一种制备并纯化4,4’‑二甲氧基三苯基氯甲烷的工业方法与流程
本发明涉及化工制备技术领域,具体是一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法。
背景技术:
4,4'-二甲氧基三苯基氯甲烷,英文别名:dmt-cl。physicalorganicchemistry(1972-1999),(7),769-78;1989报道了:以对甲氧基溴化镁格式试剂与4-甲氧基二苯甲酮为原料制备dmt-cl的方法。该法需用到乙醚或者四氢呋喃的格式试剂溶液,在制备和使用格式试剂过程中,乙醚有高温爆炸风险,四氢呋喃成本较高;同时该反应体系对水分要求非常高,故此法不易工业化方便操作。
famingzhuanlishenqinggongkaishuomingshu,1432553,30jul2003文献做了很多如温度,反应配比,反应时间条件的数据,但对纯化方法未做进一步明确阐述,且全文未提及该文献所能做到的产品dmt-cl纯度如何。dmt-cl作为生物核苷与核苷酸的羟基保护剂使用,必然对纯度有严格的要求。
indianpat.appl.,2012mu03407,06jun2014报道了以苯甲醚和三氯甲苯为主原料制备dmt-cl的方法,该法报道的案例最高纯度可达到99.73%,该法使用正庚烷或正己烷为溶剂精制粗品dmt-cl,但正庚烷及正己烷作为精制溶剂使用,成本较一般普通溶剂偏高,不易产业化使用。
因此,本发明提供一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法。
技术实现要素:
本发明的目的在于提供一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,以苯甲醚与三氯甲苯为起始原料,经过f-c反应、盐酸水解反应、氯代反应制备出dmt-cl粗品,粗品dmt-cl纯度大于98.5%,dmt-cl粗品经过溶剂结晶得到大于99.9%纯度的dmt-cl成品,dmt-cl成品收率大于80%;具体制备步骤为:
(1)将苯甲醚和三氯甲苯经过路易斯酸催化剂三氯化铝的催化,在0-30℃反应4-10h得到混合反应液ⅰ;
(2)在0-30℃下,将稀盐酸缓慢加入混合反应液ⅰ中,在此体系中搅拌水解1-3h后停止搅拌;
(3)加入溶剂a萃取水相,分出有机相;水层再用溶剂a萃取2次,合并有机相,并用无水硫酸钠干燥2h;
(4)减压浓缩回收溶剂a,回流状态下滴加氯代试剂ⅱ反应4-6h;
(5)加入溶剂b结晶,体系降温至0-30℃后,固液分离得到粗品dmt-cl,该粗品纯度大于98.5%;
(6)粗品dmt-cl经过混合溶剂b及c的重结晶,固液分离后得到dmt-cl湿品,分离后的产品母液经过浓缩gc定量后,溶剂回收套用,dmt-cl湿品干燥后得到收率大于80%、纯度大于99.9%成品dmt-cl。
作为本发明进一步的方案:所述苯甲醚与三氯甲苯的摩尔比为(2.0-2.6):1。
作为本发明进一步的方案:所述苯甲醚和三氯甲苯经过路易斯酸催化剂三氯化铝的催化反应温度为0-10℃。
作为本发明进一步的方案:所述溶剂a为二氯甲烷、三氯甲烷、二氯乙烷、乙酸乙酯或乙酸丁酯。
作为本发明进一步的方案:所述溶剂b为石油醚、环己烷、正己烷或正庚烷。
作为本发明进一步的方案:所述溶剂b为石油醚。
作为本发明进一步的方案:所述溶剂c为二氯甲烷、三氯甲烷、二氯乙烷、乙酸乙酯或乙酸丁酯。
作为本发明进一步的方案:所述溶剂c为二氯甲烷。
作为本发明进一步的方案:所述溶剂c为粗品dmt-cl质量体积的1.5-4.5倍,溶剂b为粗品dmt-cl质量体积的8-20倍。
作为本发明进一步的方案:所述溶剂c为粗品dmt-cl质量体积的2-3倍,溶剂b为粗品dmt-cl质量体积的10-16倍。
作为本发明进一步的方案:所述氯代试剂ⅱ为二氯亚砜或草酰氯。
与现有技术相比,本发明的有益效果是:
该制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法设计合理,发明中所用的萃取溶剂可回收套用,结晶过程中的溶剂也可回收套用,该方法能避免使用格式试剂和较昂贵的精制溶剂,达到安全环保工业化生产高纯度dmt-cl的目的,满足生物核苷与核苷酸的高质量标准的羟基保护剂使用;该法生产的dmt-cl纯度应大于99.9%,是一种大量生产并能有效纯化4,4'-二甲氧基三苯基氯甲烷(dmt-cl)新方法。
附图说明
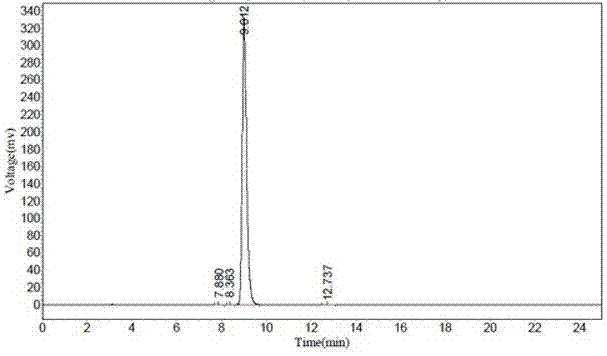
图1为实施例1制备的4,4’-二甲氧基三苯基氯甲烷的hplc图谱。
图2为实施例3制备的4,4’-二甲氧基三苯基氯甲烷的hplc图谱。
具体实施方式
下面结合具体实施方式对本专利的技术方案作进一步详细地说明。
4,4’-二甲氧基三苯基氯甲烷(dmt-cl)的结构式为:
实施例1
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
a.在1l三口瓶中加入150g三氯甲苯(m=195.48,n=0.767mol)和174.1g的苯甲醚(m=108.14,n=1.61mol),加入催化剂三氯化铝106.67g(m=133.34,n=0.80mol),反应温度控制在15℃,搅拌反应5.5h,得到混合反应液ⅰ;
b.在8℃下,将3150g5%稀盐酸加入混合反应液ⅰ中,水解反应1h,加入二氯甲烷400g,搅拌静置分层,分出有机相,再用二氯甲烷400g*2次萃取水相,合并二氯甲烷有机相,用无水硫酸钠干燥2h;
c.浓缩二氯甲烷有机相,回收溶剂二氯甲烷,加入二氯亚砜132.65g(m=118.97,n=1.115mol),在回流状态下反应4h,滴加石油醚260g,析晶完全后降温至25℃,固液分离后得到粗品dmt-cl,粗品dmt-cl的hplc纯度为98.72%;
d.粗品dmt-cl加入2倍质量体积的二氯甲烷、10倍质量体积石油醚进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl213.15g,纯度99.94%,收率82.02%。回收混合溶剂,分离后的产品母液经过浓缩gc定量后,实现溶剂回收套用。成品dmt-cl的hplc图谱见图1。
可以看出,该批次产品纯度>99.9%,单个杂质<0.03%。
实施例2
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
a.在100l反应釜中加入15kg三氯甲苯(m=195.48,n=76.73mol)和19.08kg的苯甲醚(m=108.14,n=176.48mol),加入催化剂三氯化铝10.67kg(m=133.34,n=80.02mol),反应温度控制在25℃,搅拌反应7.5h,得到混合反应液ⅰ。
b.在5℃下,将315kg5%稀盐酸加入到装有混合反应液ⅰ的500l反应釜中,水解反应2.5h,加入乙酸乙酯40kg,搅拌静置分层,分出有机相乙酸乙酯,再用乙酸乙酯40kg*2次萃取水相,合并乙酸乙酯有机相,用无水硫酸钠干燥2h。
c.浓缩乙酸乙酯有机相,回收溶剂乙酸乙酯。加入草酰氯14.15kg(m=126.93,n=111.5mol),在回流状态下反应5h,滴加环己烷26kg,析晶完全后降温至20℃,固液分离后得到粗品dmt-cl。该粗品hplc纯度为98.57%。
d.粗品dmt-cl加入1.6倍质量体积二氯乙烷、12倍质量体积环己烷进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl21.87kg,成品dmt-cl纯度99.92%,成品dmt-cl收率84.12%。回收混合溶剂,用气相色谱gc定量后再套用。
实施例3
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
a.在500l反应釜中加入75kg三氯甲苯(m=195.48,n=383.67mol)和103.72kg的苯甲醚(m=108.14,n=959.17mol),加入催化剂三氯化铝53.35kg(m=133.34,n=400.1mol),反应温度控制在8℃,搅拌反应6.5h,得到混合反应液ⅰ。
b.在15℃下,将1420kg5%稀盐酸加入到装有混合反应液ⅰ的1000l反应釜中,水解反应3h,加入二氯甲烷200kg,搅拌静置分层,分出有机相二氯甲烷,再用二氯甲烷200kg*2次萃取水相,合并二氯甲烷有机相,用无水硫酸钠干燥2h。
c.浓缩二氯甲烷有机相,回收溶剂二氯甲烷。加入草酰氯70.74kg(m=126.93,n=557.32mol),在回流状态下反应6h,滴加石油醚130kg,析晶完全后降温至15℃,固液分离后得到粗品dmt-cl。该粗品hplc纯度为98.87%。
d.粗品dmt-cl加入2.5倍质量体积二氯甲烷、15倍质量体积石油醚进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl108.58kg,成品dmt-cl纯度99.92%,成品dmt-cl收率83.52%。回收混合溶剂,用气相色谱gc定量后再套用。成品dmt-cl的hplc图谱见图2。
可以看出,该批次产品纯度>99.9%,单个杂质<0.05%。
实施例4
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
在500l反应釜中加入75kg三氯甲苯(m=195.48,n=383.67mol)和103.72kg的苯甲醚(m=108.14,n=959.17mol),加入催化剂三氯化铝53.35kg(m=133.34,n=400.1mol),反应温度控制在25℃以下,搅拌反应9.5h,得到混合反应液ⅰ。
在0-20℃下,将475kg5%稀盐酸加入到装有混合反应液ⅰ的1000l反应釜中,水解反应2.5h,加入实施例三中回收的二氯甲烷123kg,搅拌静置分层,分出有机相二氯甲烷,再用二氯甲烷200kg*2次萃取水相,合并二氯甲烷有机相,用无水硫酸钠干燥2h。
浓缩二氯甲烷有机相,回收溶剂二氯甲烷。加入草酰氯70.74kg(m=126.93,n=557.32mol),在回流状态下反应5h,滴加石油醚130kg,析晶完全后降温至10℃,固液分离后得到粗品dmt-cl。该粗品hplc纯度为98.17%。
粗品dmt-cl加入3倍质量体积二氯甲烷、18倍质量体积石油醚进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl104.41kg,成品dmt-cl纯度99.91%,成品dmt-cl收率80.32%。回收混合溶剂,用气相色谱gc定量后再套用。
实施例5
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
a.在500l反应釜中加入75kg三氯甲苯(m=195.48,n=383.67mol)和103.72kg的苯甲醚(m=108.14,n=959.17mol),加入催化剂三氯化铝53.35kg(m=133.34,n=400.1mol),反应温度控制在0℃,搅拌反应4h,得到混合反应液ⅰ。
b.在0℃下,将475kg5%稀盐酸加入到装有混合反应液ⅰ的1000l反应釜中,水解反应1h,加入二氯甲烷200kg,搅拌静置分层,分出有机相二氯甲烷,再用二氯甲烷200kg*2次萃取水相,合并二氯甲烷有机相,用无水硫酸钠干燥2h。
c.浓缩二氯甲烷有机相,回收溶剂二氯甲烷。加入草酰氯70.74kg(m=126.93,n=557.32mol),在回流状态下反应4h,滴加石油醚130kg,析晶完全后降温至0℃,固液分离后得到粗品dmt-cl。该粗品hplc纯度为98.65%。
d.粗品dmt-cl加入1.5倍质量体积二氯甲烷、8倍质量体积石油醚进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl104.67kg,成品dmt-cl纯度99.91%,成品dmt-cl收率80.52%。回收混合溶剂,用气相色谱gc定量后再套用。
实施例6
一种制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法,包括以下步骤:
a.在500l反应釜中加入75kg三氯甲苯(m=195.48,n=383.67mol)和103.72kg的苯甲醚(m=108.14,n=959.17mol),加入催化剂三氯化铝53.35kg(m=133.34,n=400.1mol),反应温度控制在30℃,搅拌反应10h,得到混合反应液ⅰ。
b.在30℃下,将475kg5%稀盐酸加入到装有混合反应液ⅰ的1000l反应釜中,水解反应3h,加入二氯甲烷200kg,搅拌静置分层,分出有机相二氯甲烷,再用二氯甲烷200kg*2次萃取水相,合并二氯甲烷有机相,用无水硫酸钠干燥2h。
c.浓缩二氯甲烷有机相,回收溶剂二氯甲烷。加入草酰氯70.74kg(m=126.93,n=557.32mol),在回流状态下反应6h,滴加石油醚130kg,析晶完全后降温至30℃,固液分离后得到粗品dmt-cl。该粗品hplc纯度为98.91%。
d.粗品dmt-cl加入4.5倍质量体积二氯甲烷、20倍质量体积石油醚进行重结晶,固液分离后得到dmt-cl湿品,dmt-cl湿品干燥后得到成品dmt-cl110.66kg,成品dmt-cl纯度99.95%,成品dmt-cl收率85.13%。回收混合溶剂,用气相色谱gc定量后再套用。
该制备并纯化4,4’-二甲氧基三苯基氯甲烷的工业方法设计合理,发明中所用的萃取溶剂可回收套用,结晶过程中的溶剂也可回收套用,该方法能避免使用格式试剂和较昂贵的精制溶剂,达到安全环保工业化生产高纯度dmt-cl的目的,满足生物核苷与核苷酸的高质量标准的羟基保护剂使用;该法生产的dmt-cl纯度应大于99.9%,是一种大量生产并能有效纯化4,4'-二甲氧基三苯基氯甲烷(dmt-cl)新方法。
上面对本专利的较佳实施方式作了详细说明,但是本专利并不限于上述实施方式,在本领域的普通技术人员所具备的知识范围内,还可以在不脱离本专利宗旨的前提下做出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!