一种异紫草酸的制备方法与流程
本发明涉及一种药物化合物的制备,属于医药化工领域,具体涉及一种异紫草酸的制备方法。技术背景4-(2-羧基乙烯基)-2-(3,4-二羟基苯基)-2,3-2h-7-羟基-3-苯骈呋喃羧酸-3-[1-羧基-2-(3,4-二羟基)乙基]酯,又名异紫草酸,为紫草酸的同分异构体,其结构见结构式ⅰ:异紫草酸的由来,该化合物最早是利用液质检测到的丹酚酸b水溶液的降解产物,(上述相关信息见文献:yong-xueguoetc.,kineticsandmechanismofdegradationoflithospermicacidbinaqueoussolution,journalofpharmaceuticalandbiomedicalanalysis43(2007)1249–1255)后由韩国学者在丹酚酸b水溶液的降解产物中分离得到并报道了其核磁数据,其制备方法为:将丹酚酸b溶解在水中,并用hcl调节ph值为4.9。然后将上述水溶液100℃加热24小时,再在39℃下减压浓缩。浓缩液用sephadexlh-20柱色谱(70-230目,2.0*132cm,柱床体积480ml)进行纯化,洗脱液为甲醇/水(8:2,v/v)。所得流份利用薄层色谱或液相分析进行合并,最终得到23个流份。洗脱体积和柱床体积比为1.30-2.40和3.96-4.58的流份合并后使用制备液相(色谱柱:20*250mm,流速:9.9ml/min,检测波长:280nm,流动相:三氟乙酸调节ph值为2.65,a乙腈/水10:90v/v,b乙腈/水10:90v/v,洗脱梯度在70分钟内100%a→100%b,100%b维持15分钟)进行纯化,得到异紫草酸(tr35.33min)。该文献明确了异紫草酸的结构,如结构式ⅰ所示。(上述相关信息见文献:hyoungjaeleeetc.,chemicalconversionsofsalvianolicacidbbydecoctioninaqueoussolution,fitoterapia83(2012)1196–1204)。该工艺极其复杂、用到多种有机试剂,并且对高压分离设备的依赖性高,制备周期长,成本高。丹参酚酸提取物,包括符合2015版中国药典标准的市售丹参总酚酸提取物(《中国药典》2015年版第1部中397-398页)和现有市场上销售的注射用丹参多酚酸的制剂原料丹参多酚酸提取物(其方法很多种,例如cn105588885a,申请号为201410572435.3
背景技术:
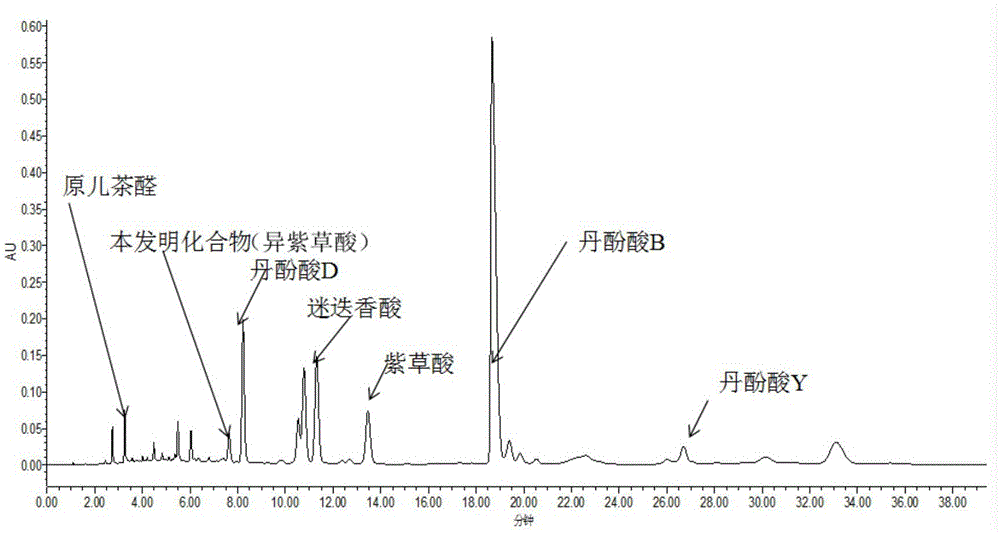
:中提到的制备方法),其中符合2015版中国药典标准的市售丹参总酚酸提取物制备方法为:取丹参、切成小段,加水于80℃提取两次,合并提取液,滤过,滤液于60℃减压浓缩至相对密度为1.18-1.22(50℃)的清膏,放冷,加乙醇使得含醇量为70%,静置12小时,取上清液,减压回收乙醇,并浓缩至稠膏,干燥,即得。经过进一步分析发现丹参酚酸提取物指纹图谱的建立方法为uplc法,色谱条件如下:仪器:watersuplc-pda色谱柱:watersacquityhsst31.8um流动相:a:0.1%甲酸水,b:乙腈梯度洗脱:表1:梯度洗脱表结果见图1,丹参酚酸提取物包括原儿茶醛、丹酚酸d、丹酚酸b、迷迭香酸、丹酚酸y、紫草酸和异紫草酸等,丹参总酚酸提取物和注射用丹参多酚酸的原料(即丹参多酚酸提取物)二者的图谱均一致。基于上述发现,发明人试图从丹参酚酸提取物分离异紫草酸,但是:首先,该化合物在丹参酚酸提取物中的含量很低;第二,该化合物在指纹图谱中紧挨丹酚酸d,表明这两个化合物极性非常接近;第三、酚酸类化合均含有若干个酚羟基,呈弱酸性,易被氧化分解,对分离条件比较苛刻。因此,从丹参酚酸提取物中分离该化合物的难度很大,采用现有技术如上述文献中的方法均无法将丹酚酸d和异紫草酸完全分离。本发明的目的是提供一种可以将从丹参酚酸提取物中分离得到异紫草酸的方法,分离制备得到高纯度单体异紫草酸对于进一步研究其药理活性、完善其质量标准,以及相关产品的标准提升具有重要意义。技术实现要素:本发明提供了一种丹参中异紫草酸的制备方法,步骤如下:步骤1、异紫草酸的富集步骤2:异紫草酸粗品的制备步骤3:异紫草酸纯品的制备其中,所述步骤1、异紫草酸的富集,方法如下:取丹参酚酸提取物,用水溶解,加入反向色谱柱中,用乙醇水溶液洗脱,再次用反向色谱柱固相萃取,用95%乙醇洗脱,洗脱液浓缩至干得到异紫草酸富集物;其中,所述步骤2:异紫草酸粗品的制备,方法如下:异紫草酸富集物去除色素,得到异紫草酸富集物粗品;其中,所述步骤3:异紫草酸纯品的制备,方法如下:采用ods色谱柱进一步分离纯化得到高纯度异紫草酸单体,所得纯品经干燥即得。优选的,所述步骤1、异紫草酸的富集,方法如下:取丹参酚酸提取物,用1-3倍量的水溶解,加入反向色谱柱中,然后用8-15%乙醇水溶液洗脱,下接液调节ph值为酸性,再次用反向色谱柱固相萃取,然后用90-95%乙醇洗脱,收集下接液,浓缩至干得到异紫草酸富集物;所述步骤2:异紫草酸粗品的制备,方法如下:异紫草酸富集物去除色素,得到异紫草酸富集物粗品;所述步骤3:异紫草酸纯品的制备,方法如下:用乙腈溶解粗品,采用ods色谱柱分离,流动相为乙腈和含0.01%甲酸的水溶液,体积比为23:77-30:70,流速为4-8bv/h,比照丹参酚酸指纹图谱,收集含异紫草酸的洗脱液,减压浓缩干燥,即得。上述方法中:其中步骤1中,反向色谱柱为mcigel色谱柱、聚酰胺色谱柱、ods-c18色谱柱或lh-20凝胶色谱柱,优选为mcigel色谱柱;所述过柱为两次过柱,第一次,提取物与填料的重量体积比为1:60-1:20;第二次,提取物与填料的重量体积比为1:6-1:3;所述两次下接液,第一次柱子洗脱液需要检测,用hplc法检测,同时与丹参酚酸提取物的指纹图谱对照,收集含有异紫草酸的洗脱液;第二次直接收集乙醇洗脱液。其中,所述调节ph值的调节剂为甲酸或盐酸;ph值至3.0-4.5;上述方法中:其中步骤2中,去除色素方法为lh20凝胶柱层析或活性炭脱色,优选为lh-20凝胶柱层析;当lh20凝胶柱层析去除色素时,用1-3倍质量体积的45-55%乙醇水溶液完全溶解,加入lh20凝胶柱层析中,用45-55%乙醇洗脱,流速:0.08-0.12bv/h,参考丹参酚酸提取物指纹图谱对照,合并含有异紫草酸的粗品。本发明所述方法,具体可以包括以下步骤:步骤1、异紫草酸的富集取丹参酚酸提取物,用1-3倍量的水溶解,加入反向色谱柱中,上样量与色谱柱填料的体积比为1:60-1:20,上样速度为1-3bv/h,然后用8-15%乙醇水溶液洗脱,流速为1-3%,下接液调节ph值2.5-5,再次用反向色谱柱固相萃取,提取物与色谱柱填料的体积比为1:6-1:3,用90-95%乙醇洗脱,流速1-3bv/h,收集洗脱液,浓缩至干得到异紫草酸富集物;步骤2:异紫草酸粗品的制备异紫草酸富集物去除色素,得到异紫草酸富集物粗品;步骤3:异紫草酸纯品的制备用乙腈溶解粗品,乙腈和粗品的质量体积比为1-3:1,采用ods色谱柱分离,流动相为乙腈和含0.01%甲酸的水溶液,体积比为23:77-30:70,流速为4-8bv/h,比照异紫草酸指纹图谱,收集含丹酚酸y的洗脱液,减压浓缩干燥。更具体的,本发明所述方法,可以包括以下步骤:步骤1:异紫草酸的富集取丹参酚酸提取物,2倍质量体积的水溶解,加入mcigel色谱柱中(每次上样提取物量-mci-gel填料1:60-1:20),流速:2bv/h(2倍柱床体积/小时),上样结束后用10%乙醇水溶液洗脱,流速:2bv/h(2倍柱床体积/小时),每1/10bv(1/10柱床体积)接一个样品;用hplc法检测,同时与丹参酚酸提取物的指纹图谱对照,合并含有异紫草酸的洗脱液。调节洗脱液ph值至3.0-4.5,再次加入mcigel色谱柱(提取物-mci-gel填料1:6-1:3)中,用水洗涤5bv,洗脱液弃去,流速:2bv/h(2倍柱床体积/小时),之后用95%乙醇洗脱,流速:2bv/h(2倍柱床体积/小时),收集洗脱液2bv,回收溶剂,浓缩至干得到异紫草酸富集物;步骤2:异紫草酸粗品的制备异紫草酸富集物用2倍质量体积的50%乙醇水溶液完全溶解,加入lh20凝胶柱层析中,用50%乙醇洗脱,流速:0.1bv/h,每500ml接一个洗脱液,用hplc法检测,同时与丹参酚酸提取物指纹图谱对照,合并含有异紫草酸的粗品;步骤3:异紫草酸纯品的制备异紫草酸粗品用2倍质量体积的乙腈水溶液完全溶解,ods色谱柱分离,流动相:乙腈-含0.01%甲酸的水溶液,体积比为23:77-30:70(),流速6bv/h,每1/4bv接一个样品,用hplc法检测,同时与丹参酚酸指纹图谱对照,合并含有异紫草酸的洗脱液,减压浓缩干燥得到异紫草酸单体,60℃减压干燥12小时。本发明的制备方法具有以下优点:1、发明人在研究中发现丹参酚酸提取物中含有少量的化合物异紫草酸,为了从丹参酚酸提取物中得到该化合物,发明人做了大量的实验。丹参酚酸类化合物大多经过硅胶柱粗分离,然后反复经过反相c18柱分离。所用多种有机试剂,对高压分离设备的依赖性高。化合物异紫草酸在制备工艺开发过程中借鉴酚酸类化合物常规工艺,比如对照例1,先用硅胶柱分离,再用ods(c18填料)色谱柱分离,最后只能得到化合物异紫草酸与丹酚酸d的混合物(该混合物的hplc检测图谱见图2)。且该方法分离周期长,使用的有机溶剂多,对操作人员的安全带来隐患。为了将该化合物与丹酚酸d分离,实验人员筛选了不同类型的ods柱,均未达到目的,最后重回原点重新筛选方法,最终确定采用凝胶柱色谱先化合物与丹酚酸d分离,再用ods纯化地方法得以解决。2、现有技术采用大孔吸附树脂柱、ods反向色谱、反复hplc分离酚酸类化合物,但是发明人在借鉴该方法从丹参酚酸提取物中分离异紫草酸和丹酚酸d时发现,因为原料不同,同样该方法,比如采用ods柱或hplc分离的同时,丹酚酸d极易转化成另一种未知化合物,总是混入异紫草酸的馏分中,从而得不到单一的异紫草酸,因此本方法利用丹酚酸d和异紫草酸分子量差异,先采用凝胶柱将丹酚酸d去除,再用ods柱分离得到单一的化合物。3、异紫草酸富集物经lh20凝胶柱层析分离得到异紫草酸粗品,再经过c18反相色谱柱纯化即得到高纯度异紫草酸,其含量大于98.5%。本方法步骤少,纯度高,操作简单,可以用于心脑血管系统新药的开发。附图说明图1:丹参酚酸提取物的指纹图谱,具体建立方法见
背景技术:
;图2:异紫草酸和丹酚酸d混合物的hplc检测图;图3:富集后的异紫草酸uplc检测图;图4:异紫草酸粗品uplc检测图图5:异紫草酸单体uplc检测图。四、具体实施方式下面结合实施例对本发明的药物做进一步说明,下述各实施例仅用于说明本发明而并非对本发明的限制。实施例1:异紫草酸的制备方法步骤一、异紫草酸的富集取丹参酚酸提取物500g,用1000ml水溶解,每次上样100ml,加入填装有3lmcigel玻璃色谱柱中,流速:100ml/min。上样结束后用10%乙醇水溶液洗脱,流速:100ml/min,从下接液有颜色起每300ml接一个样品。一共收集到25瓶样液,用uplc法检测,同时与丹参酚酸指纹图谱对照,合并第2-6瓶含有异紫草酸的洗脱液。调节洗脱液ph值至3.0,再次加入填装有3lmcigel玻璃色谱柱中,用水洗涤15l,洗脱液弃去,流速:100ml/min。之后用95%乙醇洗脱,流速:100ml/min,收集洗脱液6l,回收溶剂,浓缩至干得到异紫草酸富集物。平行操作以上步骤完成所有样品的分离,共得到富集物45g,批号20150530。步骤二:异紫草酸富集物粗品的制备异紫草酸富集物45g用90ml50%乙醇水溶液完全溶解,加入装有1kglh-20葡聚糖凝胶的玻璃柱中,用50%乙醇洗脱,流速:10ml/min,每500ml接一个洗脱液。用uplc法检测,同时与丹参酚酸指纹图谱对照,合并含有异紫草酸的粗品12.3g,批号20150609。步骤三:异紫草酸富集物纯品的制备异紫草酸粗品12.3g用25ml23%乙腈水溶液完全溶解,采用动态轴向加压制备液相分离,每次进样10ml,流动相:乙腈-水23:77(0.01%甲酸),流速200ml/min,每500ml接一个样品。用uplc法检测,同时与丹参酚酸指纹图谱对照,合并含有异紫草酸富集物的洗脱液。减压浓缩得到异紫草酸纯品,平行以上操作完成所有样品的分离,60℃减压干燥12小时。产物批号20150622。将得到的产物进行核磁共振测试,所得核磁数据(见表2)与文献报道(hyoungjaeleeetc.,chemicalconversionsofsalvianolicacidbbydecoctioninaqueoussolution,fitoterapia83(2012)1196–1204)一致,证明所得化合物为异紫草酸。表2:异紫草酸核磁共振数据归属实验例1:异紫草酸的含量测定1、试验方法色谱条件色谱柱:watersaqucityuplctmhsst3(2.1×150mm,1.8μm)色谱条件:λ=280nm;柱温:40℃;流速:0.4ml/min;流动相:采用梯度洗脱,见表3表3丹参中酚酸类标准品测定流动相梯度表(二)待测样品溶液的配制称取异紫草酸富集物20150530(实施例1中步骤1方法制备)30mg,至于置25ml容量瓶中,用甲醇溶解,并定溶至刻度,待测。称取异紫草酸粗品20150509(实施例1中步骤2方法制备)20mg,至于置25ml容量瓶中,用甲醇溶解,并定溶至刻度,待测。取高纯度异紫草酸20150620(实施例1中步骤1方法制备)10mg,至于置25ml容量瓶中,用甲醇溶解,并定溶至刻度,待测。(3、测定分别取以上样品溶液各2μl,进行测定,记录异紫草酸归一化法百分含量。4、含量测定结果,见表4、图3和图4。表4:异紫草酸样品含量测定结果样品名称归一化法百分含量(%)备注异紫草酸富集物2015053042.3%见图3异紫草酸粗品2015050992.6%见图5高纯度异紫草酸2015062098.7%见图4结果表明:本发明的方法得到的是异紫草酸纯度高。对照例1:分离及检测1、分离方法步骤1、异紫草酸的硅胶柱层析分离取丹参酚酸提取物50g,溶于200ml水中,用等体积的乙酸乙酯萃取3次,收集乙酸乙酯萃取液减压回收溶剂,加入原料的80g100-200目硅胶进行拌样。硅胶柱填装:取原料1000g的200-300目硅胶,用乙酸乙酯-石油醚1:1(2%甲酸)浸泡30min,装柱,用乙酸乙酯-石油醚1:1平衡3bv。分离:将样品干法上样,用乙酸乙酯-石油醚1:1(2%甲酸)洗脱,约0.2bv为单位收集馏分,tlc检测,用5%硫酸乙醇为显色剂进行显色。馏分合并:将与异紫草酸对照品斑点rf值一致的馏分合并,减压浓缩至干,得到含有异紫草酸的粗品15g。步骤2、精细分离异紫草酸粗品15g用30ml23%乙腈水溶液完全溶解,采用动态轴向加压制备液相(填装有反相c18填料)分离,每次进样10ml,流动相:乙腈-水23:77(0.01%甲酸),流速200ml/min,每500ml接一个样品。用hplc法检测,同时与丹参酚酸指纹图谱对照,合并含有富集物的洗脱液。减压浓缩得到异紫草酸,平行以上操作完成所有样品的分离,得到0.8g,批号20150305。2、检测方法对该产物进行检测,检测方法为:实验仪器和材料:2.1agilent1100高效液相色谱仪2.2hplc检测条件:2.3色谱柱:agilentzorbaxsb-c18、5um、4.6*250mm,进样量10ul,流速1ml/min,检测波长280nm,柱温30℃。流动相a为0.02%磷酸水,b为80%乙腈-20%0.02%磷酸水。洗脱梯度0min,90%a;8min,78%a;15min,74%a;35min,61%a;40min,90%a;50min,90%a。3、结果见图2结果表明:采用反相c18填料无法将异紫草酸与丹酚酸d分离,并且在分离后浓缩过程中很容易出现一个未知的化合物。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1