达沙替尼无水合物的制备方法与流程
本发明涉及药物合成领域,特别是涉及达沙替尼无水合物的制备方法。
背景技术:
达沙替尼一水合物化学名称为n-(2-氯-6-甲基苯基)-2-[[6-[4-(2-羟乙基)-1-哌嗪基]-2-甲基-4-嘧啶基]氨基]-5-噻唑甲酰胺(一水物),其结构式如下:
达沙替尼(dasatinib)是一种强效的酪氨酸激酶多靶点抑制剂,是继尼洛替尼之后另一个用于伊马替尼耐药和不耐受的cml慢性期的药物。与针对bcr-abl融合蛋白单靶点的伊马替尼和尼洛替尼相比,达沙替尼属于多靶点药物,对5种关键性致癌酪氨酸蛋白激酶有作用。该产品最早由百时美施贵宝公司研发,2006年6月在美国获准上市,11月在欧盟上市。目前现有的达沙替尼的合成路线其合成方法如下。
一种为文献j·med·chem·2004,47,6658-6661;j·med·chem·2006,49,6819-6832中提供的路线,具体如下
该工艺的不足之处在于反应条件较为苛刻且收率,该路线需用到氢化钠,反应条件苛刻,仅适合实验室规模生产,在商业化规模生产(公斤级别)中,不适合工业化生产,所以人们仍在不断探索新的合成路线。
另外一种为专利cn200580011916.6公开路线如下:
该路线为了解决化合物ii与4,6-二氯-2-甲基嘧啶在高温、强碱条件下,副产物含量较高的问题,通过控制不同的反应条件,选择碱性较弱的diea作为碱性试剂,降低反应温度减少副产物的生成,但同时也降低了主产物化合物的收率,催化剂比较贵,提高了成本。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种达沙替尼无水合物的制备方法,用于解决现有技术中存在的制备达沙替尼的反应条件苛刻,成本高,不易纯化,不适用于工业化大规模生产的问题。
为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
本发明中所述的达沙替尼无水合物的结构式如式ii所示:
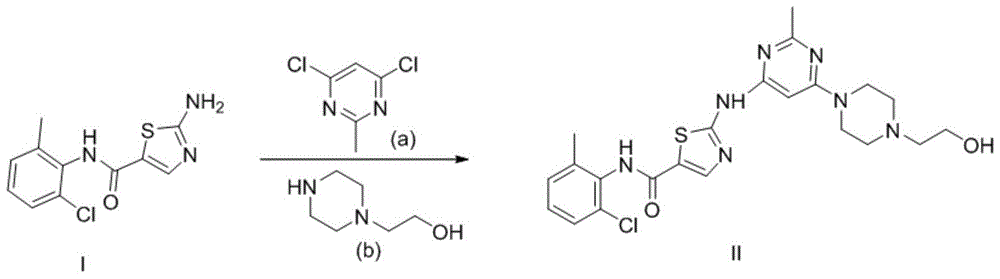
本发明公开了如上述达沙替尼无水合物的制备方法,包括采用结构式如式ⅰ所示化合物在化合物a和化合物b反应生成,合成路线如下所示:
根据上述所述的达沙替尼的制备方法,所述反应在缚酸剂存在的条件下进行。缚酸剂在反应中,可以中和反应过程终产生的氯化氢,从而使反应更好的向目标产物转化。优选地,所述缚酸剂为选自三乙胺、二异丙胺、吡啶、碳酸钾和三正丁胺中的任意一种或多种的组合。
根据上述所述的达沙替尼的制备方法,所述缚酸剂与式ⅰ化合物的摩尔比为(2.5~8):1。在此摩尔比区间可以保证反应在碱性条件下进行,否则,原料不能完全反应,影响收率,提高纯化难度。更优选地,所述所述缚酸剂与式ⅰ化合物的摩尔比为6:1。
根据上述所述的达沙替尼的制备方法,上述反应中在反应介质中进行,所述反应介质为有机溶剂。优选地,所述有机溶剂选自乙腈、正丁醇、甲苯和1.4-二氧六环中的一种或多种。
根据上述所述的达沙替尼的制备方法,结构式如式ⅰ所示化合物与化合物a的摩尔比为1:(1.0~1.2)。在此摩尔比区间内反应能够顺利进行,副产物少,若化合物a的摩尔当量小于于1.0,会有原料剩余,若化合物a的摩尔当量大于1.2,会使副产物增大,增加纯化难度。更优选地,结构式如式ⅰ所示化合物与化合物a的摩尔比为1:1.05。
根据上述所述的达沙替尼的制备方法,反应时间为5~10h。反应时间大于5h,原料基本可以反应完全,若工厂放大生产存在放大效应,延长反应时间,反应会继续进行,至原料完全反应。
根据上述所述的达沙替尼的制备方法,结构式如式ⅰ所示化合物与化合物b的摩尔比为1:(1.0~4.0)。在此摩尔比区间内均可以使反应正常进行;化合物b少于1.0摩尔当量会有原料剩余不能完全反应;化合物b大于4.0摩尔当量,会使副产物增大,增加纯化难度。经验证在2.5时反应速度相对更快且反应收率稳定,处理简单。更优选地,结构式如式ⅰ所示化合物与化合物a的摩尔比为1:2.5。
根据上述所述的达沙替尼的制备方法,反应温度为70~120℃。在此温度区间,反应能够顺利且高效进行,若温度低于70℃,反应速度会降低,反应温度低于40℃,基本不反应。
根据上述所述的达沙替尼的制备方法,还包括后处理步骤,所述后处理步骤包括分离、过滤、洗涤或干燥中的多种。更优选地,所述分离为用水淬灭反应,更优选地,所述水的体积为结构式如式i所示化合物体积至少8倍。更优选地,所述洗涤为乙醇洗涤滤饼。更优选地,所述乙醇的体积为结构式如式i所示化合物体积的1~2倍。更优选地,所述干燥为在鼓风烘箱中烘干。更优选地,所述干燥温度在50~60℃。
本发明上述技术方案的有益效果为:
本发明提供的技术方案能够获得高品质的达沙替尼无水合物,避免了繁琐的分离提纯步骤,操作简单,也避免了原料的浪费,降低了生产成本,更适用于工业化生产。
附图说明
图1为实施例中结构式如式ii所示的化合物的制备路线图
图2为实施例中达沙替尼粗品纯度图谱
具体实施方式
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
本申请中的结构式如式i所述的化合物为现有技术,可根据以下文献获得:friedrichc,arnulfv.thechemistryofhigh-energyphosphates.iii.preparationofisothioureidophosphatesandphosphoguanidines[j].chemischeberichte,1958,91:919-923。
在本申请的示例性实施例中,用如图1所示的路线合成,具体步骤如下。
结构式如式ii所示的化合物的制备方法如下
n2保护下,向机械搅拌的20l三口瓶中称取化合物a(1.3kg,4.84mol)加入7.5l乙腈,搅拌均匀,再加入化合物i(752g,4.61mol),搅拌均匀分批缓慢加入碳酸钾(3.8kg,27.66mol),室温搅拌4小时。再加入化合物b(1.5kg,11.5mol),升温回流6小时,降温到室温缓慢加入6.4l水中搅拌离心,800ml乙醇洗涤虑饼,55度烘干得白色固体1.83kg,收率:81.3%,纯度:99.3%。
本步骤产物中间体化合物ii的分析方法如下:
1.纯度检测hplc分析方法
1.1仪器
hplc:lc-20a,配紫外检测器
1.2试剂
乙腈(色谱级)
磷酸(ar级)
哇哈哈纯净水
1.3样品溶液的配制
称取样品适量,用20%乙腈/水溶解,样品浓度约0.5mg/ml。
1.4流动相配置
a相:0.01%h3po4/h2o
在1l容量瓶中加入600ml水,移取0.1ml磷酸于容量瓶中,加水稀释定容,充分混匀。
b相:乙腈
1.5色谱条件
1.6系统适应性
空白干净,在主峰的出峰未知处没有干扰。(干扰峰的信噪比﹤10或干扰峰面积小于灵敏度溶液中主峰的峰面积,即认为无干扰)
灵敏度溶液中主峰的信噪比s/n≧10。
1.7结果计算
按峰面积归一化法计算样品的纯度,如果每次的测定结果之间的rsd≦2.0%,则取两次。
测定结果的平均值报告最终结果。
本实施例中形成的达沙替尼的核磁表征结果如下:hnmr(400mhz,dmso-d6)ppmδ:2.25(s,3h,ch3),2.41(s,3h,ch3),2.43(t,j=6.2hz,2h),2.49(t,4h,ch2),3.52~3.53(m,4h,ch2),3.54~3.55(m,2h,ch2),6.06(s,1h,ch),7.27~7.28(m,1h,ch),7.29~7.31。(m,1h,ch),7.40(d,j=7.5hz,1h),8.22(s,1h,ch),9.85(s,1h,nh)。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!