一类喹诺酮羧酸化合物及其中间体、制备方法以及应用与流程
本发明涉及喹诺酮羧酸类化合物、其制备方法以及其应用,本发明还涉及用于制备该喹诺酮羧酸类化合物的中间体。
背景技术:
1962年萘啶酸的发现,为喹诺酮的研究开启了一扇大门。喹诺酮类药物因其具有抗菌谱较广﹑活性强,与其他抗菌药未明显交叉耐药,给药方便等优点,而得到药物化学家的青眯。据研究,当喹诺酮侧链为非取代哌嗪时,其显示出很强的神经毒性,而将哌嗪环烷基化修饰后,其神经毒性明显降低(如申请号为200310122707.1的中国专利申请)。
然而,由于人们用药的广泛及不合理,导致许多菌对该类药物产生了耐药性,如:革兰氏阳性菌耐甲氧西林金葡菌/表葡菌、耐青霉素链球菌等。因此,设计合成新的具有活性的或者副作用更低的喹诺酮羧酸类衍生物迫在眉睫,以应对未来对抗生素类药物的需求。
技术实现要素:
本发明的目的在于提供一类喹诺酮羧酸化合物,以解决上述技术问题中的至少一个。
本发明的另一个目的还在于提供上述喹诺酮羧酸化合物的制备方法,以解决上述技术问题中的至少一个。
本发明的另一个目的还在于提供上述喹诺酮羧酸化合物的应用,以解决上述技术问题中的至少一个。
本发明的另一个目的还在于提供一种用于制备上述喹诺酮羧酸化合物的中间体,以解决上述技术问题中的至少一个。
本发明的另一个目的还在于提供一种用于制备上述喹诺酮羧酸化合物的中间体的制备方法,以解决上述技术问题中的至少一个。
根据本发明的一个方面,本发明提供了一类喹诺酮羧酸化合物,为结构如式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)和(ⅵ)所示的化合物中的一种或两种以上,或者为如式(ⅰ)、(ⅱ)、(ⅲ)和(ⅳ)所示的化合物形成的无毒盐中的一种或两种以上:
其中,式(ⅰ)中的r1可以为2,4-f2c6h3(2,4-二氟苯基,结构式为:
式(ⅱ)中的r4可以为(s/r)-ch2oh或(s/r)-chohch3;r5可以为boc(叔丁氧羰基,结构式为:
式(ⅲ)中的r6可以为h或oh。
式(ⅳ)中的r7可以为h或oh。
式(ⅴ)中的r8可以为2,4-f2c6h3(2,4-二氟苯基,结构式为:
式(ⅵ)中的r10可以为2,4-f2c6h3(2,4-二氟苯基,结构式为:
上述喹诺酮羧酸化合物,可以制成的医学上可接受的可溶性盐。其中医学上可接受的可溶性盐包括该衍生物与无机酸如:盐酸、氢溴酸、磷酸、硫酸等,或与有机酸如:乙酸、三氟乙酸、柠檬酸、马来酸、草酸、琥珀酸、苯甲酸、酒石酸、富马酸、扁桃酸、抗坏血酸、苹果酸、甲磺酸、对甲苯磺酸、赖氨酸、天冬氨酸等形成的盐。
根据本发明的另一个方面,本发明提供了上述喹诺酮羧酸化合物的制备方法。
当r1为2,4-f2c6h3(2,4-二氟苯基,
将1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso(二甲基亚砜)中,加入tea(三乙醇胺)和1-叔丁氧羰基-2-羟甲基哌嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅰ)化合物;
将制得的式(ⅰ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至110~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加入到无水乙醇中,外温加热至90~120℃回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅰ)化合物的盐酸盐。
当r1为2,4-f2c6h3(2,4-二氟苯基,
将1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入n-甲基吗啡啉和1-叔丁氧羰基-2-(1-羟乙基)哌嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅰ)化合物;
将制得的式(ⅰ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至110~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅰ)化合物的盐酸盐。
当r1为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和1-叔丁氧羰基-2-(1-羟乙基)哌嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅰ)化合物;
将制得的式(ⅰ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至110~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅰ)化合物的盐酸盐。
当r1为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和1-叔丁氧羰基-2-羟甲基哌嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅰ)化合物;
将制得的式(ⅰ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至110~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅰ)化合物的盐酸盐。
当r4为(s/r)-ch2oh,r5为boc(叔丁氧羰基,
将左氧氟沙星羧酸溶于dmso,加入tea和1-叔丁氧羰基-2-羟甲基哌嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅱ)化合物;
将式(ⅱ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅱ)化合物的盐酸盐。
当r4为(s/r)-chohch3,r5为boc(叔丁氧羰基,
将左氧氟沙星羧酸溶于dmso,加入tea和11-叔丁氧羰基-2-(1-羟乙基)哌嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅱ)化合物;
将式(ⅱ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅱ)化合物的盐酸盐。
当r6为h时,式(ⅲ)化合物可以由以下步骤制得:
将左氧氟沙星羧酸溶于dmso,加入tea和(r)-八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅲ)化合物。
将式(ⅲ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅲ)化合物的盐酸盐。
当r6为oh时,式(ⅲ)化合物可以由以下步骤制得:
左氧氟沙星羧酸溶于dmso,加入tea和(s/r)-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅲ)化合物。
将式(ⅲ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅲ)化合物的盐酸盐。
当r7为h时,式(ⅳ)化合物可以由以下步骤制得:
将左氧氟沙星羧酸溶于dmso,加入tea和(s/r)-八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅳ)化合物。
将式(ⅳ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~140℃回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅳ)化合物的盐酸盐。
当r7为oh时,式(ⅳ)化合物可以由以下步骤制得:
左氧氟沙星羧酸溶于dmso,加入tea和(s/r)-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应20~26h,反应完成后,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得式(ⅳ)化合物。
将式(ⅳ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至100~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤1~3次,所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤1~3次,即得式(ⅳ)化合物的盐酸盐。
当r8为2,4-f2c6h3(2,4-二氟苯基,
将1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和r-八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅴ)化合物;
将制得的式(ⅴ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅴ)化合物的盐酸盐。
当r8为2,4-f2c6h3(2,4-二氟苯基,
将1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和r-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅴ)化合物;
将制得的式(ⅴ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅴ)化合物的盐酸盐。
当r8为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和r-八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅴ)化合物;
将制得的式(ⅴ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅴ)化合物的盐酸盐。
当r8为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和s/r-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅴ)化合物;
将制得的式(ⅴ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅴ)化合物的盐酸盐。
当r8为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和s/r-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅴ)化合物;
将制得的式(ⅴ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅴ)化合物的盐酸盐。
当r10为2,4-f2c6h3(2,4-二氟苯基,
将1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和s/r-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅵ)化合物;
将制得的式(ⅵ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅵ)化合物的盐酸盐。
当r10为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和s/r-八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅵ)化合物;
将制得的式(ⅵ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅵ)化合物的盐酸盐。
当r10为c-c3h5(环丙基,
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸溶于dmso中,加入tea和s/r-7-羟基八氢吡咯并[1,2-a]吡嗪,在80~100℃的条件下微波反应10~14h,冷却至室温,加入二氯甲烷,再水洗1~3次,干燥,用无水乙醇重结晶即得到式(ⅵ)化合物;
将制得的式(ⅵ)化合物加到hcl-乙醇的混合溶剂中,其中,hcl的浓度为2~8mol/l,hcl与乙醇的体积比为1:3~3:1,外温加热至90~120℃回流10~12h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤1~3次;所得固体加到无水乙醇中,外温加热至90~120℃以上回流30~50min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤,即得式(ⅵ)化合物的盐酸盐。
根据本发明的另一方面,本发明还提供了式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)或(ⅵ)所示的化合物在制备抗革兰氏阳性菌和抗革兰氏阴性菌药物中的应用,该药物可以是片剂、胶囊、粉针或注射剂型。
根据本发明的另一方面,本发明还提供了式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)或(ⅵ)所示的化合物的盐酸盐在制备抗革兰氏阳性菌和抗革兰氏阴性菌药物中的应用,该药物可以是片剂、胶囊、粉针或注射剂型。
根据本发明的另一个方面,本发明还提供了用于合成式(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)、(ⅴ)或(ⅵ)化合物的中间体,其结构如式(ⅶ)、式(ⅷ)和式(ⅸ)所示。
其中,式(ⅶ)中的r12可以为(s/r)-ch2oh或(s/r)-chohch3;
式(ⅷ)中的r13可以为h或oh;
式(ⅸ)中的r14可以为h或oh;
根据本发明的另一个方面,本发明还提供了上述中间体的制备方法:
当r12为(s/r)-ch2oh时,式(ⅶ)化合物可以由以下步骤制得:
将(s/r)-丝氨酸加到无水甲醇中,冰盐浴下,缓慢滴加socl2,滴加完毕后,改为室温反应6~15h,反应毕,反应液浓缩至干,所得残余物加水溶解,冰盐浴下,加入nahco3,待气体释放毕,滴加clch2cocl-ch2cl2溶液,其中,clch2cocl与ch2cl2体积比为1:2~1:20,滴加完毕后,改为室温反应6~15h,反应完毕后,分出有机层,ch2cl2萃取水层1~3次,合并有机层,用饱和氯化钠溶液洗涤,加无水硫酸钠干燥,过滤,减压蒸除溶剂,得淡黄色液体(s/r)-n-氯乙酰基丝氨酸甲酯。
n-氯乙酰基丝氨酸甲酯溶于无水甲醇,冰水浴,加入tea和苄胺,在外温80~120℃以上回流40~56h,冷却至室温,减压抽除溶剂,残余物加ch2cl2溶解,水洗1~3次,饱和氯化钠溶液洗涤,无水硫酸钠干燥有机层,减压抽除ch2cl2得粗产品,柱层析得白色固体手性(s/r)-1-苄基-3-羟甲基-2,5-哌嗪二酮。
lialh4悬浮于无水thf中,冰浴下,分批加入化合物手性1-苄基-3-羟甲基-2,5-哌嗪二酮,加毕,室温搅拌30~50min,氩气保护,外温90~140℃加热回流5~8h,tlc检测反应完毕,冷致室温,冰浴下依次滴加h2o、浓度为2mol/l的naoh、h2o,室温搅拌0.5~2h,抽滤,ch2cl2洗涤,合并滤液,加无水硫钠干燥,过滤,抽除溶剂得黄色液体(s/r)1-苄基-3-羟甲基哌嗪。
(s/r)-1-苄基-3-羟甲基哌嗪溶于ch2cl2中,加tea,冰浴下,滴加(boc)2o,滴毕,室温反应10~14h,tlc检测,反应毕,依次用饱和碳酸氢钠溶液、水和饱和氯化钠溶液洗涤一次,有机层加无水硫酸钠干燥,旋除溶剂,柱层析,得黄色粘状物(s/r)-1-叔丁氧羰基-2-羟甲基-4-苄基哌嗪。
(s/r)-1-叔丁氧羰基-2-羟甲基-4-苄基哌嗪溶于甲醇中,加入pd/c或/和pd(oh)2,滴加冰醋酸1~3滴,抽除空气,通入h2,室温反应10~14h,过滤,甲醇洗涤,合并滤液,减压抽出溶剂得淡黄色固体式(ⅶ)化合物(s/r)-1-叔丁氧羰基-2-羟甲基哌嗪。
当r12为(s/r)-chohch3时,式(ⅶ)化合物可以由以下步骤制得:
将(s/r)-苏氨酸加到无水甲醇中,冰盐浴下,缓慢滴加socl2,滴加完毕后,改为室温反应6~15h,反应毕,反应液浓缩至干,所得残余物加水溶解,冰盐浴下,加入nahco3,待气体释放毕,滴加clch2cocl-ch2cl2溶液,其中,clch2cocl与ch2cl2体积比为1:2~1:20,滴加完毕后,改为室温反应6~15h,反应完毕后,分出有机层,ch2cl2萃取水层1~3次,合并有机层,饱和氯化钠溶液洗涤,加无水硫酸钠干燥,过滤,减压蒸除溶剂,得淡黄色液体(s/r)-n-氯乙酰基苏氨酸甲酯。
n-氯乙酰基苏氨酸甲酯溶于无水甲醇,冰水浴,加入tea和苄胺在外温80~120℃以上回流40~56h,冷却至室温,减压抽除溶剂,残余物加ch2cl2溶解,水洗1~3次,饱和氯化钠溶液洗涤,无水硫酸钠干燥有机层,减压抽除ch2cl2得粗产品,柱层析得白色固体(s/r)-1-苄基-3-(1-羟乙基)-2,5-哌嗪二酮。
lialh4悬浮于无水thf中,冰浴下,分批加入化合物1-苄基-3-(1-羟乙基)-2,5-哌嗪二酮,加毕,室温搅拌30~50min,氩气保护,外温90~140℃加热回流5~8h,tlc检测反应完毕,冷致室温,冰浴下依次滴加h2o、浓度为2mol/l的naoh、h2o,室温搅拌0.5~2h,抽滤,ch2cl2洗涤,合并滤液,加无水硫钠干燥,过滤,抽除溶剂得黄色液体(s/r)-1-苄基-3-(1-羟乙基)哌嗪。
(s/r)-1-苄基-3-(1-羟乙基)哌嗪溶于ch2cl2中,加tea,冰浴下,滴加(boc)2o,滴毕,室温反应10~14h,tlc检测,反应毕,依次用饱和碳酸氢钠溶液、水和饱和氯化钠溶液洗涤一次,有机层加无水硫酸钠干燥,旋除溶剂,柱层析,得黄色粘状物(s/r)-1-叔丁氧羰基-2-(1-羟乙基)-4-苄基哌嗪
(s/r)-1-叔丁氧羰基-2-(1-羟乙基)-4-苄基哌嗪溶于甲醇中,加入pd/c或/和pd(oh)2,滴加冰醋酸1~3滴,抽除空气,通入h2,室温反应10~14h,过滤,甲醇洗涤,合并滤液,减压抽出溶剂得淡黄色固体式(ⅶ)化合物手性(s/r)-1-叔丁氧羰基-2-(1-羟乙基)哌嗪。
当r13和r14均为h时,式(ⅷ)和式(ⅸ)化合物可以由以下步骤制得:
将(s/r)-脯氨酸加到无水甲醇中,冰盐浴下,缓慢滴加socl2,滴加完毕后,改为室温反应6~15h,反应毕,反应液浓缩至干,所得残余物加水溶解,冰盐浴下,加入nahco3,待气体释放毕,滴加clch2cocl-ch2cl2溶液,其中,clch2cocl与ch2cl2体积比为1:2~1:20,滴加完毕后,改为室温反应6~15h,反应完毕后,分出有机层,ch2cl2萃取水层1~3次,合并有机层,饱和氯化钠溶液洗涤,加无水硫酸钠干燥,过滤,减压蒸除溶剂,得黄色油状物(s/r)-n-氯乙酰基脯氨酸甲酯。
(s/r)-n-氯乙酰基脯氨酸甲酯溶于无水甲醇,冰水浴,加入tea和苄胺在外温80~120℃回流40~56h,冷却至室温,减压抽除溶剂,残余物加ch2cl2溶解,水洗1~3次,饱和氯化钠溶液洗涤,无水硫酸钠干燥有机层,减压抽除ch2cl2得粗产品,柱层析得白色固体(s/r)-2-苄基-六氢吡咯并[1,2-a]吡嗪-1,4二酮。
lialh4悬浮于无水thf中,冰浴下,分批加入化合物(s/r)-2-苄基-六氢吡咯并[1,2-a]吡嗪-1,4二酮,加毕,室温搅拌30~50min,氩气保护,外温90~140℃加热回流5~8h,tlc检测反应完毕,冷致室温,冰浴下依次滴加h2o、浓度为2mol/l的naoh、h2o,室温搅拌0.5~2h,抽滤,ch2cl2洗涤,合并滤液,加无水硫钠干燥,过滤,抽除溶剂得(s/r)-2-苄基八氢吡咯并[1,2-a]吡嗪。
(s/r)-2-苄基八氢吡咯并[1,2-a]吡嗪溶于甲醇中,加入pd/c或pd(oh)2,滴加冰醋酸1~3滴,抽除空气,通入h2,室温反应10~14h,过滤,甲醇洗涤,合并滤液,减压抽出溶剂得淡黄色液体式(ⅷ)或式(ⅸ)化合物(s/r)八氢吡咯并[1,2-a]吡嗪。其中从r-脯氨酸为原料出发制得式(ⅷ)化合物,从s-脯氨酸为原料出发制得式(ⅸ)化合物。
当r13和r14均为oh时,式(ⅷ)和式(ⅸ)化合物可以由以下步骤制得:
将(s/r)-4-羟基脯氨酸加到无水甲醇中,冰盐浴下,缓慢滴加socl2,滴加完毕后,改为室温反应6~15h,反应毕,反应液浓缩至干,所得残余物加水溶解,冰盐浴下,加入nahco3,待气体释放毕,滴加clch2cocl-ch2cl2溶液,其中,clch2cocl与ch2cl2体积比为1:2~1:20,滴加完毕后,改为室温反应6~15h,反应完毕后,分出有机层,ch2cl2萃取水层1~3次,合并有机层,饱和氯化钠溶液洗涤,加无水硫酸钠干燥,过滤,减压蒸除溶剂,得黄色油状物手性(s/r)-n-氯乙酰基-4-羟基脯氨酸甲酯。
(s/r)-n-氯乙酰基-4-羟基脯氨酸甲酯溶于无水甲醇,冰水浴,加入tea和苄胺在外温80~120℃回流40~56h,冷却至室温,减压抽除溶剂,残余物加ch2cl2溶解,水洗1~3次,饱和氯化钠溶液洗涤,无水硫酸钠干燥有机层,减压抽除ch2cl2得粗产品,柱层析得白色固体手性(s/r)-2-苄基-7-羟基六氢吡咯并[1,2-a]吡嗪-1,4二酮。
lialh4悬浮于无水thf中,冰浴下,分批加入化合物手性(s/r)-2-苄基-7-羟基六氢吡咯并[1,2-a]吡嗪-1,4二酮,加毕,室温搅拌30min,氩气保护,外温90~140℃加热回流5~8h,tlc检测反应完毕,冷致室温,冰浴下依次滴加h2o、2nnaoh、h2o,室温搅拌0.5~2h,抽滤,ch2cl2洗涤,合并滤液,加无水硫钠干燥,过滤,抽除溶剂得(s/r)-2-苄基-7-羟基八氢吡咯并[1,2-a]吡嗪。
(s/r)-2-苄基-7-羟基八氢吡咯并[1,2-a]吡嗪溶于甲醇中,加入pd/c或pd(oh)2,滴加冰醋酸1~3滴,抽除空气,通入h2,室温反应10~14h,过滤,甲醇洗涤,合并滤液,减压抽出溶剂得淡黄色液体式(ⅷ)或式(ⅸ)化合物(s/r)-7-羟基八氢吡咯并[1,2-a]吡嗪。其中从r-4-羟基脯氨酸为原料出发制得式(ⅷ)化合物,从s-4-羟基脯氨酸为原料出发制得式(ⅸ)化合物。
附图说明
图1为化合物s1的核磁共振氢谱图。
图2为化合物s1的核磁共振碳谱图。
图3为化合物s2的核磁共振氢谱图。
图4为化合物s2的核磁共振碳谱图。
图5为化合物s3的核磁共振氢谱图。
图6为化合物s3的核磁共振碳谱图。
图7为化合物s4的核磁共振氢谱图。
图8为化合物s4的核磁共振碳谱图。
图9为化合物s5的核磁共振氢谱图。
图10为化合物s5的核磁共振碳谱图。
图11为化合物s6的核磁共振氢谱图。
图12为化合物s6的核磁共振碳谱图。
图13为化合物s7的核磁共振氢谱图。
图14为化合物s7的核磁共振碳谱图。
图15为化合物s8的核磁共振氢谱图。
图16为化合物s8的核磁共振碳谱图。
图17为化合物s9的核磁共振氢谱图。
图18为化合物s9的核磁共振碳谱图。
图19为化合物s10的核磁共振氢谱图。
图20为化合物s10的核磁共振碳谱图。
图21为化合物s11的核磁共振氢谱图。
图22为化合物s11的核磁共振碳谱图。
图23为化合物s12的核磁共振氢谱图。
图24为化合物s12的核磁共振碳谱图。
图25为化合物s1-tm的核磁共振氢谱图。
图26为化合物s2-tm的核磁共振氢谱图。
图27为化合物s3-tm的核磁共振氢谱图。
图28为化合物s4-tm的核磁共振氢谱图。
图29为化合物s5-tm的核磁共振氢谱图。
图30为化合物s6-tm的核磁共振氢谱图。
图31为化合物r1-tm的核磁共振氢谱图。
图32为化合物r2-tm的核磁共振氢谱图。
图33为化合物r3-tm的核磁共振氢谱图。
图34为化合物r4-tm的核磁共振氢谱图。
图35为化合物r5-tm的核磁共振氢谱图。
图36为化合物r6-tm的核磁共振氢谱图。
具体实施方式
下面通过实施例对本发明作进一步详细的说明。
实施例1
(s)-9-氟-2,3-二氢-3-甲基-10-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅱ)结构,以下称化合物s1)及其盐酸盐(以下称化合物s1-tm)的合成。
将丝氨酸(10.50g,0.10mol)加到无水甲醇(200ml)中,冰盐浴下,滴加socl2(29.01ml,0.40mol),滴加完毕后改为室温反应,tlc跟踪检测至反应完毕后,将反应液浓缩至干,所得残余物(丝氨酸甲酯盐酸盐,以下称化合物s1-a)直接进行下步反应。
残余物加水(120ml),冰盐浴下,加入nahco3(19.75g,0.24mol),待气体释放毕,滴加clch2cocl(8.0ml,0.11mol)的ch2cl2(120ml)溶液,30min滴毕,滴加完毕后改为室温反应,tlc跟踪检测至反应完毕,分出有机层,以ch2cl2多次萃取水层,合并有机层,加无水硫酸钠干燥,过滤,30℃旋转蒸发除去溶剂,得淡黄色液(n-氯乙酰基丝氨酸甲酯,以下称化合物s1-1)11.17g。
将化合物s1-1(9.78g,0.05mol)溶于无水甲醇(100ml),冰水浴,依次加入tea(20.6ml,0.15mol)和苄胺(6.7ml,0.06mol),在80℃的条件下回流48h,冷却至室温,加入30ml乙酸乙酯,冷冻析出大量白色固体,抽滤,再用乙酸乙酯洗涤滤饼3次(每次20ml),真空干燥后得白色固体4.36g。母液旋转蒸发除去溶剂,得淡黄色固体,以ch2cl2(30ml)溶解,柱层析得白色固体4.32g,合并共得到白色固体(1-苄基-3-羟甲基-2,5-哌嗪二酮,以下称化合物s1-2)8.68g。
将lialh4(2.88g,76mmol)悬浮于无水thf(四氢呋喃,60ml)中,冰浴下,小心分批加入化合物s1-2(4.69g,20mmol),加毕,室温搅拌30min,氩气保护下外温90℃加热回流4h,tlc检测反应完毕,冷却至室温,冰浴下依次滴加h2o(2.8ml)、naoh(浓度为2n,9.0ml)、h2o(2.8ml),室温搅拌1h,抽滤,再用ch2cl2洗涤3次(每次10ml),合并滤液,加无水硫酸钠干燥,过滤,抽滤除去固体,得4.06g黄色液体(1-苄基-3-羟甲基哌嗪,以下称化合物s1-3)。冷藏转化成为半固体,在空气中很容易吸潮。
取化合物s1-3(2.06g,10mmol)溶于15mlch2cl2中,加tea(1.53ml,11mmol),冰浴下,滴加(boc)2o(2.40g,11mmol),滴加完毕,室温反应12h,tlc检测反应完毕,依次用10ml饱和碳酸氢钠溶液、10ml水、10ml饱和氯化钠溶液各洗涤1次,有机层加无水硫酸钠干燥,旋转蒸发除去溶剂,柱层析,得黄色粘状物(1-叔丁氧羰基-2-羟甲基-4-苄基哌嗪,以下称化合物s1-4)2.68g。
取化合物s1-4(2.00g,6.5mmol)溶于20ml甲醇中,加入pd/c(0.20g),pd(oh)2(0.10g),滴加冰醋酸1滴,抽除空气,通入h2,室温反应12h。过滤,再用甲醇洗涤3次(每次5ml),合并滤液,旋干溶剂得淡黄色固体(1-叔丁氧羰基-2-羟甲基哌嗪,以下称化合物s1-5)1.38g。
将左氧氟沙星羧酸(225.0mg,0.8mmol)溶于dmso(二甲基亚砜,5.0ml),加入tea(三乙醇胺,0.44ml,3.2mmol)和化合物s1-5(259.5mg,1.2mmol),在90℃的条件下微波反应24h。反应完成后,冷却至室温,加入二氯甲烷(15ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得黄色固体105.1mg,计算得收率为27.51%,测得mp(熔点):221-223℃。
以上操作中利用的原料是l-丝氨酸,获得的是s构型的产物。
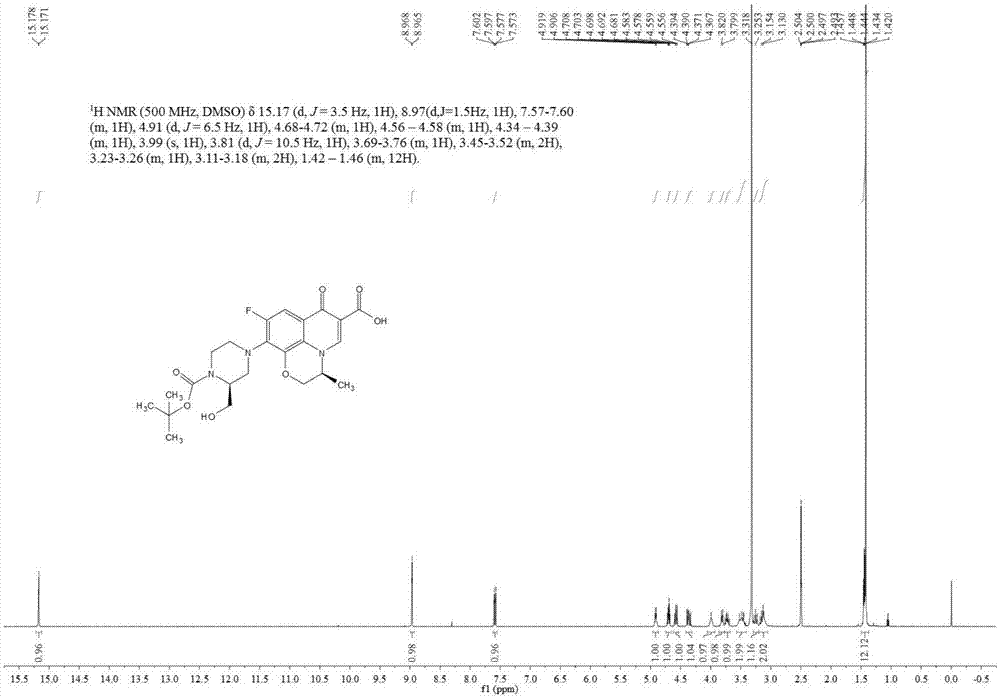
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图1、图2所示,具体的谱图数据如下所示,由此确定产物为化合物s1。
1hnmr(500mhz,dmso)δ15.17(d,j=3.5hz,1h),8.97(d,j=1.5hz,1h),7.57-7.60(m,1h),4.91(d,j=6.5hz,1h),4.68-4.72(m,1h),4.56-4.58(m,1h),4.34-4.39(m,1h),3.99(s,1h),3.81(d,j=10.5hz,1h),3.69-3.76(m,1h),3.45-3.52(m,2h),3.23-3.26(m,1h),3.11-3.18(m,2h),1.42-1.46(m,12h);
13cnmr(126mhz,dmso)δ176.36,165.99,155.59,154.26,146.17,140.70,132.04,124.75,120.07,106.72,103.15,78.86,68.08,57.87,54.83,52.87,49.9,49.5,28.04,17.87;
hrms-esi(m/z):calcd.forc23h28fn3nao7(m+na)+:500.18090;found:500.17975。
将化合物s1(29.1mg,0.061mmol)加到hcl(浓度3n,2ml)-乙醇(5ml)混合溶剂中,外温95℃加热回流10h,冷却至室温,析出白色固体,抽滤,滤渣再用无水乙醇洗涤3次(每次1ml)。所得固体加到5ml无水乙醇中,外温95℃加热回流30min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤3次(每次1ml),得黄色固体13.2mg,计算得收率为52.29%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图25所示,具体的谱图数据如下所示,由此确定产物为化合物s1-tm。
1hnmr(400mhz,dmso)δ15.10(s,1h),9.54(s,1h),9.20(s,1h),8.99(d,j=5.6hz,1h),7.59-7.64(m,1h),5.47(s,1h),4.94(s,1h),4.61(d,j=8.4hz,1h),4.40(d,j=11.2hz,1h),3.68(s,2h),3.39-3.50(m,4h),3.13(s,1h),1.46(s,3h);
hrms-esi(m/z):calcd.forc18h21fn3o5(m+h)+:378.14653;found:378.14553。
实施例2
(s)-9-氟-2,3-二氢-3-甲基-10-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅱ)结构,以下称化合物s2)及其盐酸盐(以下称化合物s2-tm)的合成。
将苏氨酸(11.95g,0.10mol)加到无水甲醇(200ml)中,冰盐浴下,滴加socl2(29.01ml,0.40mol),滴加完毕,改为室温反应,tlc跟踪检测至反应完毕,将反应液浓缩至干,所得残余物(苏氨酸甲酯盐酸盐,以下称化合物s2-a)直接进行下步反应。
所得残余物加水(120ml),冰盐浴下,加入nahco3(19.75g,0.24mol),待气体释放毕,滴加clch2cocl(8.0ml,0.11mol)的ch2cl2(120ml)溶液,30min滴加完毕,改为室温反应,反应毕,分出有机层,ch2cl2多次萃取水层。合并有机层,加无水硫酸钠干燥,过滤,30℃减压抽滤除去溶剂,得淡黄色固体(n-氯乙酰基苏氨酸甲酯,以下称化合物s2-1)17.05g。
将化合物s2-1(14.67g,70mmol)溶于无水甲醇(140ml)中,冰水浴,依次加入tea(28.8ml,0.21mol)和苄胺(9.34ml,85.4mmol),外温80℃回流48h,冷却至室温,析出大量白色固体,抽滤,再用乙酸乙酯洗涤滤饼3次(每次20ml),真空干燥后得白色固体10.96g。母液再减压抽除溶剂,得黄色固体,柱层析得白色固体1.18g,共得到白色固体(1-苄基-3-(1-羟乙基)-2,5-哌嗪二酮,以下称化合物s2-2)12.14g。
将lialh4(2.88g,76mmol)悬浮于无水thf(60ml)中,冰浴下,小心分批加入化合物s2-2(4.97g,20mmol),加毕,室温搅拌30min,氩气保护下外温90℃加热回流4h,tlc检测反应完毕,冷却至室温,冰浴下依次滴加h2o(2.8ml)、naoh(浓度2n,9.0ml)、h2o(2.8ml),室温搅拌1h,抽滤,再用ch2cl2洗涤3次(每次10ml),合并滤液,加无水硫酸钠干燥,过滤,减压抽除溶剂得4.63g黄色液体(1-苄基-3-(1-羟乙基)哌嗪,以下称化合物s2-3)。
将化合物s2-3(2.20g,10mmol)溶于15ml的ch2cl2中,加入tea(1.53ml,11mmol),冰浴下,滴加(boc)2o(2.40g,11mmol),滴加完毕后,室温反应12h,tlc检测反应完毕,依次用10ml饱和碳酸氢钠溶液、10ml水、10ml饱和氯化钠溶液各洗涤1次,有机层加无水硫酸钠干燥,减压抽除溶剂,柱层析,得黄色粘状物(1-叔丁氧羰基-2-(1-羟乙基)-4-苄基哌嗪,以下称化合物s2-4)2.70g。
取化合物s2-4(2.15g,6.7mmol)溶于15ml甲醇,加入pd/c(0.22g)和pd(oh)2(0.10g),滴加冰醋酸1滴,抽除空气,通入h2,室温反应12h,过滤,再用甲醇洗涤3次(每次5ml),合并滤液,减压抽滤除去溶剂得淡黄色固体(1-叔丁氧羰基-2-(1-羟乙基)哌嗪,以下称化合物s2-5)1.54g。
参照实施例1中化合物s1的合成方法,将化合物s1-5替换成化合物s2-5(276.4mg,1.2mmol)与左旋氧氟沙星羧酸反应即可。得黄色固体78.5mg,计算得收率为19.96%,测得mp:210-212℃。
以上操作中利用的原料是l-苏氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图3、图4所示,具体的谱图数据如下所示,由此确定产物为化合物s2。
1hnmr(500mhz,dmso)δ15.12(s,1h),8.98(s,1h),7.58(d,j=12.0hz,1h),4.92-4.94(m,1h),4.51-4.57(m,2h),4.39(dd,j=11.4,2.1hz,1h),4.23-4.27(m,1h),3.85(d,j=10.0hz,1h),3.77(s,1h),3.25-3.29(m,2h),3.19-3.22(m,3h),1.44(d,j=7.0hz,3h),1.41(s,9h),1.15(d,j=6.5hz,3h);
13cnmr(126mhz,dmso)δ176.77,166.37,156.44,155.39,146.61,141.66,132.22,125.08,121.04,107.25,103.64,78.89,68.60,63.28,57.85,55.25,51.53,50.60,28.55,21.79,18.20;
hrms-esi(m/z):calcd.forc24h30fn3nao7(m+na)+:514.19655;found:514.19542。
参照实施例1中s1-tm的合成方法,将化合物s1替换为化合物s2(49.1mg,0.1mmol)进行反应即可,得黄色固体31.1mg,计算得收率为72.50%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图26所示,具体的谱图数据如下所示,由此确定产物为化合物s2-tm。
1hnmr(400mhz,dmso)δ15.11(s,1h),9.21(s,1h),9.00-9.05(m,2h),7.62(d,j=12.0hz,2h),5.65(s,1h),4.94(d,j=6.8hz,1h),4.60(d,j=11.6hz,1h),4.41(d,j=11.2hz,1h),3.85(t,j=13.2hz,1h),3.25-3.53(m,5h),3.09(s,2h),1.45(d,j=6.8hz,3h),1.18(d,j=6.4hz,3h);
hrms-esi(m/z):calcd.forc19h23fn3o5(m+h)+:392.16217;found:392.16138。
实施例3
(s)-9-氟-2,3-二氢-3-甲基-10-(八氢吡咯并[1,2-a]吡嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅳ)结构,以下称化合物s3)的合成。
将脯氨酸(11.51g,0.10mol)加到无水甲醇(200ml)中,冰盐浴下,滴加socl2(29.01ml,0.40mol),滴加完毕,改为室温反应,tlc跟踪检测至反应完毕,反应液浓缩至干,所得残余物(脯氨酸甲酯盐酸盐,以下称化合物s3-a)直接进行下步反应。
往上步骤所得的残余物中加水(80ml),冰盐浴下,加入nahco3(19.74g,0.24mol),待气体释放毕,滴加clch2cocl(9.50ml,0.12mol)的ch2cl2(50ml)溶液,30min滴加完毕,改为室温反应6h,反应完毕后分出有机层,再用ch2cl2萃取水层3次(每次20ml),合并有机层,用饱和氯化钠溶液(30ml)洗涤,再加无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,得黄色油状物(n-氯乙酰基脯氨酸甲酯,以下称化合物s3-1)11.64g。
将化合物s3-1(11.34g,55mmol)溶于无水甲醇(120ml),冰水浴,加入tea(9.15ml,66mmol)和苄胺(7.07g,66mmol),外温80℃回流48h,冷却至室温,减压抽滤除去溶剂,滤渣加ch2cl2(100ml)溶解,先用水洗2次(每次60ml),然后用饱和氯化钠溶液(30ml)洗涤,最后用无水硫酸钠干燥有机层,减压抽滤除去ch2cl2得粗产品,柱层析得白色固体(2-苄基-六氢吡咯并[1,2-a]吡嗪-1,4二酮,以下称化合物s3-2)11.15g。
将lialh4(4.33g,114mmol)悬浮于无水thf(100ml)中,冰浴下,分批加入化合物s3-2(7.33g,30mmol),加完后,室温搅拌30min,氩气保护下外温90℃加热回流5h,tlc检测反应完毕,冷却至室温,冰浴下依次滴加h2o(8.4ml)、naoh(浓度为2n,27.0ml)、h2o(8.4ml),室温搅拌1h,抽滤,滤渣用ch2cl2洗涤后再抽滤,合并滤液并加无水硫钠干燥,抽滤除去溶剂得6.42g液体(2-苄基八氢吡咯并[1,2-a]吡嗪,以下称化合物s3-3)。
将化合物s3-3(4.33g,20mmol)溶于甲醇(50ml)中,加入pd/c(0.45g),pd(oh)2(0.20g),滴加冰醋酸1滴,抽除空气,通入h2,室温反应12h,过滤,滤渣用甲醇洗涤3次(每次5ml),合并滤液,减压抽滤除去溶剂得淡黄色液体(八氢吡咯并[1,2-a]吡嗪,以下称化合物s3-4)1.83g。
参照实施例1中化合物s1的合成方法,将化合物s1-5替换成化合物s3-4(151.4mg,1.2mmol)与左旋氧氟沙星羧酸反应即可,得黄色固体147.7mg,计算得收率为47.66%,测得mp:237-240℃。
以上操作中利用的原料是l-脯氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图5、图6所示,由此确定产物为化合物s3。
1hnmr(500mhz,dmso)δ15.19(s,1h),8.95(s,1h),7.54(d,j=12.5hz,1h),4.91(q,j=6.5hz,1h),4.57-4.95(m,1h),4.33-4.37(m,1h),3.46(d,j=10.5hz,1h),3.32(d,j=7.0hz,1h),3.22(t,j=11.0hz,1h),2.97-3.03(m,3h),2.25-2.30(m,1h),2.00-2.14(m,2h),1.63-1.78(m,3h),1.45(d,j=6.5hz,3h),1.27-1.35(m,1h);
13cnmr(126mhz,dmso)δ176.82,166.49,155.91,146.55,140.54,132.77,125.24,120.00,107.09,103.69,68.53,62.87,55.68,55.30,53.59,52.37,50.04,27.39,21.22,18.33;
hrms-esi(m/z):calcd.forc20h23fn3o4(m+h)+:388.16726;found:388.16545。
实施例4
(s)-9-氟-2,3-二氢-3-甲基-10-(7-羟基八氢吡咯并[1,2-a]吡嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅳ)结构,以下称化合物s4)的合成。
将4-羟基脯氨酸(19.67g,0.15mol)加到无水甲醇(300ml)中,冰盐浴下,滴加socl2(43.8ml,0.60mol),滴加完毕,改为室温反应,tlc跟踪检测至反应完毕后将反应液浓缩至干,所得残余物(4-羟基脯氨酸甲酯盐酸盐,以下称化合物s4-a)直接进行下步反应。
所得的残余物加水(240ml),冰盐浴下,加入nahco3(29.61g,0.35mol),待气体释放毕,滴加clch2cocl(12.42ml,0.17mol)的ch2cl2(180ml)溶液,30min滴加完毕,改为室温反应6h,反应完毕后分出有机层,用ch2cl2萃取水层3次(每次20ml),合并有机层,加无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,得黄色油状物(n-氯乙酰基-4-羟基脯氨酸甲酯,以下称化合物s4-1)25.26g。
将化合物s4-1(22.16g,0.10mol)溶于无水甲醇(200ml)中,冰水浴下,加入tea(16.63ml,0.12mol)和苄胺(11.79g,0.12mol),外温80℃回流48h,冷却至室温,减压抽滤除去溶剂,残余物加ch2cl2(100ml)使其溶解,柱层析,得白色固体(2-苄基-7-羟基六氢吡咯并[1,2-α]吡嗪-1,4二酮,以下称化合物s4-2)21.5g。
取lialh4(1.71g,45mmol)悬浮于无水thf(40ml)中,冰浴下,分批加入化合物s4-2(3.90g,15mmol),加完后室温搅拌30min,氩气保护下外温90℃加热回流5h,tlc检测反应完毕,冷却至室温,冰浴下依次滴加h2o(2.1ml)、2nnaoh(9.0ml)、h2o(2.1ml),磁力搅拌1h,抽滤,滤渣用ch2cl2洗涤3次(每次10ml),合并滤液,加入无水硫酸钠干燥,过滤,抽滤除去溶剂得粗产品,柱层析得1.94g液体,静置固化得淡黄色固体(2-苄基-7-羟基八氢吡咯并[1,2-a]吡嗪,以下称化合物s4-3)。
将化合物s4-3(1.80g,7.74mmol)溶于15ml甲醇中,加入pd/c(0.18g)和pd(oh)2(0.10g),滴加冰醋酸1滴,抽除空气,通入h2,室温反应12h。过滤,滤渣用甲醇洗涤3次(每次5ml),合并滤液,减压抽滤除去溶剂得淡黄色固体(7-羟基八氢吡咯并[1,2-a]吡嗪,以下称化合物s4-4)0.98g。
参照实施例1中化合物s1的合成方法,将化合物s1-5替换成s4-4与左旋氧氟沙星羧酸反应即可。将左氧氟沙星羧酸(112.5mg,0.4mmol)溶于dmso(3.0ml)中,加入tea(0.22ml,1.6mmol)和化合物s4-4(85.3mg,0.6mmol),在90℃的条件下微波反应24h,冷却至室温,加入二氯甲烷(15ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得黄色固体63.5mg,收率39.35%,测得mp:229-232℃。
以上操作中利用的原料是l-4-羟基脯氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱、质谱图如图7、图8所示,具体的谱图数据如下所示,由此确定产物为化合物s4。
1hnmr(500mhz,dmso)δ15.21(s,1h),8.95(s,1h),7.56(d,j=12.5hz,1h),4.91(d,j=7.0hz,1h),4.78(d,j=4.5hz,1h),4.57-4.59(m,1h),4.35-4.37(m,1h),4.22(s,1h),3.44(d,j=12.0hz,1h),3.19(t,j=11.0hz,1h),2.90-2.96(m,2h),2.32-2.38(m,2h),2.01(dd,j=9.0,5.5hz,1h),1.56-1.60(m,2h),1.45(d,j=6.5hz,3h);
13cnmr(126mhz,dmso)δ176.82,166.51,155.90,146.55,140.56,132.72,125.25,120.01,107.09,103.70,68.52,68.02,63.42,61.07,55.49,55.30,52.16,50.14,39.05,18.34;
hrms-esi(m/z):calcd.forc20h23fn3o5(m+h)+:404.16218;found:404.16134。
实施例5
(s)-1-(2,4-二氟苯基)-6,8-二氟-7-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物s5)及其盐酸盐(以下称化合物s3-tm)的合成。
将2,3,4,5-四氟苯甲酰乙酸乙酯(11.89g,45mmol)加到入原甲酸三乙酯(11.24ml,67.5mmol)和醋酐(20ml)中,升温到140℃反应1h,减压蒸馏除去溶剂得淡黄色液体。将所得淡黄色液体溶于二氯甲烷(150ml)中,在氩气保护下,用冰浴冷却,加入2,4-二氟苯胺(6.87ml,67.5mmol),室温反应1h,减压旋除溶剂得黄色固体,石油醚重结晶得白色固体(2-(2,3,4,5-四氟苯甲酰基)-3-(2,4-二氟苯基)-丙烯酸乙酯,以下称化合物mh3-1)15.44g。
将化合物mh3-1(14.92g,37mmol)溶于80ml的dmf中,加入无水k2co3(15.34g,111mmol),升温到110℃反应5h,tlc检测反应完毕后冷却到室温,再倒入200ml的冰水中,剧烈搅拌10min,抽滤,滤渣用水洗3次(每次30ml),再进行干燥,用乙酸乙酯/石油醚重结晶得白色固体(1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸乙酯,以下称化合物mh3-2)8.61g。
将化合物mh3-2(1.53g,4.0mmol)加到15mlhcl(浓度为3n)和15ml醋酸的混合溶液中,外温100℃回流3h,冷却至室温,抽滤,滤渣用水洗3次(每次5ml),真空干燥得白色固体(1-(2,4-二氟苯基)-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸,以下称化合物mh3-3)1.19g,测得其mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,具体的谱图数据如下所示,由此确定产物为化合物mh3-3。
1hnmr(400mhz,dmso)δ13.97(s,1h),8.83(d,j=4.9hz,1h),8.24(s,1h),7.97(s,1h),7.66(s,1h),7.36(s,1h);
ms(esi,m/z):378(m+h)+。
将化合物mh3-3(284.2mg,0.8mmol)溶于dmso(5.0ml)中,加入tea(0.2ml,1.44mmol)和化合物s1-5(259.5mg,1.2mmol),在90℃的条件下微波反应12h,冷却至室温,加入二氯甲烷(15ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得白色固体243.8mg,计算得收率为55.26%,测得mp:218-219℃。
以上操作中利用的原料是l-丝氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图9、图10所示,具体的谱图数据如下所示,由此确定产物为化合物s5。
1hnmr(500mhz,dmso)δ14.44(s,1h),8.70(s,1h),7.94(d,j=11.0hz,2h),7.63(t,j=8.8hz,1h),7.35(t,j=7.5hz,1h),4.74(s,1h),3.95(s,1h),3.76(d,j=10.0hz,1h),3.58(s,1h),3.40(s,2h),3.19-3.25(m,2h),3.05(d,j=10.0hz,2h),1.39(s,9h);
13cnmr(126mhz,dmso)δ176.47,165.29,163.10,157.47,155.12,154.58,152.07,146.05,134.63,130.54,128.62,127.91,120.32,112.88,108.56,107.55,105.48,79.48,57.95,53.02,50.56,49.73,28.45;
hrms-esi(m/z):calcd.forc26h25f4n3nao6(m+na)+:574.15772;found:574.15637。
将化合物s5(113.0mg,0.2mmol)加到hcl(浓度为3n,4ml)-乙醇(10ml)混合溶剂中,外温100℃加热回流10h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤3次(每次1ml)。所得固体加入到10ml无水乙醇中,外温90℃加热回流30min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤3次(每次2ml),得白色固体75.8mg,计算得收率为77.69%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图27所示,具体的谱图数据如下所示,由此确定产物为化合物s3-tm。
1hnmr(400mhz,dmso)δ14.34(s,1h),9.27(br,2h),8.72(s,1h),7.97(t,j=11.2hz,2h),7.65(t,j=9.6hz,1h),7.36(t,j=8.2hz,1h),5.43(s,1h),3.60(s,2h),3.39-3.45(m,3h),3.24-3.28(m,3h),3.07(s,1h);
hrms-esi(m/z):calcd.forc21h18f4n3o4(m+h)+:452.12334;found:452.12232。
实施例6:
(s)-1-(2,4-二氟苯基)-6,8-二氟-7-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物s6)及其盐酸盐(以下称化合物s4-tm)的合成。
参照实施例5中化合物s5的合成方法,将tea替换成n-甲基吗啡啉(0.21ml,1.92mmol),将化合物s1-5替换成化合物s2-5(具体合成过程见实施例2,368.5mg,1.6mmol)与化合物mh3-3反应,反应后加入的二氯甲烷为30ml,反应得黄色固体203.3mg,计算得收率为42.18%,测得mp:210-211℃。
以上操作中利用的原料是l-苏氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图11、图12所示,具体的谱图数据如下所示,由此确定产物为化合物s6。
1hnmr(500mhz,dmso)δ14.21(s,1h),8.70(s,1h),7.93(d,j=11.5hz,2h),7.63(s,1h),7.35(s,1h),4.54(s,1h),4.01(s,1h),3.74-3.81(m,2h),3.09-3.23(m,5h),1.38(s,9h),1.03(d,j=5.5hz,3h);
13cnmr(126mhz,dmso)δ176.46,165.27,163.16,157.52,156.27,155.30,152.06,146.32,134.47,130.58,128.58,127.87,120.61,112.93,108.62,107.63,105.52,79.00,62.87,58.06,51.34,50.59,28.49,21.61;
hrms-esi(m/z):calcd.forc27h27f4n3nao6(m+na)+:588.17337;found:588.17224。
将化合物s6(187.8mg,3.4mmol)加到hcl(浓度为3n,4ml)-乙醇(1ml)混合溶剂中,外温100℃加热回流10h,冷却至室温,析出白色固体,抽滤,再用无水乙醇洗涤3次(每次3ml)。所得固体加到15ml无水乙醇中,外温90℃加热回流30min,冷却至室温,抽滤,滤渣再用无水乙醇洗涤3次(每次1ml),得白色固体139.3mg,计算得收率为81.64%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图28所示,具体的谱图数据如下所示,由此确定产物为化合物s4-tm。
1hnmr(400mhz,dmso)δ14.35(s,1h),9.36(s,1h),9.08(s,1h),8.73(d,j=2.8hz,1h),7.96(t,j=9.2hz,2h),7.65(s,1h),7.36(s,1h),5.63(s,1h),3.80(s,1h),3.43(d,j=14.0hz,3h),3.23(d,j=13.6hz,2h),3.04(s,2h),1.12(s,3h);
hrms-esi(m/z):calcd.forc22h20f4n3o4(m+h)+:466.13899;found:466.13788。
实施例7
(s)-1-(2,4-二氟苯基)-6,8-二氟-7-(八氢吡咯并[1,2-a]吡嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅵ)结构,以下称化合物s7)的合成。
参照实施例5中化合物s5的合成方法,将化合物s1-5替换成化合物s3-4(具体合成过程见实施例3,151.4mg,1.2mmol)与化合物mh3-3反应,反应后加入的二氯甲烷为30ml,得黄色固体239.8mg,计算得收率为64.96%,测得mp:213-215℃。
以上操作中利用的原料是l-脯氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图13、图14所示,具体的谱图数据如下所示,由此确定产物为化合物s7。
1hnmr(500mhz,cdcl3)δ14.41(s,1h),8.47(s,1h),7.94(d,j=10.0hz,1h),7.46–7.51(m,1h),7.06-7.10(m,2h),3.42(t,j=10.0hz,1h),3.28-3.36(m,1h),3.09(t,j=7.5hz,1h),2.96-3.03(m,2h),2.33(t,j=10.0hz,1h),2.17(dd,j=10.0,5.0hz,1h),2.09(s,1h),1.68–1.87(m,3h),1.38-1.41(m,1h);
13cnmr(126mhz,cdcl3)δ176.77,165.93,163.35,157.46,155.13,150.39,145.34,134.87,128.68,128.22,127.66,119.78,112.55,108.77,108.06,105.40,62.60,55.72,53.57,52.09,50.22,27.17,20.97;
hrms-esi(m/z):calcd.forc23h20f4n3o3(m+h)+:462.14408;found:462.14303。
实施例8
(s)-1-(2,4-二氟苯基)-6,8-二氟-7-(7-羟基八氢吡咯并[1,2-a]吡嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅵ)结构,以下称化合物s8)的合成。
参照实施例5中化合物s5的合成方法,将化合物s1-5替换成化合物s4-4(具体合成过程见实施例4)与化合物mh3-3反应即可。将化合物mh3-3(142.1mg,0.4mmol)溶于dmso(3.0ml)中,加入tea(0.11ml,0.8mmol)和化合物s4-4(85.3mg,1.2mmol),在90℃的条件下微波反应12h,冷却至室温,加入二氯甲烷(30ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得白色固体73.3mg,收率38.40%,测得mp:199-202℃。
以上操作中利用的原料是l-4-羟基-脯氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图15、图16所示,具体的谱图数据如下所示,由此确定产物为化合物s8。
1hnmr(500mhz,dmso)δ14.49(s,1h),8.68(s,1h),7.91-7.97(m,2h),7.61-7.65(m,1h),7.34(t,j=8.0hz,1h),4.77(d,j=5.0hz,1h),4.18(s,1h),3.23(d,j=12.5hz,1h),3.14(t,j=11.0hz,1h),2.88(d,j=11.0hz,1h),2.82(t,j=11.0hz,1h),2.21-2.30(m,2h),1.96-1.99(m,1h),1.52-1.58(m,2h);
13cnmr(126mhz,dmso)δ176.49,165.33,163.06,157.41,155.01,151.97,145.60,134.62,130.54,128.64,127.93,119.71,112.88,108.45,107.54,105.48,67.90,63.21,60.83,55.41,51.84,50.38,38.93;
hrms-esi(m/z):calcd.forc23h20f4n3o4(m+h)+:478.13960;found:478.13780。
实施例9
(s)-1-环丙基-6,8-二氟-7-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物s9)及其盐酸盐(以下称化合物s5-tm)的合成。
取2,3,4,5-四氟苯甲酰乙酸乙酯(3.96g,15mmol),加到原甲酸三乙酯(3.8ml,22.5mmol)和醋酐(6.4ml)的混合液中。在140℃的条件下反应2.5h,减压蒸馏除去溶剂得淡黄色液体。往该淡黄色液体中加入t-buoh(15ml),在氩气的保护下,冰浴冷却,加入环丙胺(0.90g,15.8mmol)的t-buoh(10ml)溶液,反应30min,在45℃的条件下反应过夜,旋转除去溶剂得黄色液体,柱层析得黄色固体(2-(2,3,4,5-四氟苯甲酰基)-3-环丙氨基丙烯酸乙酯,以下称化合物mh1-1)4.50g。
将化合物mh1-1(2.25g,6.8mmol)溶于10ml的t-buoh,加入t-buoh(0.76g,6.8mmol),在90℃的条件下反应5h,反应完毕后冷却至室温,析出黄色固体,抽滤,滤渣用水洗3次(每次20ml),干燥后以1,4-二氧六环重结晶得1.31g白色固体(1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸乙酯,以下称化合物mh1-2)。
将化合物mh1-2(1.00g,3.2mmol)加到15ml的hcl(浓度为3n)和15ml醋酸的溶液中,外温100摄氏度回流3h,冷却至室温,抽滤,滤渣用水洗3次(每次5ml),真空干燥得白色固体(1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸,以下称化合物mh1-3)0.71g,测得mp:223-225℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,具体的谱图数据如下所示,由此确定产物为化合物mh1-3。
1hnmr(400mhz,dmso)δ14.28(s,1h),8.72(s,1h),8.14(d,j=8.8hz,1h),4.14(s,1h),1.20-1.40(m,4h)。
ms(esi,m/z):306(m+na)+。
将1-环丙基-6,7,8-三氟-1,4-二氢-4-氧代喹啉-3-羧酸(以下称mh1-3,283.2mg,0.10mmol)溶于dmso(6.0ml)中,加入tea(0.21ml,1.5mmol)和化合物s2-5(具体合成过程见实施例2,345.5mg,1.5mmol),在90℃的条件下微波反应12h,冷却至室温,加入二氯甲烷(30ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得黄色固体270.5mg,计算得收率为54.81%,测得mp:200-201℃。
以上操作中利用的原料是l-苏氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图17、图18所示,具体的谱图数据如下所示,由此确定产物为化合物s9。
1hnmr(400mhz,dmso)δ14.66(s,1h),8.66(s,1h),7.81(d,j=12.0hz,1h),4.59(s,1h),4.31-4.35(m,1h),4.13-4.16(m,2h),3.82-3.89(m,2h),3.41-3.47(m,2h),3.22(d,j=7.2hz,2h),1.42(s,9h),1.14-1.21(m,7h);
13cnmr(126mhz,dmso)δ176.17,165.70,155.35,155.38,150.70,147.61,134.4,129.29,121.22,107.11,79.04,62.96,57.95,56.50,51.53,50.82,40.83,28.54,21.82,19.00,8.91;
hrms-esi(m/z):calcd.forc24h30f2n3o6(m+h)+:494.21027;found:494.20854。
参照实施例1中s1-tm的合成方法,将化合物s1替换为化合物s9(49.3mg,0.1mmol)进行反应即可,得黄色固体32.7mg,计算得收率为76.07%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图29所示,由此确定产物为化合物s5-tm。
1hnmr(400mhz,dmso)δ14.64(s,1h),9.39(s,1h),9.13(s,1h),8.70(d,j=5.2hz,1h),7.88(t,j=5.8hz,1h),5.69(s,1h),4.13(s,1h),3.88(s,1h),3.56(s,3h),3.32-3.42(m,2h),3.14(s,2h),1.20(m,7h);
hrms-esi(m/z):calcd.forc19h22f2n3o4(m+h)+:394.15784;found:394.15711。
实施例10
(s)-1-环丙基-6,8-二氟-7-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物s10)及其盐酸盐(以下称化合物s6-tm)的合成。
参照实施例9中化合物s9的合成方法,将化合物s2-5替换成化合物s1-5(具体合成过程将实施例1,324.4mg,1.5mmol)与化合物mh1-3反应即可,得白色固体287.1mg,计算得收率为59.88%,测得mp:210-212℃。
以上操作中利用的原料是l-丝氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图19、图20所示,具体的谱图数据如下所示,由此确定产物为化合物s10。
1hnmr(500mhz,dmso)δ14.71(s,1h),8.68(s,1h),7.83(d,j=11.0hz,1h),4.79(t,j=5.5hz,1h),4.13(d,j=5.5hz,1h),4.03(s,1h),3.85(d,j=11.5hz,1h),3.68-3.72(m,1h),3.56(d,j=12.5hz,1h),3.50(s,1h),3.41(d,j=11.0hz,1h),3.33(s,1h),3.13-3.22(m,2h),1.43(s,9h),1.17-1.21(m,4h);
13cnmr(126mhz,dmso)δ176.13,165.7,155.07,154.69,150.58,147.23,134.6,129.27,120.82,107.04,106.96,79.50,58.08,53.38,50.73,50.02,40.82,28.50,19.00,8.91;
hrms-esi(m/z):calcd.forc23h27f2n3nao6(m+na)+:502.17656;found:502.17514。
参照实施例1中s1-tm的合成方法,将化合物s1替换为化合物s10(48.0mg,0.1mmol)进行反应即可,得黄色固体31.6mg,计算得收率为76.0%,测得mp>250℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图30所示,具体的谱图数据如下所示,由此确定产物为化合物s6-tm。
1hnmr(400mhz,dmso)δ14.64(s,1h),9.36(br,2h),8.70(d,j=5.2hz,1h),7.88(d,j=7.6hz,1h),5.50(d,j=4.8hz,1h),4.13(s,1h),3.56-3.68(m,6h),3.18(s,1h),1.21(d,j=12.4hz,4h);
hrms-esi(m/z):calcd.forc18h20f2n3o4(m+h)+:380.14219;found:380.14139。
实施例11
(s)-1-环丙基-6,8-二氟-7-(八氢吡咯并[1,2-a]吡嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅵ)结构,以下称化合物s11)的合成。
参照实施例9中化合物s9的合成方法,将化合物s2-5替换成化合物s3-4与化合物mh1-3反应即可。将化合物mh1-3(141.0mg,0.5mmol)溶于dmso(3.0ml)中,加入tea(0.1ml,0.75mmol)和化合物s1-4(94.7mg,0.75mmol),在90℃的条件下微波反应12h,冷却至室温,加入二氯甲烷(15ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得白色固体106.1mg,收率54.50%,测得mp:205-207℃。
以上操作中利用的原料是l-脯氨酸,获得的是s构型的产物。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱如图21、图22所示,具体的谱图数据如下所示,由此确定产物为化合物s11。
1hnmr(500mhz,cdcl3)δ14.59(s,1h),8.75(s,1h),7.86(dd,j=12.0,2.0hz,1h),3.97-4.02(m,1h),3.55(d,j=11.5hz,1h),3.41-3.48(m,2h),3.09-3.16(m,3h),2.40-2.45(m,1h),2.15-2.25(m,2h),1.83-1.88(m,2h),1.74-1.78(m,1h),1.42-1.50(m,1h),1.29-1.30(m,2h),1.16(m,2h);
13cnmr(126mhz,cdcl3)δ176.58,166.49,155.22,149.45,146.69,135.03,128.88,120.67,108.05,107.97,62.83,56.07,53.78,52.37,50.49,40.22,27.37,21.16,9.22;
hrms-esi(m/z):calcd.forc20h22f2n3o3(m+h)+:390.16293;found:390.16168。
实施例12
(s)-1-环丙基-6,8-二氟-7-(7-羟基八氢吡咯并[1,2-a]吡嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅵ)结构,以下称化合物s12)的合成。
参照实施例9中化合物s9的合成方法,将化合物s2-5替换成化合物s4-4与化合物mh1-3反应即可。将化合物mh1-3(141.0mg,0.5mmol)溶于dmso(3.0ml),加入tea(0.1ml,0.75mmol)和化合物s2-4(106.6mg,0.75mmol),在90℃的条件下微波反应12h,冷却至室温,加入二氯甲烷(15ml),再水洗3次(每次15ml),干燥,用无水乙醇重结晶得白色固体69.7mg,计算得收率为34.39%,测得mp:223-226℃。
利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱、核磁共振碳谱、质谱图如图50-52所示,具体的谱图数据如下所示,由此确定产物为化合物s12。
1hnmr(500mhz,dmso)δ14.75(s,1h),8.66(s,1h),7.80(d,j=10.0hz,1h),4.81(d,j=5.0hz,1h),4.23(d,j=5.0hz,1h),4.13(d,j=5.0hz,1h),3.51(d,j=10.0hz,1h),3.33-3.40(m,2h),3.27(t,j=12.5hz,1h),2.95(t,j=10.0hz,2h),2.31-2.43(m,2h),2.03(dd,j=10.0,5.0hz,1h),1.57-1.65(m,2h),1.17-1.21(m,4h).;
13cnmr(126mhz,dmso)δ176.10,165.71,154.88,150.44,146.69,134.57,129.30,120.17,107.00,106.96,67.95,63.29,60.94,55.60,51.99,50.50,40.80,39.02,8.91;
hrms-esi(m/z):calcd.forc20h22f2n3o4(m+h)+:406.15784;found:406.15634。
实施例13
(r)-9-氟-2,3-二氢-3-甲基-10-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅱ)结构,以下称化合物r1)及其盐酸盐(以下称化合物r1-tm)的合成。
将侧链原料替换成d-丝氨酸,其它操作完全参照实施例1,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图31所示,具体的谱图数据如下所示,由此确定产物为化合物r1-tm。
1hnmr(400mhz,dmso)δ15.11(s,1h),9.48(br,1h),9.15(s,1h),9.00(s,1h),7.62(d,j=11.6hz,1h),5.46(s,1h),4.94(d,j=6.4hz,1h),4.61(d,j=11.2hz,1h),4.40(d,j=11.2hz,1h),3.67(s,2h),3.50(s,3h),3.37-3.43(m,2h),3.29(s,1h),3.12(s,1h),1.46(d,j=6.8hz,3h);
hrms-esi(m/z):calcd.forc18h21fn3o5(m+h)+:378.14653;found:378.14580
实施例14
(r)-9-氟-2,3-二氢-3-甲基-10-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-7-氧代-(3s)-7h-吡啶并[1,2,3-de]-1,4-苯并噁嗪-6-羧酸(为式(ⅱ)结构,以下称化合物r1)及其盐酸盐(以下称化合物r2-tm)的合成。
将侧链原料替换成d-苏氨酸,其它操作完全参照实施例2,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图32所示,具体的谱图数据如下所示,由此确定产物为化合物r2-tm。
1hnmr(400mhz,dmso)δ15.12(s,1h),9.29(s,1h),9.02(m,2h),7.62(dd,j=12.0hz,j=5.6hz,2h),5.66(s,1h),4.94(s,1h),4.60(d,j=7.6hz,1h),4.41(d,j=8.8hz,1h),3.86(t,j=6.4hz,1h),3.37-3.52(m,4h),3.35(d,j=10.0hz,1h),3.09(s,2h),1.45(d,j=5.6hz,3h),1.18(d,j=5.6hz,3h);
hrms-esi(m/z):calcd.forc19h23fn3o5(m+h)+:392.16217;found:392.16152
实施例15
(r)-1-(2,4-二氟苯基)-6,8-二氟-7-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物r5)及其盐酸盐(以下称化合物r3-tm)的合成。
将侧链原料替换成d-丝氨酸,其它操作完全参照实施例5,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图33所示,具体的谱图数据如下所示,由此确定产物为化合物r3-tm。
1hnmr(400mhz,dmso)δ14.36(s,1h),9.50(br,2h),9.22(br,1h),8.72(s,1h),7.92-7.99(m,2h),7.64(t,j=10.0hz,1h),7.35(t,j=8.8hz,1h),5.43(s,1h),3.61(s,2h),3.40-3.47(m,3h),3.26(d,j=19.6hz,3h),3.06(s,1h);
hrms-esi(m/z):calcd.forc21h18f4n3o4(m+h)+:452.12334;found:452.12219.
实施例16
(r)-1-(2,4-二氟苯基)-6,8-二氟-7-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物r6)及其盐酸盐(以下称化合物r4-tm)的合成。
将侧链原料替换成d-苏氨酸,其它操作完全参照实施例6,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图34所示,具体的谱图数据如下所示,由此确定产物为化合物r4-tm。
1hnmr(400mhz,dmso)δ14.35(s,1h),9.21(br,2h),8.73(d,j=2.8hz,1h),7.94-8.00(m,2h),7.65(s,1h),7.36(s,1h),5.63(d,j=4.4hz,2h),3.78(s,1h),3.41(s,3h),3.23(d,j=12.8hz,2h),3.05(s,2h),1.11(s,3h);
hrms-esi(m/z):calcd.forc22h20f4n3o4(m+h)+:466.13899;found:466.13754.
实施例17
(r)-1-环丙基-6,8-二氟-7-(4-叔丁氧羰基-3-(1-羟乙基)-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物r9)及其盐酸盐(以下称化合物r5-tm)的合成。
将侧链原料替换成d-苏氨酸,其它操作完全参照实施例9,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图35所示,由此确定产物为化合物r5-tm。
1hnmr(400mhz,dmso)δ:9.49(d,j=9.6hz,1h),9.17(s,1h),8.68(s,1h),7.86(d,j=11.6hz,1h),4.13(d,j=4.4hz,1h),3.88(t,j=6.8hz,1h),3.56-3.63(m,3h),3.39-3.42(m,2h),3.32(d,j=11.6hz,1h),3.12(s,2h),1.20(t,7h);
hrms-esi(m/z):calcd.forc19h22f2n3o4(m+h)+:394.15784;found:394.15668
实施例18
(r)-1-环丙基-6,8-二氟-7-(4-叔丁氧羰基-3-羟甲基-1-哌嗪基)-1,4-二氢-4-氧代喹啉-3-羧酸(为式(ⅰ)结构,以下称化合物r10)及其盐酸盐(以下称化合物r6-tm)的合成。
将侧链原料替换成d-丝氨酸,其它操作完全参照实施例10,获得的是r构型的产物。利用mat-95型高分辨质谱仪和mercuryplus400核磁共振谱仪对所得产物进行分析,所得核磁共振氢谱如图36所示,具体的谱图数据如下所示,由此确定产物为化合物s6-tm。
1hnmr(400mhz,dmso)δ14.64(s,1h),9.60(s,1h),9.34(s,1h)8.69(s,1h),7.87(d,j=11.6hz,1h),5.50(s,1h),4.12(s,1h),3.56-3.69(m,5h),3.35-3.45(m,3h),3.16(s,1h),1.19(d,j=9.4hz,4h);
hrms-esi(m/z):calcd.forc18h20f2n3o4(m+h)+:380.14219;found:380.14121
体外抗菌活性测定试验
1、实验菌株:r-鲍曼氏不动杆菌132025355、s-鲍曼氏不动杆菌130781636、r-大肠埃希菌132076367、s-大肠埃希菌131914426、r-铜绿假单胞菌131374625、s-铜绿假单胞菌132083811、r-肺炎克雷伯菌131636980、s-肺炎克雷伯菌132099499、r-表皮葡萄球菌131001574、r-表皮葡萄球菌gzcm、s-表皮葡萄球菌132249588、r-金黄色葡萄球菌132070039、r-金黄色葡萄球菌byj、r-金黄色葡萄球菌shr、s-型金黄色葡萄球菌131667100、r-粪肠球菌131996907、s-粪肠球菌132076451、r-无乳链球菌131021203、s-无乳链球菌131636909、r-溶血葡萄球菌132085699、s-溶血葡萄球菌120283459、r-乙型副伤寒沙门菌131667947、s-阴沟肠杆菌131127315、s-嗜水气单胞菌131931294、s-化脓性链球菌131241647。其中r代表耐药型;s代表敏感型。
2、mueller-hinton培养基(英国oxoid公司生产,批号为301651)。
3、待测化合物:根据实施例1-18制备的化合物化合物s-1、s-2、s-4、s-5、s-6、s-7、s-8、s-9、s-10、s-11、s-12、s1-tm、s2-tm、s3-tm、s4-tm、s5-tm、s6-tm、r1-tm、r2-tm、r3-tm、r4-tm、r5-tm、r6-tm。待测化合物以磷酸缓冲溶液稀释,浓度梯度由多次实验为0.03125μmol/l、0.0625μmol/l、0.125μmol/l、0.25μmol/l、0.5μmol/l、1μmol/l、2μmol/l、4μmol/l、8μmol/l、16μmol/l、32μmol/l、64μmol/l、128μmol/l。
4、阳性对照物:
对照品1:左旋氧氟沙星盐酸盐;
对照品2:环丙沙星盐酸盐。
5、试验方法
体外抗菌活性测定采用世界卫生组织推荐的对一般细菌最适宜的mueller-hinton培养基,加入1.6%琼脂粉作为药物稀释的基质。实验菌在m-h肉汤培养基中过夜培养并稀释使接种量控制为5×103-5×104cfu。药物浓度采取连续倍数稀释法,加入到50-60℃的培养基中混合均匀后制备双碟培养皿,最高浓度128μmol/l,最低浓度0.03125μmol/l。实验菌悬液用多头接种仪接种于上述含各种化合物及不同浓度的培养皿中。将培养皿在35-37℃培养16-18小时,肉眼观察实验结果,继续培养至42-44小时,再观察其结果。抑制试验细菌生长的最低浓度为最低抑菌浓度(mic)。本发明提供的侧链为手性单取代基哌嗪的喹诺酮羧酸衍生物的体外抗菌活性(mic,μmol/l)测定结果见表1和表2。
表1本发明部分化合物及阳性对照物的体外抗菌活性(micμmol/l)
——部分革兰氏阴性菌
表2本发明部分化合物及阳性对照物的抗菌活性(micμmol/l)
——部分革兰氏阳性菌
表3本发明部分化合物及阳性对照物的体外抗菌活性(micμmol/l)
——部分革兰氏阴性菌
表4本发明部分化合物及阳性对照物的体外抗菌活性(micμmol/l)
——部分革兰氏阳性菌
由表1和表2可以看出:在体外抗菌活性测试中,本发明中的化合物都表现出对耐药菌具有与对照品相当的抗菌活性。在革兰氏阴性菌抗菌活性测试中,本发明中的化合物都表现出对敏感菌具有较好的抗菌活性,其中对敏感型嗜水气单胞菌和敏感型的化脓性链球菌的抗菌活性与对照品环丙沙星(或左旋氧氟沙星)相当。在革兰氏阳性菌抗菌活性测试中,个别化合物如s6-tm及r1-tm表现出对耐药型溶血葡萄球菌具有更强的抗菌活性,其中s6-tm为对照品环丙沙星(或左旋氧氟沙星)的128倍,r1-tm为对照品环丙沙星(或左旋氧氟沙星)的64倍。
比较侧链为对映体的两类化合物发现,侧链为r构型和s构型的化合物的活性多少会有差异,活性差别从2倍至128倍不等,但无规律,有时是r构型的化合物强于s构型的化合物,有时是s构型的化合物强于r构型的化合物。
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!