反式-4-氨基-环己甲酸乙酯盐酸盐的制备方法与流程
本发明属于有机化学领域,具体涉及一种活性药物中间体反式-4-氨基-环己甲酸乙酯盐酸盐的制备方法。
背景技术:
4-氨基环己烷甲酸存在两种立式异构体:顺式-4-氨基环己甲酸与反式-4-氨基环己甲酸,其中反式异构体比顺式异构体呈现出更为独特的活性与特性,作为医药中间体已引起广泛的重视,吸引着人们加大对该产品的工艺开发。反式-4-氨基环己甲酸(acca)及其衍生物是重要的医药中间体之一,主要用来合成短肽、多肽、异奎宁环酮等类药物。反式-4-氨基环己甲酸乙酯盐酸盐是acca的衍生物,是构建活性药物的重要砌块之一。
现有技术中,作为制备反式-4-氨基环己甲酸乙酯盐酸盐的原料反式-4-氨基环己甲酸的合成通常有两种方案:
第一种方案是专利tw1280232b,us7314950等所报道的用对氨基苯甲酸高压氢化,得到4-氨基环己甲酸的顺反两种异构体,然后通过氨基保护或者成盐的方式进行拆分。但是这种方法的不足之处在于拆分不彻底、收率低、效率低,得到的产品异构体纯度较差,无法满足药用要求;同时使用大量的酸碱,高毒性的有机溶剂,造成较大的三废污染。杭州化工2005,35(4),22报道了高压氢化对氨基苯甲酸得顺反异构体混合物,然后用乙醇反复结晶,逐步分离得到反式-4-氨基环己甲酸,此方法用到大量的有机溶剂,操作繁琐,不适合放大生产。cn201510570361报道了拆分消旋体的改进方法,用消旋的4-氨基环己甲酸经过成盐,异构化,中和,后处理工序得到纯度98%以上的反式4-氨基环己甲酸,这种方法不足之处在于金属纳米催化剂不易制备,异构化过程需要高温高压,存在较大的安全风险,且得到的产品含有重金属残留。
第二种方案是用对苯二甲酸进行高压氢化,得到1,4-环己二甲酸的顺反异构体混合物,利用两种异构体不同温度下的溶解度差异进行重结晶拆分,能够得到含量99%以上的较高纯度的反式异构体,顺式含量极低,完全满足药用需求,而且反式-1,4-环己二甲酸的拆分工艺已经实现工业化的大规模生产,成本较低。但是利用该原料进行合成反式-4-氨基环己甲酸乙酯盐酸的方法报道较少。已知的但未见正式报道的合成工艺方案一般为利用反式-1,4-环己二甲酸为原料和boc酸酐在碳酸氢铵、吡啶、乙腈中先生成单酰胺,单酰胺用三氯异氰尿酸重排,重排产物再用浓盐酸水解,浓缩结晶,得到反式-4-氨基环己甲酸盐酸盐,反式-4氨基环己甲酸盐酸盐再和乙醇氯化亚砜回流,浓缩后处理得到反式-4-氨基-环己甲酸乙酯盐酸盐。这种方法存在成本较高,步骤长,设备腐蚀性强、收率低的弊端,也不适合目前绿色环保生产的要求。
综上所述,目前现有技术中制备反式-4-氨基环己甲酸乙酯盐酸盐的原料反式-4-氨基环己甲酸的工艺方案存在高温高压反应,使用高毒性有机溶剂,操作复杂,效率低下,腐蚀设备,不符合绿色环保要求等不足之处。
技术实现要素:
为了解决上述现有技术制备反式-4-氨基环己甲酸乙酯盐酸盐的原料反式-4-氨基环己甲酸的工艺方案存在高温高压反应、使用高毒性有机溶剂、操作复杂、效率低下、腐蚀设备、不符合绿色环保要求的缺陷,本发明提供一种避免无需使用反式-4-氨基环己甲酸制备反式-4-氨基环己甲酸乙酯盐酸盐的方法。
为了实现上述目的,本发明采用如下技术方案:
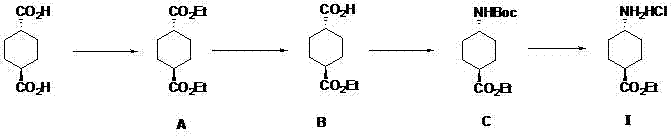
反式-4-氨基-环己甲酸乙酯盐酸盐的制备方法,其特征在于:包括以下步骤:
s1:反式-1,4-环己二甲酸和无水乙醇在催化剂条件下反应生成中间体二酯a;
s2:中间体二酯a进行皂化反应得到中间体单酸b;
s3:中间体单酸b和dppa进行curtius重排得到中间体c;
s4:中间体c脱保护基得到终产品反式-4-氨基-环己甲酸乙酯盐酸盐ⅰ;技术路线如下所示:
进一步的,s1中,对反应体系加热至回流,反应至反式-1,4-环己二甲酸反应完全。
进一步的,s1中所述催化剂为浓硫酸,所述浓硫酸的用量为反式-1,4-环己二甲酸的1%~10%。
进一步的,s1中无水乙醇的与反式-1,4-环己二甲酸的质量比为1:2~1:8。
进一步的,s2中中间体二酯a在碱和溶剂存在下进行皂化反应得到中间体单酸b,所述溶剂包括无水乙醇或/和芳香烃溶剂。
进一步的,所述无水乙醇与芳香烃的体积比为1:1~3。
说明书
进一步的,所述中间体二酯a与溶剂的质量体积比为1:2~1:8。
进一步的,所述碱为氢氧化钠,所述氢氧化钠与中间体二酯a的摩尔比为1:1~1:1.2。
进一步的,s3中,中间体单酸b反应完全后将体系冷却至室温,直接加水搅洗,即可析出中间体c。
进一步的,s4中中间体c在饱和氯化氢乙酸乙酯溶液中脱boc保护基得到目标化合物ⅰ,反应的温度范围为0~50℃。
与现有技术相比,本发明的有益效果在于:
(1)本发明的起始原料高纯的反式-1,4-环己二甲酸国内已经实现规模化生产,价格较低,本发明避免了高温高压的苛刻反应条件以及繁琐的拆分过程,同时其他的原料试剂等均为常见试剂,这些原料易得,且成本低廉;同已有的工艺路线相比,本发明大幅度降低了原辅料成本;本发明的合成反式-4-氨基环己甲酸乙酯盐酸盐的方法工艺简单,对工艺设备要求低,不需要无水无氧,高压氢化等苛刻的生产条件,易于操作,生产周期短,效率高,适合于工业规模化生产;
(2)酸的酯化有多种方法,通常采用有氯化亚砜-醇酯化法、氯化氢-醇酯化法、三氯硅烷-醇法等,本发明采用浓硫酸催化反应,一方面避免使用氯化亚砜和氯化氢催化,降低了设备腐蚀与污染,有利于环保;另一方面,选用浓硫酸催化有效解决了酯化反应的放大风险问题,提高了反应效率;
(3)二酯的选择性单皂化是个比较困难的问题,通常采用氢氧化钠或者氢氧化钾的水溶液在乙醇中进行单水解,但是此法得到的单酸收率极低,二酯大部分被皂化为二酸;本发明采用氢氧化钠在无水乙醇或/和芳香烃溶剂进行皂化,有效减少了二酸副产物的生产,单酸的收率提高到85%以上,大大提高了收率;
(4)当中间体单酸b进行curtius重排时,后处理通常采用浓缩、萃洗,浓缩、重结晶的方法得到目标产物,此方法收率低,且操作比较复杂;本发明采用将反应液冷却到室温后直接加水搅洗,即可析出产品,同时醇水体系还能将反应杂质有效去除,直接得到较纯的中间体产品。
附图说明
图1为本发明技术路线图。
具体实施方式
下面结合实施例对本发明作进一步的描述,所描述的实施例仅仅是本发明一部分实施例,并不是全部的实施例。基于本发明中的实施例,本领域的普通技术人员在没有做出创造性劳动前提下所获得的其他所有实施例,都属于本发明的保护范围。
实验例1
s1:在5升圆底烧瓶中加入2.4升乙醇和400克反式-1,4-环己烷二甲酸,充分搅拌混合,然后加入12毫升浓硫酸,升温至回流状态反应约20小时,点板检测原料反应完全,冷却降至室温,浓缩,用500毫升乙酸乙酯溶解,水洗至中性,干燥浓缩,得531克反式-1,4-环己二甲酸二乙酯,为淡黄色油状液体,收率100%;
s2:在5升三颈圆底烧瓶中,将上步得到的530克淡黄色油状液体溶于3升无水甲苯和无水乙醇的混合溶剂中(体积比1:2),室温搅拌下滴加93克氢氧化钠的无水乙醇溶液,得到白色悬浊液,继续搅拌至ph值不再变化,点板检测原料反应完全;将反应液浓缩至干回收溶剂,残余物加1.2升水溶解,水相用4mol/l盐酸调节ph值至5,用2升乙酸乙酯萃取,直至水相无产品;收集有机相,加入1.5升的0.5%碳酸钠溶液洗有机相,然后收集有机相,加入80克无水硫酸镁干燥;过滤,滤液浓缩至干得到461克反式-1,4-环己二甲酸单乙酯,为白色固体,收率87%;
s3:在三颈圆底烧瓶中依次加入上步得到的200克反式-1,4-环己二甲酸单乙酯、叔丁醇800毫升,充分搅拌,加入264克三乙胺,控制温度在30℃以下,滴加288克的dppa,滴加完毕后,将体系缓慢升温至60℃反应2小时,回流反应20小时,点板检测原料基本消失,将反应液冷却至室温,加入4升水充分搅拌,逐渐有大量固体析出,继续搅拌5小时,将析出的固体过滤,滤饼用6升水搅洗,过滤,干燥,得到233.8克反式-4-n-boc-氨基环己甲酸乙酯,为白色固体,收率86%;
s4:在2升三颈圆底烧瓶中,将上步得到的54.2g的反式-4-n-boc-氨基环己甲酸乙酯,加入271毫升的饱和氯化氢乙酸乙酯溶液中,开始时反应体系澄清,然后有大量固体析出,搅拌反应过夜,点板检测原料反应完全,过滤,滤饼用70毫升乙酸乙酯洗涤,干燥,即得到固体37.1克产品反式-4-氨基环己甲酸乙酯盐酸盐,为白色固体粉末,收率为89.3%,纯度大于99%。
实验例2
s1:在5升圆底烧瓶中加入2.5升乙醇和450克反式-1,4-环己烷二甲酸,充分搅拌混合,然后加入10毫升浓硫酸,升温至回流状态反应约20小时,点板检测原料反应完全,冷却降至室温,浓缩,用500毫升乙酸乙酯溶解,水洗至中性,干燥浓缩,得596克反式-1,4-环己二甲酸二乙酯,为淡黄色油状液体,收率100%;
s2:在5升三颈圆底烧瓶中,将上步得到的596克淡黄色油状液体溶于3.1升无水甲苯和无水乙醇的混合溶剂中(体积比1:3),室温搅拌下滴加104克氢氧化钠的无水乙醇溶液,得到白色悬浊液,继续搅拌至ph不再变化,点板检测原料反应完全,将反应液浓缩至干回收溶剂,残余物加1.2升水溶解,水相用4mol/l盐酸调节ph至5左右,用2升乙酸乙酯萃取,直至水相无产品,收集有机相,加入1.5升的0.5%碳酸钠溶液洗有机相,然后收集有机相,加入80克无水硫酸镁干燥,过滤,滤液浓缩至干得到465克反式-1,4-环己二甲酸单乙酯,为白色固体,收率89%;
s3:在三颈圆底烧瓶中依次加入上步得到的400克反式-1,4-环己二甲酸单乙酯、叔丁醇1.6升,充分搅拌,加入528克三乙胺,控制温度在30℃以下,滴加570克的dppa,滴加完毕后,将体系缓慢升温至60℃反应2小时,回流反应20小时,点板检测原料基本消失,将反应液冷却至室温,加入4升水充分搅拌,逐渐有大量固体析出,继续搅拌5小时,将析出的固体过滤,滤饼用10升水搅洗,过滤,干燥,得到477克反式-4-n-boc-氨基环己甲酸乙酯,为白色固体,收率88%;
s4:在5升三颈圆底烧瓶中,将上步得到的271g的反式-4-n-boc-氨基环己甲酸乙酯,加入1.6升的饱和氯化氢乙酸乙酯溶液中,开始时反应体系澄清,然后有大量固体析出,搅拌反应过夜,点板检测原料反应完全,过滤,滤饼用70毫升乙酸乙酯洗涤,干燥,即得到固体207克产品反式-4-氨基环己甲酸乙酯盐酸盐,为白色固体粉末,收率为90%,纯度大于99%。
实验例3
s1:在20升反应釜中加入10升乙醇和1720克反式-1,4-环己烷二甲酸,充分搅拌混合,然后加入100毫升浓硫酸。升温至回流状态反应约20小时,点板检测原料反应完全,冷却降至室温。浓缩,用5升乙酸乙酯溶解,水洗至中性,干燥浓缩,得2280克反式-1,4-环己二甲酸二乙酯,为淡黄色油状液体,收率100%。
s2:在20升反应釜中,将上步得到的2280克淡黄色油状液体溶于15升无水甲苯和无水乙醇的混合溶剂中(体积比1:1),室温搅拌下滴加400克氢氧化钠的无水乙醇溶液,得到白色悬浊液,继续搅拌至ph不再变化,点板检测原料反应完全,将反应液浓缩至干回收溶剂,残余物加10升水溶解,水相用4mol/l盐酸调节ph至5左右,用10升乙酸乙酯萃取,直至水相无产品,收集有机相,加入12升的0.5%碳酸钠溶液洗有机相,然后收集有机相,加入80克无水硫酸镁干燥,过滤,滤液浓缩至干得到1800克反式-1,4-环己二甲酸单乙酯,为白色固体,收率90%;
s3:在50升反应釜中依次加入上步得到的1800克反式-1,4-环己二甲酸单乙酯、叔丁醇7升,充分搅拌,加入2400克三乙胺,控制温度在30℃以下,滴加2600克dppa,滴加完毕后,将体系缓慢升温至60℃反应2小时,回流反应20小时,点板检测原料基本消失,将反应液冷却至室温,加入30升水充分搅拌,逐渐有大量固体析出,继续搅拌5小时,将析出的固体过滤,滤饼用10升水搅洗,过滤,干燥,得到2178克反式-4-n-boc-氨基环己甲酸乙酯,为白色固体,收率89%;
s4:在50升反应釜中,将上步得到的2178克的反式-4-n-boc-氨基环己甲酸乙酯,加入12.3升的饱和氯化氢乙酸乙酯溶液中,开始时反应体系澄清,然后有大量固体析出,搅拌反应过夜,点板检测原料反应完全,过滤,滤饼用5升乙酸乙酯洗涤,干燥,即得到固体1491克反式-4-氨基环己甲酸乙酯盐酸盐,为白色固体粉末,收率为90%,纯度大于99%。
- 还没有人留言评论。精彩留言会获得点赞!