一种猪生长抑素基因编辑位点及其应用的制作方法
本发明属于生物医药技术领域,涉及一种猪生长抑素基因编辑位点及其应用,具体地说,涉及一种猪生长抑素基因sst编辑位点(30-52)及其应用。
背景技术:
将外源基因精准敲入猪基因组中,一直是极为困难的。常见的显微注射、电穿孔、磷酸钙沉淀、逆转录病毒载体感染等方法由于其随机性无法实现外源基因在基因组特定位置的定点整合;依赖于同源重组的基因打靶技术虽然可以将外源基因导入靶细胞的特定位置,但其基因打靶效率低、操作难度大,而且在细胞筛选过程中引入的标记基因也会到来后期的生物安全隐患,这些问题都极大的限制了该技术的发展应用。直到近年来人工核酸内切酶(engineeredendonuclease,een)的出现,才彻底改变了这一现状。
人工核酸内切酶技术,包括锌指核酸内切酶(zincfingerendonuclease,zfn)、类转录激活因子效应物核酸酶(transcriptionactivator-likeeffctornuclease,talen)和crispr/cas(clusteredregularlyinterspacedshortpalindromicrepeats(crispr)/crispr-associated(cas))技术。其中,以zfn和talen为代表的技术能够实现基因组定点修饰,但具有一定的细胞毒性、操作过程复杂、脱靶率高,在哺乳动物基因组编辑中应用较少。crispr/cas技术自问世以来深受科研工作者的喜爱,该技术在基因组定点修饰方面较其它技术有无可比拟的优势,操作简单、靶向精准度高,应用面广且费用低廉,经过三年来对技术的不断改进,目前该技术已被列为活细胞及个体水平最有效、最便捷的基因组定点修饰技术。
猪生长抑素生长抑素(somatostatin,sst)是一种抑制生长激素分泌的神经调节肽,主要由下丘脑分泌,具有抑制生长激素释放的作用,同时对胰岛素、胰高血糖素、促甲状腺激素等的分泌也有抑制作用。猪与人类生活密不可分,是人类最主要的肉用家畜之一,它在组织解剖、生理代谢等方面与人更为接近,是研究人类疾病的理想动物模型。因此利用crispr/cas9基因修饰技术敲除猪生长抑素基因,不仅有利于研究sst基因对猪生长发育、代谢及表达特征等方面的影响,进而在畜牧业生产上改良育种,提高猪生长率,也可作为实验动物模型进行生长抑制等相关疾病的研究。
技术实现要素:
本发明的目的在于提供一种猪生长抑素基因编辑位点及其应用,提供一个可高效敲除猪肌抑素基因(sst)的编辑位点(30-52)。该编辑位点可用于cas9介导的猪sst基因打靶,将生长抑素基因功能失活,本发明针对sst基因设计筛选高效切割的sgrna,并分析其突变效率,为构建sst基因内源性敲除或定点敲入外源基因的转基因猪提供新途径。
一种猪生长抑素基因编辑位点,是一段包含23个脱氧核糖核苷酸的dna序列,其核苷酸序列如seqidno:1所示,该位点在猪基因组的准确定位是13号染色体sst编码区第一外显子上,染色体坐标为125337589-125337611,编码区坐标为30-52。
一种猪生长抑素基因编辑位点在cas9核酸内切酶特异识别过程中的应用。
进一步,具体包括以下步骤:
(1)从目的基因序列中通过pam寻找合适的sgrna靶位点,靶位点序列为:5’-ggccgcgctctccatcgtcctgg-3’。
(2)构建含有编辑靶位点(30-52)的sgrna表达载体;
(3)将上述载体进行体外转录获得sgrna;
(4)将上述sgrna和cas9mrna按一定比例混合后共注射到猪激活后的孤雌胚中;
(5)利用t7ne1酶切检测和ta克隆测序确认sst基因被修饰。
与现有技术相比,本发明的有益效果为:
利用本发明提供的编辑位点(30-52),可以在核酸内切酶cas9的介导下以较高效率进行双链断裂,然后通过非同源重组的方式在断裂位点出删除或增加碱基达到基因定点修饰的目的。本发明对促进内源基因敲除或外源基因定点整合技术在转基因猪研究和生产中的应用具有十分重要的作用。
统计结果表明,该位点经过ssaluciferase检测体外酶切活性为65%,并能够在胚胎水平上面敲除猪sst基因,在细胞内形成sst的点突变。统计结果表明,本发明提供了针对猪生长抑素基因的sgrna,为对该基因进行有效编辑提供了依据,为研制高瘦肉率的生猪新品种提供了新策略,也为探明生长抑素的分子机制和信号通路提供了可靠的手段和素材。
附图说明
图1pdr274-sgrna表达载体示意图;
以pdr274载体为骨架,用限制性内切酶bsaⅰ线性化pdr274质粒,与sgrna寡核苷酸双链连接形成特异性表达载体pdr274-sgrna;
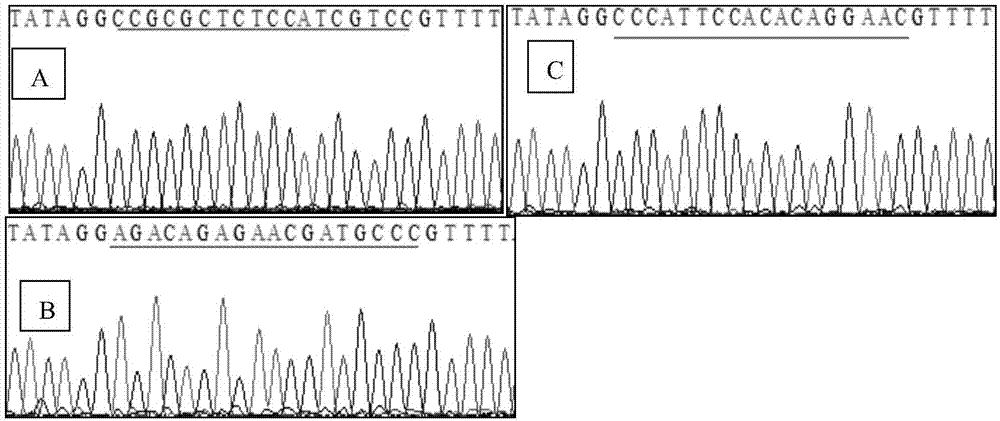
图2pdr274-sgrna质粒测序结果;
由pdr274-sgrna质粒的测序结果可知,sgrna寡核苷酸双链插入的位置和方向均正确,也即pdr274-sgrna质粒构建成功;
图3sgrna和cas9的体外转录;
a图为sst-sgrna体外转录图,100bpdnaladder(takara);
b图为cas9mrna体外转录图,5000bpdnaladder(takara);
图4t7en1酶切结果;
图5显微镜下各个时期的卵母细胞
a:50倍显微镜下未培养的卵母细胞;b:50倍显微镜下48h后成熟卵母细胞;c:400倍显微镜下具有第一极体的卵母细胞;
图6是显微镜下各个时期胚胎图;
图中a:二细胞;b:四细胞;c:八细胞。激活后的卵母细胞经ncsu-23洗涤3次后转入覆盖有石蜡油的胚胎培养液中,培养皿置于39℃,5%co2,100%湿度的恒温培养箱中。培养2天后,体视显微镜下观察记录细胞卵裂情况,统计分别处于不同发育时期的胚胎个数。分别是孤雌激活培养2天后二细胞和四细胞以及3天后的八细胞。
具体实施方式
下面结合附图和具体实施例对本发明的技术方案作进一步详细地说明。
实施例1crispr/cas9特异性敲除猪sst基因中用于特异性靶向sst基因的sgrna的设计和合成
1.靶向猪sst基因的sgrna的设计
利用网站设计sst-sgrna,具体步骤为:①登陆genbank网站,搜索猪sst基因序列并下载,其核苷酸序列如seqidno:2所示。②打开靶点预测网站(http://crispr.mit.edu/),利用靶位点在线设计工具“crisprdesigntool”,在文本框中输入基因序列,选择物种,即可得出该基因序列所有的靶位点,综合考虑各项参数后选取几组靶位点。③长20nt的寡核苷酸sgrna核心序列按gg(n)19ngg序列进行设计。
2.构建sgrna的寡聚核苷酸
根据选择的sgrna,在正义链模板的5'端添加aaacaccg,与载体质粒黏性末端互补;反义链模板的5'端添加ctctaaaac,与载体质粒黏性末端互补。分别合成上述正向寡核苷酸和反向寡核苷酸,将合成的sgrna寡聚核苷酸成对变性、退火,退火之后形成可以连入
本试验中,针对sst基因设计1个sgrnas作用靶点,其核苷酸序列见表1。
表1
实施例2sgrna、cas9mrna的准备
1.主要试剂:
pmlm3613(addgeneno.42251)、bsai、pmei酶均购自neb公司;megashortscriptkit(am1354)sgrna体外转录试剂盒、mmessagemmachinekit(am1344)cas9体外转录试剂盒、poly(a)tailingkit均购自ambion公司;pdr274(addgeneno.42250)质粒由动物胚胎工程及分子育种湖北省重点实验室提供;其他常用试剂均为国产分析纯;引物合成和dna测序由深圳华大基因公司完成。
2.操作步骤:
(1)体外转录获得sgrna:
1)表达sgrna的质粒pdr274构建(如图2所示)。
①合成寡聚核苷酸链双链:靶点活性高的sgrna-g1位点分别在其正义链模板的5'端添加tagg,与载体质粒黏性末端互补,反义链模板的5'端添加caaa,与载体质粒黏性末端互补。合成的寡聚核苷酸链单链经退火形成双链。
②线性化pdr274质粒:用限制性内切酶bsaⅰ酶切环状质粒pdr274,酶切体系为:10×nebbuffer4,5ul;100×bsa,0.5ul;bsaⅰ,2ul;pdr274环状质粒,25ul;加ddh2o水到50ul。37℃酶切2-3h,65℃失活20min。
③质粒dna酶切纯化:将酶切后的质粒利用highpurepcrproductpurificationkit纯化。
④连接:将线性化的pdr274质粒与sgrna寡核苷酸双链连接形成特异性打靶载体pdr274-sgrna。连接体系:10×buffer,2ul;t4连接酶,0.5ul;sgrna寡核苷酸双链,1ul;线性化pdr274质粒,20ng;加ddh2o水到20ul。22℃恒温培养箱内连接过夜,65℃失活20min。
⑤转化、单克隆挑选、质粒提取。
⑥pdr274-sgrna质粒构建好后测序验证,挑选正确克隆的质粒大量提取用于体外转录。
2)sgrna-pcr产物的获得
以pdr274-sgrna质粒为模板进行pcr扩增,获得sgrna-pcr产物。pcr反应体系50ul:pdr274质粒,10ng;t7-sgrna-fpg(10um),1.5ul;sgrna-rp(10um),1.5ul;2×pfumix,25ul加depch2o至50ul。pcr程序为:95℃,3min;35cycles×(94℃30s;58℃30s;72℃30s);72℃,10min;4℃,+∞。1%琼脂糖凝胶电泳检测pcr产物准确性,产物经纯化回收,最后用depc水回溶,测浓度需达到500ng/ul以上。
3)sgrna的体外转录、回收、纯化。
以获得的sgrna-pcr产物为模板进行体外转录,转录体系为10×transcriptionbuffer,2ul;rntp,2ul;t7rnapolymerasemix,2ul;sgrnapcr产物,1ug;加depch2o至20ul。37℃恒温培养箱培养2h,反应结束后加入2uldnasei,37℃反应30min。取2ul转录产物进行2%琼脂糖凝胶电泳检测。sgrna回收和提取的具体步骤为:①向转录好的sgrna内加入115uldepc水,15ul3m醋酸钠(ph5.2)混匀后,加入等体积(130ul)的1:1苯酚/异戊醇混合物,充分混合均匀后,12000-15000r/min4℃离心15min,将上清液转移到新的ep管中。②向上清液内加入等体积的氯仿,充分混匀,12000-15000r/min4℃离心15min,吸取上清液到新的ep管中。③加入等体积异丙醇沉淀rna,-80℃静置30min后,12000-15000r/min4℃离心20-30min收集沉淀。④去除上清,加入500ul的70%冷乙醇洗沉淀。乙醇晾干后,根据所需sgrna浓度,加入相应体积depc水溶解rna沉淀。注意上述全部步骤需在无rna酶的环境下进行,防止提取的sgrna被降解掉。(2)体外转录获得cas9mrna:
1)pmlm3613质粒线性化:环状质粒经pmei酶切形成线状质粒,酶切体系为:10×neb4buffer,10ul;100×bsa,1ul;pmei,5ul;质粒pmlm3613,10ug;加ddh2o至100ul;37℃酶切过夜,65℃失活20min,跑1%琼脂糖凝胶电泳检测酶切完整性。然后用琼脂糖凝胶回收试剂盒回收线状pmlm3613质粒,作为下步体外转录形成cas9mrna的模板。
2)体外转录形成cas9mrna:利用试剂盒mmessagemmachinekit进行体外转录,转录体系为:10×reactionbuffer,2ul;2×ntp/cap,10ul;enzymemix,2ul;nuclease-freewater,1ul;线状pmlm3613质粒,3ug;37℃水浴2h后加入2uldnase酶。之后跑1%琼脂糖凝胶电泳检测。
3)cas9mrna加polya尾:因为cas9mrna需要转录成蛋白行使功能,故而需加polya尾巴,使其完整。反应体系为:5×e-papbuffer,20ul;25mm;mncl210ul;10mmatp,10ul;cas9(-polya),20ul;nuclease-freewater,36ul,混匀后取0.5ul作为对照,再加入4ule-pap;37℃水浴2h,跑1%琼脂糖凝胶电泳检测。
4)cas9mrna的回收、提取:同上实验。
实施例3卵母细胞的采集和体外成熟培养
1.主要溶液配制
(1)卵母细胞体外成熟培养液的配制:成熟培养液是由改良的tcm-199构成,
也即向tcm-199培养基中加入3.05mmol/l葡萄糖、1mmol/ll-谷氨酰胺、0.91mmol/l丙酮酸钠、0.57mmol/ll-半胱氨酸、75ug/ml青霉素、50ug/ml硫酸链霉素。按照以上组分配好后,调节ph至7.2-7.4,过滤器于超净工作台上过滤后,4℃保存备用。实验2h前在此培养基基础上添加10iu/mlhcg、10iu/mlpmsg、10%pff,恒温培养箱内预热待用。
(2)胚胎培养液的配制:由ncsu-23基础培养基+4mg/mlbsa组成。ncsu-23组分为:108.73mmol/lnacl、1.19mmol/lmgso4、25.07mmol/lnahco3、4.78mmol/lkcl、1.70mmol/lcacl2、5.55mmol/l葡萄糖、1.19mmol/lkh2po4、1.00mmol/l谷氨酰胺、7.00mmol/l牛磺酸、5.00mmol/l氨乙基亚磺酸、65ug/ml青霉素、50ug/ml硫酸链霉素。配好的培养液调节ph值到中性并用过滤器过滤,4℃保存待用。
(3)dpbs洗卵液的配制:向dpbs缓冲液内加入0.1mg/mlpva和双抗(75ug/ml青霉素和50ug/ml硫酸链霉素)。
(4)猪卵泡液(pff)的配制:用16号针头注射器抽吸直径为2-6mm卵泡的卵泡液到灭菌离心管内,2000×g离心30min,吸取上清液到新的灭菌离心管中,过滤器分次过滤收集的上清液,将其分装到2ml离心管内,封口膜密封离心管,做好标记后-20℃保存待用。
(5)脱卵液的配制:向改良的tcm199中加入1mg/ml透明质酸酶。
(6)电激活液的配制:组分为0.5mmol/l氯化钙、0.5mmol/lhepes、0.1mmol/lmgso4、0.28mol/l甘露醇和0.1%bsa。
2.操作步骤
从江夏中粮屠宰场收集新鲜猪卵巢,放于37℃含青霉素和链霉素的生理盐水中,尽快送至实验室。采回的卵巢再用生理盐水冲洗3-5遍直至清洗干净,将卵巢转移到灭菌烧杯内并置于37℃恒温水浴锅中。用连接16号针头的一次性注射器,抽取卵巢上3-6mm的卵泡,收集卵泡液到50ml离心管中。待卵母细胞沉降后,吸取沉淀在体视显微镜下挑选卵丘致密完整、包裹层数较多以及胞质均匀的卵丘细胞-卵母细胞复合体(cocs)。用添加pva的dpbs将挑选出的cocs清洗3次,再用提前在培养箱内平衡2h左右的猪卵母细胞成熟培养液清洗3次。为保证卵母细胞的活力,清洗过程需在37℃热台上操作。将清洗干净的cocs放于39℃、5%co2恒温培养箱内培养48h左右,其中在培养的后20h,将卵母细胞转入未添加hcg和pmsg的成熟培养基中培养。48h后观察细胞,用0.1%的透明质酸酶消化扩散较好的卵母细胞周围的颗粒细胞,再用dpbs洗卵液清洗2-3次,最后用mtcm199将卵母细胞清洗干净。挑选细胞周围间隙明显清楚、细胞膜完整且排出第一极体的卵母细胞用来胞质显微注射。
实施例4rna胞质显微注射猪卵母细胞及rna最佳注射浓度的选择
将体外转录好的sgrna和cas9mrna测定浓度后,分别稀释到250ng/ul和1ug/ul。考虑到rna注射浓度可能会影响到体外成熟培养的卵母细胞的后期孤雌发育,为此实验中设定了不同rna浓度,通过观察统计孤雌胚发育的卵裂率,找出最佳注射浓度。实验中,对照组的成熟卵母细胞未经rna显微注射,实验组设定两两混合的sgrna浓度不变,均为12.5ng/ul,cas9mrna浓度设定100ng/ul、150ng/ul、200ng/ul和300ng/ul四个梯度。显微注射rna后立即进行体外孤雌激活,激活后的实验组和对照组细胞均置于39℃、5%co2恒温培养箱内培养。挑选发育良好的、已排出第一极体的卵母细胞进行rna胞质显微注射。
具体步骤为:
①将显微注射针插入混合好的rna注射液中,利用毛细引力将混合液吸入针内,针装入显微注射器内。
②在凹玻片中央滴20uldpbs,覆盖石蜡油,将发育成熟的卵母细胞移至液滴中。低倍显微镜下将孤雌胚胎排成一条线,调节显微注射针与持卵针,使其和孤雌胚同时聚焦在视野中央。
③低倍镜下持卵针持住1枚卵后,转至高倍镜下显微注射。在显微镜(≥400x)下,用持卵针反复吹吸卵母细胞,将细胞调至清晰、位置合适为止,然后吸持胚胎。
④将已吸有rna溶液的显微注射针快速刺入卵母细胞胞质内,注入后迅速退出注射针。将注射过的卵移至一边,继续注射下一枚卵。
⑤注射完成后将孤雌胚胎移入新的tcm-199培养基中,培养基表面用矿物油覆盖。培养皿置于39℃、5%c02恒温箱内培养。
实施例5猪卵母细胞孤雌激活
将上述各组经胞质显微注射rna的卵母细胞用电转激活液清洗3次后,口吸管依次吸取细胞,将细胞均匀摆放在已添加激活液的电击槽中。调整融合仪电压和电击时间(1.3kv/cm、30us),在体外孤雌激活卵母细胞。激活后的卵母细胞经ncsu-23洗涤3次后转入覆盖有石蜡油的胚胎培养液中,培养皿置于39℃,5%co2,100%湿度的恒温培养箱中。培养2天后,体视显微镜下观察记录细胞卵裂情况,统计分别处于不同发育时期的胚胎个数。
实施例6突变效率的检测
(1)np-40单囊胚裂解液的配制(以配制1ml为例):
具体步骤为:
①稀释10×tagbuffer为1×tagbuffer:取100ul10×tagbuffer加入900ulddh2o,共1ml,取657.6ul;
②稀释np-40(10倍):np-40原液浓度很高,取20ul原液+180ul1×tagbuffer,吸取原液时要轻吸,然后将其悬空滴入1×tagbuffer中,混匀。取42.4ul稀释后的np-40加入到657.6ul1×tagbuffer的离心管中;
③取300ul蛋白酶k到上述离心管中。将三者混匀,即可使用。
(2)卵裂卵母细胞dna的提取:
①卵母细胞孤雌激活后体外发育48h,在体视显微镜下观察各组孤雌胚发育情况,统计分别处于不同发育时期的细胞数,通过各组卵裂情况,选最佳注射浓度下的卵裂细胞将其按单个细胞用dpbs清洗3次;
②在实体显微镜下用口吸管将单个卵母细胞依次捡入装有5ulnp-40裂解液中,在裂解液表面滴加1ul的石蜡油。
③将装有细胞的裂解液按照37℃60min;55℃60min;95℃30min的程序在pcr仪内裂解。裂解完的细胞放4℃保存待用。
(3)t7en1酶切检测突变效率
①选取靶位点上下游各200bp-400bp序列,设计引物,进行pcr扩增,pcr扩增程序为:95℃,5min;(95℃30s,56℃30s,72℃30s)×30cycles;72℃,5min;4℃保存。纯化获得pcr回收产物,统一稀释到20ng/μl进行酶切鉴定。鉴定sst突变效果的引物序列如下,片段大小621bp。
sst-pl1:5’-cttcaggagtcgcgaggttc-3’
sst-pr1:5’-tgggtgggaggtactgctta-3’
(2)在15μl体系中加入10×buffer1.5μl,t7核酸内切酶0.5μl,底物12.5μl,37℃酶切30min,加入2μl10×loadingbuffer,用2%的琼脂糖凝胶电泳检测。如表2不同rna注射浓度对猪卵母细胞孤雌胚胎发育的影响中sg1样本所示,通过琼脂糖凝胶电泳可以发现,发生断裂末端连接修复的基因组会因为与原基因组不完全匹配而被t7en1切割,显示出较小的条带。
表2
体外成熟培养的卵母细胞,各组经rna显微注射,孤雌激活48h后观察卵裂情况显示:实验组各组胚胎卵裂率(65.87%、63.43%、65.88%和56.43%)显著低于对照组(83.41%)(p<0.05);100ng/ul组卵裂率与150ng/ul组和200ng/ul组间无显著差异(p>0.05),但100ng/ul组和150ng/ul组、200ng/ul组卵裂率与300ng/ul间差异显著(p<0.05)。由此得出rna胞质显微注射会影响孤雌胚的体外发育潜力,并且随着浓度增加到一定程度时,卵裂率会显著降低,由此可知cas9mrna在100-200ng/ul浓度范围内均可获得较高卵裂率的孤雌胚胎,利于后续的突变检测;
(3)ta克隆测序
①将t7en1酶切检测获得的pcr产物取1μl与pmd18-t载体连接并转化dh5α感受态细胞(transgen,cd201),挑取单克隆菌落进行测序,根据测序结果(如序列表)发现,靶基因sst缺失了靶序列的一段,基因敲除成功。
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到的技术方案的简单变化或等效替换均落入本发明的保护范围内。
序列表
<110>湖北省农业科学院畜牧兽医研究所
<120>一种猪生长抑素基因编辑位点及其应用
<141>2017-09-06
<160>2
<170>siposequencelisting1.0
<210>2
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>2
ggccgcgctctccatcgtcctgg23
<210>2
<211>1183
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgctgtcctgccgcctccagtgcgcgctggccgcgctctccatcgtcctggctctgggc60
ggtgtcactggcgcgccctcggatccccgactccgtcagtttctgcagaagtccctggct120
gctgccgctgggaagcaggtaaggagactccctcgacgccttctttcccctctcgcgaat180
cccctaaccttaccttagccttgcccctcctcccttgggtggacttaggaggtggtccca240
aagagtatcggtgcttttctgggtcccttaggcaccaaatctctcaggaaaactttcaaa300
gtccagaattcctttttacctctttgttttttccctctttgatcagcgcagtaggtcaca360
gttcaggtgagttctttggctttcaagaaaattctaagatctggggaactgagctcgagg420
ggatgatggcatctatccgcggtgctgaccatgggaggtgctgacccaggtgctgaaagc480
gcggacctctgaagcttcctaagcagtacctcccacccatgcagcagggctgggggctga540
agggcacaacagctagaacacaatatgtttacgactgtgaaaagtcttgtttcccacagt600
tgatttagtaaaaatggtaagaacaattctattttgtagctcatgatatggaaattgagt660
taaataaaatgttggcatatattttacctttgctaactaatgatgttctatttctttcta720
tgtgtgagttctaaacctgtagacactaaaaccttgcagaaacatttgagtttttaaagc780
ttccttgtgtaacttggtaaattataacaacagccctgtttttatatccttaacctattt840
taagcattacacaaagtgcacatagaaaattgggggttggatgtgattaaatctgttttt900
taaaccaagcttttccatttctttttcactatcctcattctcatccccctccccctccca960
ttccacacaggaactggccaagtacttcttggcggagctgctctctgaacccaaccagac1020
agagaacgatgccctggagcctgaagatttgtcccaggctgctgagcaggatgaaatgag1080
gctggagctgcagagatcagctaactcaaacccggccatggcaccccgagaacgcaaagc1140
tggctgcaagaatttcttctggaagactttcacatcctgttag1183
- 还没有人留言评论。精彩留言会获得点赞!