密集峰,这是由于MS中在1734.67处的峰和dom中在1709.09cm-1处的峰的重叠,这就证明在药物存在于载药聚合物中。与此同时,药物中的特征峰发生了移动,从3523cm-1移到3377cm-1,1695cm-1移到1647cm-1,1483cm-1移到1431cm-1,1434cm-1移到1372cm-1,1380cm-1移到1323cm-1,这种红移现象表明样品中氢原子氧原子之间发生氢键作用。因此,FTIR光谱结果表明,MS和药物dom有强烈的相互作用是由于氢键的形成。这就证实了如图2所示的药物与聚合物之间的物理相互作用。这可以由XRD和TG/DSC的结果进一步例证。
改性纤维素MS与多潘立酮可能的相互作用的结构式如下:
晶体结构和结晶度是衡量化合物的重要指标。原始MCC和载药及未载药的改性MS的结晶性能在图2中进行比较,结晶度大小是MS﹤MS/dom﹤MCC。对于原始纤维素(曲线a),其XRD衍射图案在2θ为15.27°,16.46°,22.66°,34.57°处表现出四个强峰,其相应的晶面分别为(101)(10ī)(002)和(040),这是纤维素Ⅰ的结构特征峰。对于改性纤维素MS(曲线b),XRD衍射图样在2θ为21.97°处只有一个宽峰。清楚地观察到改性纤维素的结晶度下降,它可以被解释为在离子液体溶解过程中分子间氢键被破坏。这些XRD衍射数据说明了纤维素改性的成功。同时,负载药物多潘立酮的改性样品的结晶特性呈递在曲线d中。可以看出,XRD衍射图案在2θ为22.73°处表现出一个强烈的峰,并且与未载药的改性纤维素样品相比,结晶度略有增加。这是由于改性纤维素和药物中的O和H原子之间形成的氢键,加强氢键相互作用导致MS/dom的结晶度的提高。
图3为原始纤维素,载药和未载药的改性纤维素的热重(TG,左图)和微分热重(DTG,右图)。从TG和DTG曲线可以看出,始终的第一阶段出现在50~100℃,这是由于纤维素样品中水分的挥发。对于原始纤维素,开始分解的温度为281.7℃,最高分解温度达到了337.3℃。然而,对于改性纤维素,初始分解温度大约为240.3℃,最大温度为312.6℃,这表明与原始纤维素相比,改性样品有较低的热稳定性。其原因是由于在改性反应中分子间氢键被破坏及分子间的弱相互作用而引起的。热性能也证明了纤维素的成功改性。至于载药样品,从TG和DTG可以看出,它与MCC和MS是明显不同的。它的初始分解温度为240.3℃,而最大分解温度达到了360.1℃。并且分解温度的范围较宽,这一结果表明载药样品的热稳定性提高,甚至高于原始纤维素。也许,MS和药物之间氢键的形成及载药过程中样品的聚集导致了这一结果。此外,载药样品的DTG图显示了两个峰,表明了药物通过与MS形成氢键而负载成功。
吸水性分析
样品膜被剪成矩形且试样大小为15mm×10mm,在测定吸水性之前,样品在20℃条件中放于含有硅胶的干燥器(RH 20%±5%)三天恒重(W0)。然后,膜样品在20℃下转移到相对湿度为100%的干燥器(CuSO4·5H2O饱和盐溶液)中,吸收水分一周直至达到平衡的重量。吸附时间为t时,样品的重量被指出为Wt。用方程(1)来计算不同时间间隔下及平衡时吸附的水的量,至少进行三次测试:
纤维素高分子链上有许多亲水基团。水分子在亲水基质中的扩散会影响体系的物理性质,影响药物的释放。因此,研究复合材料的吸水行为是十分重要的。ηMS膜在相对湿度为98%的条件下相对于时间的吸水性行为被研究研究,并在图4中显示。结果表明,与纯纤维素膜相比,吸湿行为增强。从图中可以看出,当η从1:0变到1:3时,随着SAD含量的增加,膜吸收的水分增加,当1:3MS吸水180h时吸水达到最大值226.72%,然后,吸水性降低,但是所有的数据都高于原始纤维素膜的。这是由于SAD改性纤维素后,增加了水分子与MS中氢原子与氧原子形成氢键的作用位点。当SAD浓度达到一定值后,吸水性降低,这主要是由于来自于交联产物的疏水基团酯基和亲水基团羧基的共同作用,且疏水基团起主导作用。这一事件表明将SAD引入到纤维素中为水分子扩散到聚合物基质中提供了有效的通道,因此吸水能力增加。用SAD改性纤维素显着提高了吸湿程度。此外,还研究了改性纤维素膜的溶胀性能(ηMS),结果表明,溶胀现象符合水分吸收行为的结果。
阻光性能和透明度
膜(1厘米×2厘米)的紫外可见阻光性能用紫外可见分光光度计(uv-7504c、上海、中国)在选择波长为从200到800nm下测定。膜的透明度用以下方程(2)计算:
Transparency=-logT/x (2)
在这个方程中,T是在每个波长时的透过率;x是膜厚度(mm)。根据方程,透明度的值越高,膜越不透明。
表1显示了在紫外可见光波长范围200到800nm内在选定波长处所有薄膜的透明度和透过率。从所有的数据可以看出,纯纤维素膜的透射率从0.3%增大到57.1%。改性纤维素膜在200nm和280nm处的透光率变化不大,此外,所有的改性纤维素膜在400~800nm范围的任何波长的透光率略高于纯纤维素膜。与此同时,随着SAD含量的增加,透光率先增大,然后下降。这些数据表明,加入SAD改善了纤维素膜的透光率,这是由于氢键的形成引起的。根据表中的信息也可以看出,无论SAD的含量的多少,透明度都比较大,这表明所有的复合纤维素膜是不透明的,这有利于它的应用。
表1 MS膜的阻光性能和透明度值
机械性能
用配备500N拉力载荷单元的微机控制电子万能试验机((WDL-005,济南,中国)进行测试,室温下干燥后,改性纤维素膜的机械性能的三个参数抗张强度、断裂伸长率、杨氏模量被测试。样品在测试之前,十字针探头被设为5mm/min,膜的初始握持长度、宽度和厚度分别用游标卡尺(0.02mm/150mm,上海,中国)和千分尺(0.01mm)测量。
进行拉伸试验以评估调整交联剂浓度如何影响改性纤维素复合材料的机械性能。根据不同SAD含量改性的纤维素复合膜的机械性能,包含抗张强度(TS)、断裂伸长率(EB)和杨氏模量(EM)被研究。厚度和机械性能的这三个参数总结在表2。结果表明,加入SAD表现出改性的MS薄膜的机械性能得到改善。从表2中可以看出,随着SAD含量的增加,TS大幅降低,纯纤维素膜的TS为5.58MPa,而1:5MS样品的只有3.08MPa,降低了差不多2倍,这表明,与纯纤维素样品相比,改性纤维素膜产生较低的应力。这也证明了在改性系统中氢键相互作用是减弱的。对于EB和EM,从表2中可以看出,1:5MS样品的EB为21.9,大约为纯纤维素膜的3倍,而EM从178.879MPa降到35.315MPa,与初纤维素相比大约减小了5倍,所有的数据表明纤维素膜在TS、EB和EM上发生了重大的变化,这表明纤维素矩阵由于自加固材料的刚度使其有足够的应力转移。同时,这种材料表现出弹性和灵活性且不易碎,原因是改性使得纤维素获得了更稳定的结构,此外,所有新形成的键使分子之间的相互作用减弱。一般来说,每个聚合物不仅有助于复合膜的性能,但也引起聚合物-聚合物的相互作用,影响整体系统的机械性能。与此同时,甘油也会影响薄膜的TS和灵活性。
此外,表2还显示了不同的改性MS膜的厚度,结果表明,SAD浓度越高,薄膜表现出的厚度越大。随着SAD含量的增加改性膜的EM得到提高,这将有助于增加生物膜的厚度和机械阻力。总之,膜材料的机械性能在一定程度上得到了提高,改善的机械性能拓宽了改性纤维素膜的应用范围。
表2 MS膜的机械性能
体外降解研究
纤维素膜的体外降解研究是在含有溶菌酶的磷酸盐缓冲液(PBS)(pH 5.20)中在37℃下在不同时间间隔(1,2,4,6,8,10,12,24和48h)中孵育进行的,这与Zheng[37]的描述类似。溶菌酶降解PBS缓冲液是来自于PBS,用醋酸调节pH至5.2,再加入0.3w/v%鸡蛋清(溶菌酶)制备的。改性纤维素膜剪成2cm×2cm长,使用前在60℃下干燥衡重并记为M0。将膜浸入到降解液中相应的时间间隔后取出,用蒸馏水洗涤,用真空泵抽滤并在50℃下干燥衡重并重新称量为Mt。降解性能通过方程(3)中的降解率进行了检测:
用不同SAD含量改性的纤维素膜在有溶菌酶的磷酸盐缓冲溶液中的体外降解被研究,结果显示在图5中。可以观察到,复合材料膜在第一个1小时内很快降解,随后缓慢下降,甚至在48小时后达到平衡。纯纤维素降解非常迅速是由于降解机理是水解反应,在最终降解时间48小时时,其降解率为89.01%,如果降解时间延长,降解率将增加,甚至达到最大值100%。与此同时,无论在什么时间阶段,所有改性膜的降解率都随着SAD含量的增加而不断下降。具有最低降解速率的样品为1:5MS,它们的降解率为49.66%,约为原始样品的一半。因此,用SAD改性的纤维素膜材料很在生态环境中延长存在。系统的所有数据表明,加入SAD后,改善了样品的生物降解行为并延长了其使用寿命,这是由于纤维素改性后侧链暴露于基质外,阻碍了酶进入纤维素中的反应位点。
形态学评价
SEM研究的目的是获得用SAD改性的纤维素膜的形态特征。不同放大倍率下拍摄的空白纤维素,改性纤维素MS和载药样品MS膜的断面面和表面的SEM照片显示于图6,第Ⅰ行的照片是在相同的放大倍数下样品的微观表面结构,第Ⅱ行显示的是样品的断裂面结构。从第I行样品的表面可以看出,纯纤维素样品a1的照片中呈现了具有许多气泡的光滑且均匀的表面,没有任何沉淀物,这种现象表明纤维素完全溶解在AMIMCl离子液体中。对于改性纤维素MS样品b1,它也获得了光滑的表面,然而,有许多具有不同尺寸的固体颗粒存在,这是由于改性过程中含有残余的交联剂。该结果说明纤维素的表面形态通过SAD改性纤维素而破坏。与此同时,载药的改性纤维素膜呈现出粗糙的微观表面。从图中可以看出,药物出现粘附于改性样品的表面,并且一些药物嵌入到纤维素基质中。第Ⅱ行中的断面图片也提供了证据确保这一过程。空白纤维素膜材料中断面结构是分层的,然而样品MS和载药膜却存在紧凑和致密的结构,对于载药样品,可以发现药物粘附在横截面上。所有数据意味着药物与聚合物基质作为物理相互作用而不是化学物相互作用。同时,这种现象也可以表明药物装载的成功,这一特性的所有结果为药物释放提供了理论依据。
此外,图7显示了改性纤维素MS和载药MS膜在不同倍率下的表面形貌的SEM图像。从图中A1和A2可以看出,改性纤维素膜表面有一些大小不同的沉淀。对载药MS膜样品,可以观察到,多潘立酮药物位于改性纤维素纤维的表面上,甚至包裹在纤维素基质中。并且事实上从这些图像中可以推导出药物和改性纤维素材料之间的强相互作用。所有结果表明,该改性纤维素膜对药物控释是有利的。
多潘立酮浓度的测量:药物释放研究
多潘立酮浓度用紫外-可见分光光度计用10mm石英比色皿在287nm处测定,首先,多潘立酮的吸收光谱被记录并检测到最大吸收出现在287nm处。校准曲线是使用已知多潘立酮浓度的不同溶液(浓度范围为0.01–0.1mg/mL)建立的,药多潘立酮浓度和吸光度的标准曲线方程在Fig.S2中显示,校准曲线方程是y=0.11333x-0.0116,y代表多潘立酮浓度,x是吸光度,相关系数R2=0.999。
负载多潘立酮药物的MS复合膜的体外释放通过含有不同SAD含量的MS膜在磷酸盐缓冲液(PBS,pH 7.4)中的静态释放进行研究,这是根据Ciolacu和Shao[38-39]在文献中描述的做了部分修改而得。称量一定量的含多潘立酮药物量为0.25mg/g的载药样品,放于锥形瓶中,然后,向里加入50mL溶液介质,在37℃下保持。在不同的时间间隔(最初5小时每0.5h收集依次,后6小时每1小时收集一次)取出5毫升的样品溶液,另外立即更换5mL新鲜的磷酸盐缓冲液,以保持一个恒定的溶液体积。在不同的时间得到的样品用紫外可见分光光度计(UV-7504C、上海、中国)在最大波长287nm时测定。所有的研究进行了三次测试。
包封的药物从聚合物基质的释放行为对药物的活性是至关重要的,此外,药物的稳定性必须在负载和释放后保持。为了理解载药MS膜的体外药物释放行为,实验在pH 7.4缓冲介质中进行,并在37℃下孵育,以模拟人体的条件。图8比较了MS膜中SAD含量对体外释放曲线的影响,随着SAD含量的增加,药物的最终的累积释放量分别为98.88%,95.25%,92.55%,88.24%,81.08%和78.2%。从图中可以看出,体外释放曲线分为两个阶段,第一阶段是前5h的体外释放,这是由于存在于MS膜表面的药物发生突释。表面药物溶解产生的空隙为溶解介质渗透到膜内部提供了曲折的通路,这进一步增强了药物释放直到其完全释放。从图中可以看出,载药纯纤维素的累积释放速率最高,数据为98.88%,而在ηMS/dom样品中,释放较缓慢,特别是1:5MS/dom样品,仅为78.2%,说明改性纤维素膜可以达到缓释效果。与空白载药的纤维素膜相比,随SAD含量的增加累积释放率降低,这是由于MS和药物之间形成氢键相互作用的位点增加,这加强了膜基质的稳定性,因此,减慢了药物的释放。因此,SAD的浓度是可以控制多潘立酮释放速率的重要因素。总而言之,这种现象的原因是该体系中氢键的增加。从这项测试中可以发现,改进的药物递送系统保护了药物多潘立酮,并确保了其及时在人体环境中发挥治疗效果,这可以延长药物释放周期和提高药物耐受性。
释放机理研究
为了研究多潘立酮药物从改性纤维素膜中的释放,体外释放数据均符合
Korsmeyer-Peppa方程:
Q/Q0=Ktn (4)
在上述公式中,Q/Q0是在时间t时的药物释放率;K为速率常数;n为扩散指数,依赖于释放机制。
如果n≤0.5、释放机制遵循Fickian扩散,如果0.5<n<1,释放为非Fickian扩散或不规则传输,如果n=1,药物释放遵循零级药物释放和案例II传输,当n>1,释放机制为超级Ⅱ型传输。此模型用于聚合物剂型时,释放机制是未知的或超过一个释放现象存在于制剂中。
采用Korsmeyer-Peppas模型研究了ηMS基质中dom的释放机制,并将数据拟合到表3中。从表中可以看出,样品MCC和1:1MS/dom显示指数值(n)在1.095至1.032的范围内(>1),表明释放机制是超级II型传输。而对于1:2MS/dom至1:5MS/dom的样品,表中的指数值(n)在0.98至0.73范围内(0.5<n<1),表明释放机制遵循非Fickian扩散或不规则传输。
表3药物释放率参数
(速率常数(K),释放指数(n),相关系数(R2),药物释放80%的时间(单位:h)(T80%))
本发明成功制备了SAD改性纤维素膜,通过FTIR,XRD和TG/DSC证明了药物的改性和负载的成功。并且膜的性能得到改善。1:3MS膜的吸水性能最高,为226.72%,透射率也最好。此外,该膜还表现出优越的弹性和柔韧性并且不易碎,因为其EB从8.4%增加至21.9%,并且EM从178.879MPa降低至35.315MPa。所有薄膜的降解率数据表明,改性纤维素膜是抗降解的。对于载药样品,SEM照片证明,药物的负载是由于物理相互作用。释放结果表明,该膜延长了药物的释放时间,是药物缓释的良好原料,并且药物释放遵循超级Ⅱ型传输和非Fickian扩散机制,这表明扩散负责dom药物的控制释放。总之,改性纤维素膜是形成缓释材料的理想选择。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!