具有纳米原纤化表面特征的高表面积纤维介质的制作方法
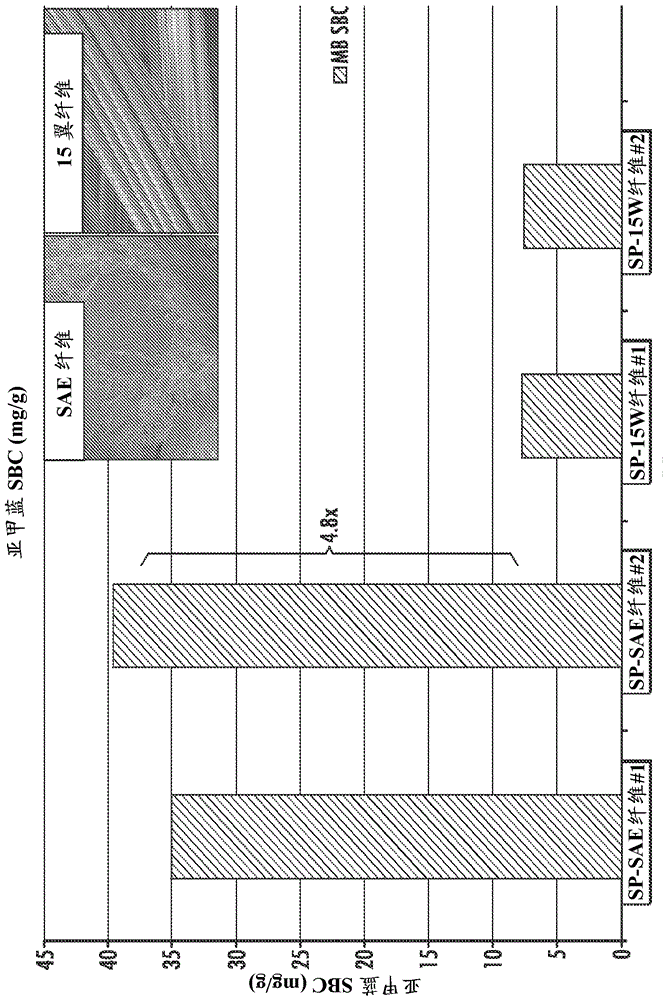
发明领域本文公开的实施方案涉及多孔的高表面积纤维,例如以阳离子交换层析法模式,其适用于用作用于结合/洗脱纯化蛋白质的层析法固定相。背景商业规模纯化各种治疗生物分子(诸如单克隆抗体)当前使用基于珠的层析树脂来完成。单克隆抗体作为治疗和诊断剂持续获得重要性。用于候选mAbs的筛选杂交瘤库的过程既耗时又为劳动密集型的。一旦建立表达合适的mAb的杂交瘤细胞系,必须开发纯化方法学以生产足够的mAb用于进一步表征。用于纯化的常规的方法涉及使用蛋白质A或蛋白质G亲和层析法以及离子交换层析法。将经纯化的抗体脱盐,并且使用渗析交换成为生物学缓冲剂。整个过程通常需要若干天来完成,并且如果多个mAbs待并联评价,则可能特别繁重。层析树脂当前使用能够使珠以亲和力、阳离子交换或阴离子交换模式起作用的各种配体结构来制备。这些树脂证明高多孔性和大表面积,其提供具有足够吸附能力的材料,用于以生产规模(例如,10,000升)批量加工生物分子。层析树脂通常呈现球状结构,其能够使柱有效填充有最小流动非均匀性。珠之间的间隙空间提供流动通道,用于通过层析柱而对流输送。这能够使层析柱以高的线性速度以大的床深度运行,且具有最小压降。这些因素的组合能够使层析树脂呈现所需的效率、高渗透性和大规模纯化生物分子所需的足够的结合能力。在基于珠的层析法中,用于吸附的大多数可用的表面积在珠的内部。因此,分离过程固有地缓慢,这是由于传质速率通常通过孔扩散来控制。为了使该扩散抵抗力最小化和伴随使动态结合能力最大化,可采用小直径珠。然而,使用小直径珠的代价是柱的压降提高。因此,制备型层析法分离的优化通常涉及在效率/动态能力(优先小珠)和柱压降(优先大珠)之间的折衷。层析介质通常具有非常高的成本(>$1000/L),并且需要显著量用于大规模生产柱。结果是,生物药物制造商再循环层析树脂数百次。这些再生循环每次消耗显著量的缓冲剂介质,并且每一个步骤招致与每一次清洁、灭菌和柱填充操作的批准关联的另外的成本。在专利文献中描述了若干技术,并且在商业上销售用于基于官能化的纤维介质和/或复合材料的生物药物分离。大多数依赖于在纤维基质中掺入多孔凝胶,所述凝胶提供所需的表面积,以获得合理的结合能力。然而,在这样的结构中,凝胶位置和质量的差的均匀性通常导致差的效率(浅贯穿和洗脱前缘)。此外,即使对于短的床深度,对流动的抵抗力可能高,通过在适度的压力负载下的凝胶压缩,问题通常加重。采用的另一种方法是在纤维基质内掺入微粒,所述微粒通常为多孔的并且具有天然的吸附官能团,其实例为活性炭和硅胶。近来,EMDMillipore已开发基于纤维的层析介质,用于生物分子纯化应用,其利用表面官能化的翼形纤维作为吸附介质。比起类似尺寸的普通圆形纤维,在纤维表面上的翼形突起提供高得多的表面积。比起缺乏这样的翼形突起的类似官能化的纤维,所得到的表面官能化的纤维介质还具有高得多的蛋白质结合能力。其它现有技术当前开发用于蛋白质纯化应用,且这些包括膜吸附剂、整体料和使用商业树脂体系的流通吸附剂纯化方法。虽然膜吸附剂和整体料对于这些应用可提供可接受的结合能力,但是这些技术通常具有它们自身的规模限制,并且这样的纯化介质的极高的成本可进一步限制它们在使用现有纯化过程模板的价格敏感的行业中的采用。概述为了解决本领域当前已知的纯化技术的许多限制,本文公开的实施方案涉及层析介质,其包含低成本、高表面积热塑性纤维和在该纤维表面上的离子交换配体官能团。在某些实施方案中,离子交换配体能选择性结合来自生物学进料流的蛋白质。当溶液条件变化时,已结合的蛋白质可随后从层析介质释放,例如,通过使用具有较高离子强度的洗脱缓冲剂。在某些实施方案中,将表面侧基官能团加入到为高表面积纤维提供阳离子交换或阴离子交换官能团的介质中。该侧基官能团可用于离子交换层析法纯化生物分子,诸如重组体融合蛋白质、含有Fc的蛋白质、ADC's(抗体药物缀合物、疫苗、血浆蛋白质(IgM、血液凝固因子等)和单克隆抗体(mAbs)。在某些实施方案中,基于纤维的固定相为多孔的,并且显示旋绕的结构,其由高度缠结的纳米纤丝的离散束构成。在某些实施方案中,位于所述纳米纤丝束内的每一个纳米纤丝具有小于或等于1微米的直径。这些纤维通常呈现在1-12平方米/克范围的表面积。在某些实施方案中,多孔纤维被原纤化或成脊状(ridged)。在某些实施方案中,纳米纤维束可通过两种不混溶的聚合物的共混物的熔融挤出而制备,所述聚合物诸如尼龙(包括聚酰胺6、聚酰胺6,6、聚酰胺4,6、聚酰胺、聚酰胺12、聚酰胺6,12和各种聚酰胺的共聚物或共混物),以及聚(乳酸)PLA。在熔融挤出后,将纤维拉伸至约20微米的目标直径。PLA聚合物致孔剂组分随后通过用氢氧化钠溶液处理而提取,留下贯穿尼龙微纤维的伸长的腔或通道。所得到的纤维介质具有高度缠结的尼龙纳米纤维束的外观,其以共线排列而松散对准。这些束具有普通微纤维的流动性质,并且以填充床形式,还证明高渗透性。与此相反,不成束的单个纳米纤维以填充床形式证明非常低的渗透性。该纳米纤维的独特排列提供高渗透性、高表面积基材,在使用适当的离子交换配体表面修饰后,其能够具有高蛋白质结合能力。被侧基离子交换官能团修饰的纤维可用于层析法纯化蛋白质,诸如单克隆抗体。在某些实施方案中,表面积增强的(SAE)纤维被表面官能的磺丙基(SP)配体修饰,并且用于结合/洗脱阳离子交换层析法应用,用于纯化IgG。SAE纤维介质可被表面修饰,以安装侧基离子交换配体,诸如磺丙基(SP)基团。可在合适的设备(诸如层析柱)中填充官能化的介质,并且压缩至目标填充密度。待纯化的蛋白质溶液可随后经过该纤维填充,通过离子交换过程,在其上,关注的蛋白质可在SAE纤维的表面上与配体结合。例如,在pH5下,磺丙基带强负电荷,并且将结合蛋白质,诸如IgG,其pI大于约7。在结合靶蛋白质(例如,IgG)后,柱通常用合适的缓冲剂洗涤,诸如50mM乙酸盐缓冲剂(pH5),以清除任何未结合的杂质。随后,诸如使用0.5M氯化钠/50mM乙酸盐(pH5)溶液,提高缓冲剂的离子强度,以从SAE纤维柱洗脱已结合的IgG。通过诸如使用5-10柱体积的0.5M氢氧化钠和5-10柱体积的50mM乙酸盐缓冲剂(pH5)洗涤,纤维柱可随后再生。SAE纤维介质现在已准备好用于另一个CEX结合/洗脱循环。因此,本文公开的实施方案涉及使用包含高表面积官能化的多孔纤维的介质隔离、纯化或分离生物分子的方法。附图简述图1(a)为根据某些实施方案,20微米纤维直径的普通非多孔尼龙单丝的SEM图,4000倍放大;图1(b)为根据某些实施方案,在完全除去皮材料后,15微米纤维直径的15翼尼龙纤维的SEM图,5000倍放大;图1(c)为根据某些实施方案,具有共混物组合物PA6/Albafil®CaCO375/25的挤出的单丝在完全除去嵌入的致孔剂和皮材料后的SEM图,2:1拉伸比,4000倍放大;图1(d)为根据某些实施方案,具有芯共混物组合物尼龙/PLA70/30的15翼纤维在完全除去嵌入的致孔剂和皮材料后的冷冻SEM横截面图,10,000倍放大;图1(e)为根据某些实施方案,具有芯共混物组合物尼龙/PLA60/40的表面积增强的(SAE)芯/皮纤维在完全除去嵌入的致孔剂和皮材料后的冷冻SEM横截面图,5000倍放大;图1(f)为根据某些实施方案,具有“岛”组合物PA6尼龙100/0、“海洋”组合物PA6尼龙/PLA55/45的“在海洋中的连接的岛”(CIST)纤维在完全除去嵌入的致孔剂和皮材料后的冷冻SEM横截面图,i/S比率,1/1,5000倍放大;图2(a)为根据某些实施方案,PA6尼龙的挤出的单丝的SEM图,2:1拉伸比,1000倍放大;图2(b)为根据某些实施方案,PA6尼龙的挤出的单丝的SEM图,2:1拉伸比,4000倍放大;图2(c)为根据某些实施方案,在从具有PA6/Multifex-MM™CaCO375/25的共混物组合物的挤出的单丝进行矿物致孔剂提取后的SEM图,1.3:1拉伸比,1000倍放大;图2(d)为根据某些实施方案,在从具有PA6/Multifex-MM™CaCO375/25的共混物组合物的挤出的单丝进行矿物致孔剂提取后的SEM图,1.3:1拉伸比,4000倍放大;图2(e)为根据某些实施方案,在从具有PA6/Albafil®CaCO375/25的共混物组合物的挤出的单丝进行矿物致孔剂提取后的SEM图,2:1拉伸比,1000倍放大;图2(f)为根据某些实施方案,在从具有PA6/Albafil®CaCO375/25的共混物组合物的挤出的单丝进行矿物致孔剂提取后的SEM图,2:1拉伸比,4000倍放大;图3(a)为根据某些实施方案,在从具有尼龙/PLA65/35的共混物组合物的挤出的混配的细丝(混配挤出机)进行聚合的致孔剂提取后的SEM图,2000倍放大;图3(b)为根据某些实施方案,在从具有尼龙/PLA60/40的共混物组合物的挤出的混配的细丝(混配挤出机)进行聚合的致孔剂提取后的SEM图,80倍放大;图3(c)为根据某些实施方案,在从具有尼龙/PLA55/45的共混物组合物的挤出的混配的细丝(混配挤出机)进行聚合的致孔剂提取后的SEM图,75倍放大;图3(d)为根据某些实施方案,在从具有尼龙/PLA50/50的共混物组合物的挤出的混配的细丝(混配挤出机)进行聚合的致孔剂提取后的SEM图,65倍放大;图4(a)为根据某些实施方案,在具有芯共混物组合物尼龙/PLA65/35的芯/皮纤维的聚合的致孔剂和PLA皮提取后的SEM图,2000倍放大;图4(b)为根据某些实施方案,在具有芯共混物组合物尼龙/PLA60/40的芯/皮纤维的聚合的致孔剂和PLA皮提取后的SEM图,400倍放大;图4(c)为根据某些实施方案,在具有芯共混物组合物尼龙/PLA55/45的芯/皮纤维的聚合的致孔剂和PLA皮提取后的SEM图,2000倍放大;图4(d)为根据某些实施方案,在具有芯共混物组合物尼龙/PLA50/50的芯/皮纤维的聚合的致孔剂和PLA皮提取后的SEM图,400倍放大;图5(a)为根据某些实施方案,表面积增强的(SAE)纤维的冷冻SEM横截面图;图5(b)为根据某些实施方案,表面积增强的(SAE)尼龙/PLA60/40纤维的冷冻SEM横截面图;图6为根据某些实施方案,对于所选的纤维,KrBET表面积测量的图;图7(a)为根据某些实施方案,在具有芯共混物组合物PA6/PLA70/30的15翼纤维的聚合的致孔剂和PLA皮提取后的冷冻SEM横截面图,熔体泵送比率S:C2:1,10000倍放大;图7(b)为根据某些实施方案,在具有芯共混物组合物PA6/PLA70/30的15翼纤维的聚合的致孔剂和PLA皮提取后的冷冻SEM横截面图,熔体泵送比率S:C2:1,20000倍放大;图8(a)为根据某些实施方案,在具有芯共混物组合物PA6/PLA60/40的芯/皮纤维的聚合的致孔剂和PLA皮提取后的冷冻SEM横截面图,5000倍放大;图8(b)为根据某些实施方案,在具有芯共混物组合物PA6/PLA60/40的15翼纤维的聚合的致孔剂和PLA皮提取后的冷冻SEM横截面图,熔体泵送比率S:C2:1,5000倍放大;图8(c)为根据某些实施方案,在具有芯共混物组合物PA6/PLA60/40的15翼纤维的聚合的致孔剂和PLA皮提取后的冷冻SEM横截面图,熔体泵送比率S:C1:1,5000倍放大;图9为根据某些实施方案,对于SP表面修饰的SAE纤维(左)相对类似修饰的SP15翼纤维(右)的IgGSBC的图;图10为根据某些实施方案,对于SP表面修饰的SAE纤维(左)相对类似修饰的SP15翼纤维(右)的溶菌酶SBC的图;图11为根据某些实施方案,对于SP表面修饰的SAE纤维(左)相对类似修饰的SP15翼纤维(右)的亚甲蓝SBC的图;图12(a)为根据某些实施方案,具有芯共混物组合物尼龙/PLA60/40的SAE纤维的SEM图,400倍放大;图12(b)为根据某些实施方案,具有芯共混物组合物尼龙/PLA60/40的SAE纤维的冷冻SEM横截面图,5000倍放大;图12(c)为根据某些实施方案,具有“岛”组合物尼龙PA6100/0、“海洋”组合物尼龙/PLA55/45的CIST纤维的SEM图,I/S比率1/1,1000倍放大;图12(d)为根据某些实施方案,具有“岛”组合物尼龙PA6100/0、“海洋”组合物尼龙/PLA55/45的CIST纤维的冷冻SEM横截面图,I/S比率1/1,5000倍放大;图13(a)为根据某些实施方案,分形纤维1的光学显微照片;图13(b)为根据某些实施方案,分形纤维2的光学显微照片;图13(c)为根据某些实施方案,雪片纤维的光学显微照片;图13(d)为根据某些实施方案,分形纤维2的SEM显微照片;和图13(e)为根据某些实施方案,雪片纤维的SEM显微照片。详细描述本文公开的实施方案包括适用于结合/洗脱纯化蛋白质的高表面积纤维。纤维为多孔的并且可通过提取在其熔融挤出过程中制造期间嵌入纤维中的可溶解的矿物或聚合的致孔剂而制备。可溶解的矿物致孔剂可包括沉淀的碳酸钙、硅胶或任何其它可溶解的固体矿物微粒。可溶解的聚合的致孔剂的一个实例为聚(乳酸),PLA。例如,该聚合物将溶解于氢氧化钠水溶液中。以10-90重量%的负载范围,优选35-60重量%的负载范围,可将可溶解的聚合的致孔剂掺入到纤维中。小于约25重量%的可溶解的聚合的致孔剂负载仅提供纤维表面积的最小增强,并且在致孔剂提取后,这些纤维还缺乏期望的多孔或原纤化表面特征。高于约65重量%的可溶解的聚合的致孔剂负载在致孔剂提取后可损害纤维的结构完整性。以5-40重量%的负载范围,优选15-25重量%的负载范围,可将可溶解的矿物致孔剂掺入到纤维中。小于约15重量%的可溶解的矿物致孔剂负载仅提供纤维表面积的最小增强,并且在致孔剂提取后,这些纤维还缺乏期望的多孔表面特征。在挤出期间或在致孔剂提取后,高于约30重量%的可溶解的矿物致孔剂负载可损害纤维的结构完整性。经由待引入到挤出机中的不同材料的预加工干重,或通过比较在致孔剂除去前后的纤维重量,可测量负载。用于纤维的合适的材料包括尼龙PA6,尽管可使用任何其它熔体可加工的热塑性聚合物,诸如聚酰胺、聚烯烃、聚氯乙烯、聚苯乙烯、聚甲基丙烯酸甲酯、聚乳酸、共聚物、聚丙烯、聚酯、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚乙烯、或热塑性聚氨酯、聚醚聚氨酯、聚乙烯醇、聚酰亚胺、聚碳酸酯、聚醚醚酮、聚苯乙烯、聚砜、聚对苯二甲酸丙二醇酯、共聚酯或液晶聚合物。这些热塑性物质可作为粒料或粉末得到,并且借助商业熔融混配和纤维熔融挤出加工设备,可随后将这些材料加工成为产品纤维。如分别在图1a和1b中说明的,比起普通圆形微纤维或翼形纤维,这些纤维呈现高得多的表面积。在某些实施方案中,可将纤维表面修饰,以安装适当的侧基阳离子交换配体官能团,用于结合/洗脱或流通纯化蛋白质、单克隆抗体或关注的其它生物分子。可在纤维表面上展开的合适的配体包括用于阳离子交换层析法应用的磺丙基,用于阴离子交换层析法应用的四烷基卤化铵、伯胺和仲胺,以及用于反相层析法和疏水相互作用层析法应用的正烷基链、苯基、苄基或其它芳族基团。通过高铈氧化还原接枝聚合、ATRP、RAFT或通过电子束、UV或γ辐射源引发的自由基聚合,可将配体安装在纤维表面上。在某些实施方案中,将合适的热塑性聚合物与一种或多种合适的致孔剂添加剂共混,诸如使用混配挤出机。聚合物和/或致孔剂可预先干燥并且干混。可随后将共混物引入到挤出机中,从挤出机中,可在水浴中将其从单股模具挤出,随后造粒。或者,可将基础聚合物和致孔剂粒料或粉末干混,并且直接进料至纤维或细丝纺丝机,无需预混配。随后可使用适当装备的纤维纺丝机将粒料熔融纺丝成双组分细丝。共混的基础聚合物/致孔剂材料形成芯,而致孔剂聚合物形成外部皮。在纤维纺丝、拉伸和卷绕后,可使用合适的提取剂(诸如1M盐酸溶液或1.5N氢氧化钠溶液),从双组分细丝提取致孔剂,取决于所用的致孔剂的性质。在图1c至1f中,显示根据某些实施方案的高表面积纤维的若干实例。图1c为使用可溶解的矿物致孔剂方法制备的多孔微纤维的表面SEM图。由尼龙和25重量%沉淀的碳酸钙(Albafil®PCC)的熔融混配的共混物制备该挤出的单丝。在纤维纺丝、拉伸和使用盐酸提取碳酸钙致孔剂后,在纤维的表面上可观察到许多孔。KrBET表面积测量指示,相对于大约相同纤维直径的普通的非多孔尼龙纤维,对于该材料,BET表面积约300%增加。图1d显示使用可溶解的聚合的致孔剂方法制备的多孔15翼纤维的冷冻SEM横截面图。该挤出的双组分纤维由尼龙和30重量%PLA的熔融混配的共混物制备,其组成翼形纤维芯和在熔融纺丝(未显示)期间围绕纤维芯并且稳定翼形突起的可溶解的PLA皮。在纤维纺丝、拉伸和从纤维皮和从纤维芯内使用氢氧化钠溶液提取PLA聚合的致孔剂后,贯穿翼形纤维的横截面,可观察到许多孔。图1e显示使用可溶解的聚合的致孔剂方法制备的多孔芯/皮纤维的冷冻SEM横截面图。该纤维结构称为表面积增强的(SAE)类型纤维。该挤出的双组分纤维由尼龙和40重量%PLA的熔融混配的共混物制备,其组成纤维芯和在熔融纺丝(未显示)期间围绕纤维芯并且稳定材料的可溶解的PLA皮。在纤维纺丝、拉伸和从纤维皮和从纤维芯内使用氢氧化钠溶液提取PLA聚合的致孔剂后,贯穿SAE纤维的横截面,观察到松散对准的尼龙纳米纤丝的成束的排列。该表面积增强的结构大大提高纤维表面积,和KrBET表面积测量指示通过该方法可实现高达10.6m2/g的值。与此相反,非多孔的15翼纤维具有仅1.4m2/g的适度的表面积。图1f显示使用可溶解的聚合的致孔剂方法制备的另一种类型的多孔双组分纤维的冷冻SEM横截面图。该纤维结构称为“在海洋中连接的岛”(CIST)类型纤维。该挤出的双组分纤维由尼龙和45重量%PLA的熔融混配的共混物制备,其组成纤维“海洋”域和36个连续尼龙“岛”的阵列。在纤维纺丝、拉伸和使用氢氧化钠溶液从纤维“海洋”域提取PLA聚合的致孔剂后,贯穿CIST纤维的横截面,观察到松散对准的尼龙纳米纤丝和较大微米尺寸尼龙岛的排列。该“在海洋中连接的岛”结构大大提高纤维表面积,并且N2BET表面积测量指示,通过该方法可实现高达7m2/g的值。在某些实施方案中,本文公开的多孔纤维可具有规则或不规则形状的横截面。示例性形状包括圆形、椭圆形和多边形以及翼形形状,其中中心主体具有多个由此延伸的径向突起。在某些实施方案中,纤维横截面通常为翼形形状,具有包含纵轴的中间区域,其沿着纤维的中心向下行进并且具有多个从中间区域向外延伸的突起。在某些实施方案中,多个突起通常从中间区域径向延伸。由于该构造,多个通道被突起限定。突起之间的合适的通道宽度在约200-约1000纳米范围。合适的纤维公开于美国专利公布号2008/0105612,其公开的内容通过引用结合到本文中。在某些实施方案中,纤维为具有至少三个从纵轴延伸的分支突起的分形纤维,如在图13(a)中显示的。在某些实施方案中,纤维为具有至少三个从纵轴延伸的分支突起的分形纤维,每一个分支突起具有从中延伸的亚突起,如在图13(b)和(d)中显示的。在某些实施方案中,纤维为雪片纤维,具有至少六个从纵轴延伸的突起,每一个突起具有至少四个从中延伸的亚突起,如在图13(c)和(e)中显示的。在某些实施方案中,适用于结合/洗脱纯化蛋白质的高表面积纤维为具有不同形状的横截面的固体纤维。具有离子交换配体的这些成形的纤维呈现足够的表面积和可接受的流动性质,以用于层析法分离。通过BET气体吸附,这些成形的纤维具有0.5-5平方米/克的表面积。纤维作为双组分纤维生产。除去皮材料,暴露高表面积芯。该芯用离子交换配体修饰并且用于蛋白质分离。可使用的横截面的实例在图13中显示。高表面积多孔纤维的表面官能化可例如如下进行:通过在纤维表面上沉积环氧官能的聚合物涂层,接着加热以使聚合物涂层与纤维表面共价连接,和随后进行环氧开环过程,以在纤维表面上安装磺酸官能团。在其它实施方案中,可进行使用表面接枝的离子交换配体来修饰SAE类型纤维,用于结合/洗脱阳离子交换层析法应用。可进行使用交联的HPA/MBAm95/5聚合物涂层来活化SAE纤维表面,以在纤维表面上提供高度反应性羟基官能的涂层,接着诸如使用2-丙烯基酰氨基-2-甲基-1-丙磺酸钠盐进行高铈离子氧化还原聚合,以提供聚合物接枝的纤维基材。约0.1-0.4g/ml(优选约0.35g/ml)的合适的柱填充密度将提供足够的流动均匀性,用于在层析评价中可接受的性能。在某些实施方案中,不同于基于珠的介质,介质(官能化的填充的纤维)可以预填充的形式递送至使用者。纤维可通过热或化学手段熔融,以形成可封装在压力容器中的半刚性结构。通过这样的结构,介质和伴随的设备可准备好使用。基于层析珠的介质通常作为疏松的材料(湿的)递送,其中需要使用者装填压力容器(柱)和通过各种手段来产生没有空隙或通道的良好填充床。通常需要随后的测试以确保填充的均匀性。与此相反,根据某些实施方案,由于产品已准备好用于工作,使用者不需要填充。本文公开的实施方案的表面官能化的多孔纤维介质以填充床形式证明高渗透性。取决于填充密度,床渗透性可在2500mDarcy至小于100mDarcy范围。填充的纤维床在高线性速度下不压缩。本文公开的实施方案的表面积增强的纤维介质可设置为在合适的外壳内的填充床形式,诸如层析柱或其它设备。可如下制备表面积增强的短纤维的填充的纤维床:通过在层析柱中装填短纤维的稀释的含水悬浮液,随后轴向压缩层析柱的顶部溶剂分配总管至1-10cm的目标床深度。轴向压缩定义为降低位于层析柱或其它合适的外壳内的短纤维填充床深度,以提高短纤维填充的填充密度至0.1-0.4g/mL的目标值。通过机械置换流动分配总管来完成该压缩,以提供较小的柱或设备体积和相应地提高层析介质填充密度。在此上下文中,正被压缩的轴为其中填充短纤维的柱的垂直轴。由于短纤维可被压缩,当实施这样的轴向压缩时,短纤维的填充密度相应地提高。与此相反,径向压缩定义为降低在层析柱或其它合适的外壳内的短纤维填充的内径,以提高短纤维填充的填充密度至0.1-0.4g/mL的目标值。径向压缩操作不改变纤维介质填充床深度。实施例实施例1.使用矿物或聚合的致孔剂熔融混配。在该实验中,将尼龙与多种矿物或聚合的致孔剂添加剂(诸如碳酸钙、二氧化硅或聚(乳酸)聚合物(PLA))共混。还制备尼龙、矿物和PLA聚合物致孔剂的三元混合物。这些共混物随后用于纤维挤出实验。使用混配挤出机制备尼龙6和矿物填料的若干共混物。还制备含有尼龙、PLA和矿物致孔剂的三元混合物的另外的共混物。检查四种不同类型的矿物填料:得自SMI的AlbafilA-O-255-12、得自SMI的VicalityHeavy、得自SMI的Multifex-MM™和得自W.R.Grace的Syloid244FP。将预先干燥的材料称重并干混。将干混物放在进料运输机上,调节进料运输机,以充分进料microtruder。在水浴中,将材料从单股模具挤出,随后造粒。用于该研究的某些尼龙/致孔剂制剂在以下表2中汇总。表1.与各种矿物和聚合的致孔剂混配的尼龙制剂的汇总材料共混比率致孔剂类型PA6:Albafil75:25矿物(CaCO3)PA6:Vicality75:25矿物(CaCO3)PA6:Syloid75:25矿物(SiO2)PA6:Multifex-MM™75:25矿物(CaCO3)PA6:PLA75:25聚合物(PLA)PA6:PLA:Albafil65:25:10矿物(CaCO3)+聚合物(PLA)PA6:PLA:Vicality65:25:10矿物(CaCO3)+聚合物(PLA)PA6:PLA:Syloid73.1:24.4:2.5矿物(SiO2)+聚合物(PLA)实施例2.矿物致孔剂负载的单丝的熔融挤出。在该实验中,提供用于将共混的尼龙/矿物致孔剂粒料熔融纺丝成约20微米直径的单丝纤维的过程的一般性描述。使用纤维纺丝机,将共混的尼龙/矿物致孔剂粒料熔融纺丝成单丝。纤维纺丝机为得自HillsInc的LBS系统(Melbourne,FL)。将挤出的单丝纤维样品拉伸至约20微米的直径。在纤维纺丝、拉伸和卷绕后,根据以下描述的程序,随后从单丝提取矿物致孔剂。实施例3.从挤出的单丝的矿物致孔剂提取。在该实验中,描述了使用1M盐酸溶液,从挤出的单丝纤维提取矿物致孔剂的过程。随后将纤维中和,洗涤,且通过扫描电子显微镜法(SEM)检查纤维的表面。还进行KrBET表面积测量。在100mL带盖的玻璃罐中加入1.0g挤出的单丝(约20µm直径)和50mL1.0MHCl(50mmol)。将悬浮液在30℃下搅拌过夜。通过真空过滤分离纤维固体,和用0.5MTris-HCl(1×100mL)、去离子水(1×100mL)和乙醇(1×100mL)洗涤。将材料放置在烘箱中,以在40℃下干燥18小时。矿物致孔剂提取实验的结果在以下表3中显示。在提取矿物致孔剂后,进行纤维表面形态学的SEM检查,并且这些图在图2中显示。在致孔剂提取后,在纤维表面上,含有Albafil®的单丝得到大的微米尺寸的孔或腔。由于Multifex-MM™-致孔剂较小的粒径(<0.2微米),在致孔剂提取后,在纤维表面上,观察到小得多的孔。在类似处理的尼龙纤维对照样品上,明显的是没有这样的孔。相对于非多孔尼龙纤维对照样品,对于使用Albafil®致孔剂制备的材料,KrBET表面积测量揭示BET表面积显著的增加(~300%)。表2.碳酸钙矿物致孔剂提取实施例4.使用聚合的致孔剂熔融混配。在该实验中,将尼龙与各种量的聚合的致孔剂聚(乳酸)聚合物共混。这些共混物随后用于纤维挤出实验。使用混配挤出机产生多种尼龙/PLA共混物样品。制备多种PLA和尼龙6共混物,并且在以下表4中显示。将适量的干燥的粒料称重,并干混。随后将干混的混合物加入到在混配挤出机上的进料输送带。调节进料带,以充分进料混配挤出机。在水浴中,将材料从单股模具挤出,随后造粒。表3.使用PLA作为聚合的致孔剂混配的尼龙制剂的汇总。材料共混比率PA6:PLA80:20PA6:PLA75:25PA6:PLA70:30PA6:PLA65:35PA6:PLA60:40PA6:PLA55:45PA6:PLA50:50实施例5.由挤出的细丝的PLA致孔剂提取。在该实验中,描述了使用1.5N氢氧化钠溶液,从来自熔融混配机的挤出的细丝提取PLA致孔剂的过程。随后将纤维中和,洗涤,实施比重测定,通过扫描电子显微镜法(SEM)检查纤维的表面。在250mL带盖的玻璃罐中加入2.0g挤出的细丝(约2.0mm直径)和0.2L1.5NNaOH(0.75mol)。将悬浮液在室温下搅拌过夜。通过真空过滤分离纤维固体,且用去离子水(3×250mL)和乙醇(1×250mL)洗涤。将材料放置在烘箱中,以在60℃下干燥3小时。由挤出的2mm细丝的PLA聚合的致孔剂提取实验的结果在以下表5中显示。由这些数据可见,比起低PLA负载样品(35重量%PLA),对于高PLA负载样品(50重量%PLA),PLA致孔剂更容易提取。实际和预期产率之间的大的差异是由于受限制地进入这些大的2mm直径细丝的内部。在提取PLA聚合的致孔剂后,进行细丝表面形态学的SEM检查,且这些图在图3中显示。这些数据显示在从细丝的PLA致孔剂提取后,原纤化的表面形态学的外观。对于等于或大于40重量%的PLA负载,这些表面特征相当显著。预期原纤化的表面形态学(诸如这些)大大提高纤维基材的表面积。表4.由挤出的细丝的PLA致孔剂提取实施例6.聚合的致孔剂负载的纤维的熔融挤出。在该实验中,提供了将混配的尼龙/PLA聚合物致孔剂粒料熔融纺丝成约20微米直径的芯/皮或15翼双组分纤维的过程的一般性描述。使用双组分纤维纺丝机,将共混的尼龙/PLA粒料熔融纺丝成纤维。双组分纤维纺丝机为来自HillsInc.的LBS系统(Melbourne,FL)。挤出的纤维样品为芯/皮和15翼纤维,具有PLA的皮和在芯中共混的粒料(或反之亦然)。样品在以下表6中汇总。表5.含有位于纤维芯内的聚合的致孔剂或作为致孔剂负载的外部成皮的挤出的纤维的汇总表样品编号纤维类型芯共混物组合物(尼龙:PLA)皮组成芯/皮比率7895-122芯2芯/皮80:20PLA50:507895-122芯4芯/皮75:25PLA50:507895-128芯4芯/皮65:35PLA50:507895-128芯6芯/皮60:40PLA50:507895-128芯7芯/皮55:45PLA50:507895-128芯8芯/皮50:50PLA50:507895-123芯615翼70:30PLA1:27895-159芯2芯/皮100:0尼龙:PLA60:4050:507895-159芯4芯/皮100:0尼龙:PLA60:402.6:17895-159芯1芯/皮100:0尼龙:PLA60:408:17895-159芯5芯/皮100:0尼龙:PLA60:401:2实施例7.使用手动共混的样品熔融挤出。在该实验中,提供了将尼龙和PLA聚合物致孔剂粒料的手动共混的混合物熔融纺丝成约20微米直径的芯/皮双组分纤维的过程的一般性描述。使用安装有芯/皮纺丝组件的来自HillsInc.的实验室规模双组分挤出机(Melbourne,FL),通过熔融纺丝产生多种熔融挤出的纤维样品。模具的皮侧进料有聚乳酸(PLA)粒料。对于模具的芯侧,在进料至挤出机中之前,通过在袋子中简单搅拌,将聚合物粒料以各种比率混合。将纤维拉伸,且在芯上缠绕,用于后来的加工和分析。样品在以下表7中汇总。表6.位于纤维芯内的含有手动共混的聚合的致孔剂/尼龙混合物的挤出的纤维的汇总表实施例8.用于PLA提取的通用程序。在该实验中,描述了使用1.5N氢氧化钠溶液由挤出的双组分芯/皮纤维提取PLA致孔剂的过程。随后将纤维中和,洗涤,实施比重测定,且通过扫描电子显微镜法(SEM)检查纤维的表面。还进行KrBET表面积测量。在1升带盖的Pyrex瓶中加入5.0g短切纤维(1.0mm长)和0.5L1.5NNaOH(0.75mol)。将悬浮液在室温下搅拌过夜。通过真空过滤隔离纤维固体,且用去离子水(3×250mL)和乙醇(1×250mL)洗涤。将材料放置在烘箱中,以在60℃下干燥18小时。由挤出的双组分纤维(20微米直径)的PLA聚合的致孔剂提取实验的结果在以下表8中显示。由这些数据,我们发现对于所有纤维样品,从纤维皮和从纤维芯内部二者,提取PLA致孔剂。实际和预期产率之间小的差异证明从双组分纤维内完全提取PLA致孔剂。在提取PLA聚合的致孔剂后进行纤维表面形态学的SEM检查,且这些图在图4中显示。由这些数据,我们发现在从PLA负载大于35重量%的纤维提取PLA致孔剂后,原纤化的表面形态学的外观。这些纤维看起来由高度缠结的尼龙纳米纤丝的束构成。对于大于约50重量%的PLA致孔剂负载,存在纤维结构的明显解开,以得到单个尼龙纳米纤维。40重量%PLA样品的冷冻SEM横截面图在图5中显示。由这些横截面图,我们发现该纤维看起来由数百松散轴向对准的尼龙纳米纤丝构成,并且在纤维横截面内存在显著的多孔性。预期原纤化的表面形态学(诸如这些)大大提高纤维基材的表面积。在图6中,显示对于由大于25重量%的PLA聚合的致孔剂负载构成的提取的尼龙纤维样品,测量高KrBET表面积。表7.PLA致孔剂提取和皮去除实施例9.具有多孔芯的翼形纤维的熔融挤出。在该实验中,提供了将混配的尼龙/PLA聚合物致孔剂粒料熔融纺丝成约15微米直径的15翼双组分纤维的过程一般性描述。随后从纤维皮以及从纤维芯内提取PLA。结果是,这些15翼纤维呈现多孔芯结构。使用双组分纤维纺丝机,将共混的尼龙/PLA粒料熔融纺丝成纤维。双组分纤维纺丝机为来自HillsInc.的LBS系统(Melbourne,FL)。挤出的纤维样品为15翼纤维,并且使用PLA的皮和共混的粒料作为纤维芯制备。在从纤维皮和从纤维芯内提取PLA后,提供了具有多孔结构的15翼纤维。用于提取PLA聚合的致孔剂的通用程序在以上实施例中描述。使用尼龙/PLA70/30和60/40共混物作为纤维芯组合物制备的各种15翼纤维样品在图7和图8中说明。这些图显示在翼形纤维横截面内延伸的圆柱形孔或腔的外观。预期这样的特征进一步提高翼形纤维的表面积。实施例10.用于表面修饰SAE纤维的通用程序。在该实验中,描述了用于使用侧基强阳离子交换官能团表面修饰SAE纤维的通用程序。该程序涉及在纤维表面上沉积环氧官能的聚合物涂层,加热步骤以使聚合物涂层与纤维表面共价连接,和随后进行环氧开环过程,以在纤维表面上安装磺酸官能团。在30mL玻璃小瓶中制备25g聚(甲基丙烯酸缩水甘油基酯)在甲乙酮(MEK)中的1重量%溶液。在单独的30mL玻璃小瓶中,加入0.2g纤维和12.5g1%聚(甲基丙烯酸缩水甘油基酯)聚合物溶液。将悬浮液在室温下搅拌过夜。随后通过真空过滤来分离纤维固体,并且放置在100℃的烘箱中达30分钟。从烘箱取出纤维固体,并且在室温下再悬浮于40mLMEK中达1小时。通过真空过滤分离纤维固体,随后悬浮于15mL1M亚硫酸钠/0.4M四正丁基硫酸氢铵溶液中。悬浮液用氮气鼓泡5分钟,将小瓶密封,并加热至80℃过夜。将悬浮液冷却至室温。通过真空过滤分离纤维固体,且用去离子水(5×30mL)和乙醇(1×30mL)洗涤。将纤维在60℃下干燥2小时。表面积增强的(SAE)芯/皮纤维以及非多孔15翼对照样品的表面修饰的结果在以下表9中显示。使用IgG、溶菌酶和亚甲蓝分别作为大蛋白质、小蛋白质和小分子探针,对于这两种磺酸官能化的纤维,还进行静态结合能力测量。对于所有这些静态结合能力试验,采用标准阳离子交换结合条件,且结果在以下图9、10、11和表10中汇总。由这些数据,显示相对于15翼纤维,对于SAE纤维,静态结合能力提高,以及随着分子尺寸降低,SAE纤维的结合能力优势提高。表8.SAE尼龙和15翼纤维的表面修饰、BET表面积和回收率数据。样品编号纤维量(g)纤维描述KrBET表面积得到%产率7895-179A0.20gSAE10.57m2/g0.19g95%7895-179B0.20g15翼1.43m2/g0.17g85%表9.对于SP修饰的SAE和15翼纤维,静态结合能力数据的汇总。IgG溶菌酶亚甲蓝分子量160KDa14.3KDa374Da15W纤维SBC(mg/g)25218SAE纤维SBC(mg/g)586238SAE纤维(SBCgain)2.3x2.9x4.8x估计可接近的表面积13.2m2/g4.2m2/g6.7m2/g%的BET表面积31%40%64%1基于15W纤维,1.4m2/g的100%可接近的表面积。实施例11.SAE纤维(7895-136)的表面修饰。在该实验中,描述了使用表面接枝的离子交换配体来修饰SAE类型纤维的程序,用于结合/洗脱阳离子交换层析法应用。该过程的第一步骤涉及使用交联的HPA/MBAm95/5聚合物涂层活化SAE纤维表面。该步骤在纤维表面上提供高度反应性羟基-官能的涂层,纤维表面良好适用于随后的聚合物接枝过程。在第二步骤中,HPA/MBAm-修饰的纤维经历与2-丙烯基酰氨基-2-甲基-1-丙磺酸钠盐的高铈离子氧化还原聚合,以提供聚合物接枝的纤维基材。接枝的聚合物提供侧基磺酸官能团,用于阳离子交换层析法应用。使用HPA/MBAm95/5的SAE尼龙纤维表面修饰。在500mLPyrex瓶中加入羟丙基丙烯酸酯(HPA,4.9g,38mmol)、N,N′-亚甲基双(丙烯酰胺)(MBAm,0.28g,2mmol)和水(253mL)。将6.0g表面积增强的(SAE)尼龙纤维加入到混合物中。加入过硫酸铵(0.63g,3mmol)。将湿固体加热至80℃达4小时。在冷却至室温后,将固体转移至布氏漏斗,且用热水(4×200mL)和乙醇(1×200mL)洗涤。让材料真空干燥20分钟。将材料转移至烘箱,且在60℃下干燥18小时。得到6.46g白色纤维。HPA/MBAm修饰的尼龙纤维的接枝聚合。在3×125mL玻璃罐中加入2-丙烯基酰氨基-2-甲基-1-丙磺酸钠盐(AMPS-Na)、水、HPA/MBAm修饰的SAE尼龙纤维(参见以上)和1MHNO3溶液(其量在下表中描述)。将硝酸铈(IV)铵(CAN)在1MHNO3中的0.4M溶液加入到每一个瓶子中。将反应瓶加盖,用氮气鼓泡,且将混合物加热至35℃达18小时。在冷却至室温后,固体用0.2M抗坏血酸在0.5M硫酸中的溶液(3×80mL)、去离子水(3×80mL)、0.5M氢氧化钠溶液(3×80mL)、去离子水(3×80mL)和乙醇(1×80mL)洗涤。将材料放置在烘箱中,以在60℃下干燥18小时。得到白色纤维固体的样品(参见表的回收率和重量添加数据)。表10.铈氧化还原接枝聚合组合物和回收率数据实施例12.静态结合能力测量。在该实验中,呈现以阳离子交换模式的SP表面修饰的SAE纤维的IgG静态结合能力。对于SP修饰的SAE尼龙纤维,IgG静态结合能力测量的结果在以下表12中提供。由这些数据,对于SP表面修饰的SAE纤维,显示实质的IgG静态结合能力,且这些IgGSBC值与商业基于珠的阳离子交换层析树脂可比。表11.对于SP修饰的SAE尼龙纤维,IgG静态结合能力(SBC)数据。挑战:2g/L多克隆人IgG(SeraCareLifeSciences,Milford,MA),在50mM乙酸钠(pH5)中。样品纤维量(mg)结合的IgG(mg)SBC(mg/g)SBC(mg/mL)17895-136A-1568.0143477895-136A-2324.6143477895-136B-1448.1185617895-136B-25910177587895-136C-16212197657895-136C-2694.261201基于0.33g/mL纤维填充密度实施例13.动态结合能力测量。在该实验中,描述了在层析柱中SP表面修饰的SAE纤维的填充和已填充的纤维床的渗透性。还呈现以阳离子交换模式的SP表面修饰的SAE纤维的IgG动态结合能力。在以下表13中提供用于实施例7895-136B的SP-官能化的SAE纤维介质的IgG动态结合能力测量的结果。在11mm内径Vantage柱中填充1.0g介质,且压缩至3.0cm的床深度(2.85mL柱体积,0.35g/mL纤维填充密度)。使用50mM乙酸盐缓冲剂(pH5),测得在0.35g/mL下,填充的纤维渗透性为200mDa。经200cm/hr-60cm/hr范围的线性速度,进行动态结合能力测量。这些速度对应于54秒-3分钟的停留时间。实施例7895-136B的纤维介质证明在50mg/mL范围的IgG动态结合能力。表12.在不同的线性速度(RT=停留时间)下,以1、5、10和50%贯穿,对于SP官能化的SAE纤维阳离子交换介质的IgGDBC值。挑战:2.0g/L多克隆人IgG(SeraCareLifeSciences,Milford,MA),在50mM乙酸盐(pH5)中。实施例14.“在海洋中连接的岛”(CIST)纤维的熔融挤出。在该实验中,描述了用于将尼龙和PLA聚合物致孔剂粒料的手动共混的混合物熔融纺丝成约20微米直径的“在海洋中连接的岛”(CIST)类型纤维的过程的一般性描述。随后将纤维中和,洗涤,实施比重测定,且通过扫描电子显微镜法(SEM)检查纤维的表面。还进行N2BET表面积测量。使用安装有“在海洋中的岛”纺丝组件(36岛构造)的来自HillsInc.的实验室规模双组分挤出机(Melbourne,FL),通过熔融纺丝,产生多种熔融挤出的纤维样品。模具的“岛”侧进料尼龙6粒料。对于模具的“海洋”侧,在进料至挤出机中之前,通过在袋子中简单搅拌,将聚合物粒料以各种比率混合。对于该实施例,共混物组分为PLA和尼龙6。将纤维拉伸,且在芯上缠绕,用于后续加工和分析。样品在下表中汇总。在从挤出的双组分纤维的“海洋”域内提取PLA聚合的致孔剂后,提供了具有多孔结构的CIST纤维。CIST类型纤维的若干实例在以下表15中汇总。用于提取PLA聚合的致孔剂的通用程序在以上实施例中描述。制备了使用多种尼龙/PLA致孔剂共混物作为纤维“海洋”域组合物制备的各种CIST纤维样品。CIST纤维和SAE类型纤维的表面和冷冻SEM横截面图在图12中提供。由横截面图可见,大通道或裂缝贯穿CIST纤维的内部延伸,并且比起SAE类型纤维,这些特征可使得内表面显著更加可接近。预期这样的特征改进大蛋白质和生物分子与这样的纳米原纤化的纤维支撑体的内部表面积的接近。表13.含有36尼龙PA6“岛”和手动共混的聚合的致孔剂/尼龙混合物作为纤维“海洋”组分的挤出的“在海洋中连接的岛”(CIST)纤维的汇总表。样品编号纤维类型岛组成海洋共混物组成(尼龙:PLA)岛/海洋比率7895-188DCISTPA6PA6:PLA40:601:17895-188ACISTPA6PA6:PLA60:403:27895-189DCISTPA6PA6:PLA40:601:17895-189ACISTPA6PA6:PLA60:401:17993-8ACISTPA6PA6:PLA55:452.1:17993-8BCISTPA6PA6:PLA55:451:17993-8CCISTPA6PA6:PLA55:450.6:17993-8DCISTPA6PA6:PLA60:402.1:17993-8ECISTPA6PA6:PLA60:401:17993-8FCISTPA6PA6:PLA60:400.6:1CISTPA6PA6:PLA65:352.1:1CISTPA6PA6:PLA65:351:1表14.对于“在海洋中连接的岛”(CIST)类型纤维,PLA致孔剂提取和N2BET数据。实施例15.SAE、CIST、15翼和圆形尼龙纤维的表面修饰。在该实验中,描述了使用表面接枝的离子交换配体来修饰SAE、CIST和15翼类型纤维的程序,用于结合/洗脱阳离子交换层析法应用。在该过程中,使用高铈离子氧化还原聚合与3-磺基丙基甲基丙烯酸钾盐,在单一步骤中修饰纤维表面,以提供聚合物接枝的纤维基材。接枝的聚合物提供侧基磺酸官能团,用于阳离子交换层析法应用。在125mL瓶中加入3-磺基丙基甲基丙烯酸钾盐(3-SPMA)、水、CIST尼龙纤维和1MHNO3溶液(其量在以下表中描述)。将硝酸铈(IV)铵(CAN)在1MHNO3中的0.4M溶液加入到瓶中。将反应瓶加盖,且将混合物加热至35℃达5小时。在冷却至室温后,来自瓶的纤维固体用0.2M抗坏血酸在0.5M硫酸中的溶液(3×50mL)、去离子水(3×50mL)、0.5M氢氧化钠溶液(3×50mL)、去离子水(3×50mL)和乙醇(1×50mL)洗涤。将材料放置在烘箱中,以在60℃下干燥18小时。得到白色纤维固体的样品(参见表的回收率和重量添加数据)。表15.对于表面积增强的(SAE)、“在海洋中连接的岛”(CIST)、非多孔15翼和圆形对照纤维(15µm直径),铈氧化还原接枝聚合组合物和回收率数据。实施例16.静态结合能力测量。在该实验中,呈现以阳离子交换模式的SP表面修饰的SAE、CIST、15翼和简单的圆形纤维的IgG静态结合能力。对于SAE纤维(样品#7895-142A和7895-142B),我们发现,在接枝步骤中,将3-SPMA单体从7.5mmol提高至19mmol,得到IgG静态结合能力从47mg/g显著提高至212mg/g。在低3-SPMA单体负载条件(7.5mmol)下,CIST纤维(条目7993-2A-1,7993-2A-2)得到与SAE类型纤维可比的IgGSBC值。15翼纤维(条目7895-62D)使用38mmol3-SPMA单体修饰,且该样品得到130mg/g的IgGSBC值。与之相比,在所有评价的接枝条件下,简单的15微米圆形纤维提供非常低的IgGSBC值。这可归因于缺乏突起或内部多孔结构的圆形纤维的非常低的表面积。对于SP修饰的表面积增强的(SAE)、“在海洋中连接的岛”(CIST)、非多孔15翼和圆形对照纤维(15µm直径),IgG静态结合能力测量结果在以下表17中提供。表16.对于SP修饰的表面积增强的(SAE)、“在海洋中连接的岛”(CIST)、非多孔15翼和圆形对照纤维(15µm直径),IgG静态结合能力数据。挑战:2g/L多克隆人IgG(SeraCareLifeSciences,Milford,MA),在50mM乙酸钠(pH5)中。实施例17.成形的纤维的熔融挤出。使用双组分熔融纺丝过程制备成形的纤维。双组分纤维具有一种材料的芯和第二种聚合物的皮。这些芯和皮材料可为在本领域中研究的那些人已知的任何类型的可熔融加工的热塑性材料。一系列模具板用于将两种聚合物进料流分开和再引导成为给定数量的纤维和期望的横截面形状。在熔融纺丝后,将纤维拉伸至适当的尺寸。纤维特性在表17中汇总。表17.成形的纤维形状、BET表面积和纤维直径的汇总。纤维形状BET表面积(m2/g)纤维直径(微米)分形1没有数据14.8分形22.1115.1雪片1.2526.9实施例18.用于成形的纤维的表面修饰的通用程序。根据实施例10制备成形的纤维。参见表中IgG静态结合能力数据。表1817.对于SP修饰的成形的和15翼纤维,静态结合能力数据的汇总。纤维类型IgGSBC(mg/g)15W纤维SBC(mg/g)25分形1NA分形290雪片29.5实施例19.成形的尼龙纤维的表面修饰。根据实施例11制备成形的纤维。参见表中的回收率和重量添加数据。表19.对于分形纤维,铈氧化还原接枝聚合组合物和回收率数据。实施例20.动态结合能力测量。根据在实施例13中描述的方法填充来自以上实施例19的表面修饰的分形纤维。经200cm/hr至600cm/hr范围的线性速度,进行动态结合能力测量。这些速度对应于54秒至18秒的停留时间。实施例19的纤维介质证明IgG动态结合能力在72mg/mL范围。表20.在不同的线性速度(RT=停留时间)下,以1、5、10和50%贯穿,对于SP官能化的成形的纤维阳离子交换介质的IgGDBC值。挑战:2.0g/L多克隆人IgG(SeraCareLifeSciences,Milford,MA),在50mM乙酸盐(pH5)中。实施例21.未修饰的SAE纤维的接枝聚合。使用四烷基铵(Q型)聚合的配体官能团表面修饰SAE纤维,用于阴离子交换层析法(AEX)应用。在500mL瓶中加入甲基丙烯酸缩水甘油基酯(GMA,1.70g,12mmol)和水(232.8mL)。将5gSAE纤维加入到溶液中。将1MHNO3溶液(7.22mL,7.2mmol)加入到反应混合物中,接着加入硝酸铈(IV)铵在1MHNO3(0.602mL,0.240mmol)中的0.4M溶液。将反应混合物加热至35℃达1小时。在冷却至室温后,固体用去离子水(3×100mL)洗涤,且潮湿的材料(12.21g)立即用于以下步骤。环氧官能化的SAE纤维的Q-官能化。在250mL瓶中加入来自以上实施例的潮湿的GMA-官能化的SAE纤维和50重量%三甲基胺(水性)在甲醇中的溶液(其量在以下表21中描述)。将混合物在室温下搅拌18小时。纤维固体随后用0.2M抗坏血酸在0.5M硫酸中的溶液(3×50mL)、去离子水(3×50mL)、1M氢氧化钠溶液(3×50mL)、去离子水(3×50mL)和乙醇(1×50mL)洗涤。将材料放置在烘箱中,以在40℃下干燥12小时。得到白色纤维固体的样品。表21.用于使用三甲基胺修饰环氧官能化的SAE纤维的组合物。实施例22.未修饰的SAE纤维的接枝聚合。使用聚(羟乙基甲基丙烯酸酯)聚合物官能团表面修饰SAE纤维,用于疏水相互作用层析法(HIC)应用。在500mL瓶中加入羟乙基甲基丙烯酸酯(HEMA,1.69g,13mmol)和水(232.5mL)。将5.00gSAE纤维加入到溶液中。将1MHNO3溶液(7.21mL,7.2mmol)加入到反应混合物中,接着加入硝酸铈(IV)铵在1MHNO3(0.601mL,0.240mmol)中的0.4M溶液。将反应混合物加热至35℃达1小时。在冷却至室温后,固体用0.2M抗坏血酸在0.5M硫酸中的溶液(3×100mL)、去离子水(3×100mL)、1M氢氧化钠溶液(3×100mL)、去离子水(3×100mL)和乙醇(1×100mL)洗涤。将材料放置在烘箱中,以在40℃下干燥12小时。实施例23.使用重组体蛋白质A亲和配体rSPA的SAE纤维表面修饰。使用重组体蛋白质A亲和配体表面修饰SAE纤维,用于亲和层析法应用。在250mL瓶中加入1M碳酸氢钠(100mL)、重组体蛋白质A(rSPA#RN091139,150mg,作为47.5mg/mL水溶液)和水(90mL)。将来自以上实施例21的GMA-接枝的SAE纤维(350mg)加入到反应混合物中。将混合物在37℃下加热2.5小时。在冷却至室温后,将固体转移至布氏漏斗,且用0.1M碳酸氢钠(3×100mL)洗涤。将湿的纤维固体悬浮于100mL硫代甘油在0.2M碳酸氢钠/0.5M氯化钠溶液中的10重量%溶液中。将混合物在室温下搅拌过夜。将固体转移至布氏漏斗,且用具有0.15M氯化钠(1×75mL)、0.05M乙酸溶液(1×75mL)的0.1MTRIZMA碱的溶液洗涤。将TRIZMA碱和乙酸洗涤周期再重复两次。SAE纤维固体最后用去离子水(1×75mL)和20重量%乙醇(1×75mL)洗涤。将SAE纤维固体在20重量%乙醇溶液中储存。实施例24.环氧官能化的纤维的聚(烯丙基胺)修饰。使用聚(烯丙基胺)聚合的配体官能团表面修饰SAE纤维,用于阴离子交换层析法(AEX)应用。在30mL瓶中加入来自以上实施例21的GMA接枝的SAE纤维(0.5g)、水(10mL)、40重量%聚(烯丙基胺)盐酸溶液(1.25g40重量%溶液)和1.0M氢氧化钠(10mL)。将反应混合物加热至35℃达18小时。在冷却至室温后,固体用去离子水(3×50mL)和丙酮(1×50mL)洗涤。将潮湿的材料放置在烘箱中,以在40℃下干燥12小时。实施例25.流通宿主细胞蛋白质清除。采用流通抛光模式,评价根据实施例21制备的Q-官能化的SAE纤维介质的HCP去除活性。在14.5mm内径柱中填充0.34gQ-官能化的纤维介质,并且压缩至0.6cm的床深度(1.00mL柱体积,0.34g/mL纤维填充密度)。使含有单克隆抗体的细胞培养培养基澄清,且随后使用蛋白质A柱层析法分离,且将溶液的pH调节至pH5。随后使用TRIZMA碱,将蛋白质A洗脱的pH调节至pH8,且随后通过0.2微米膜过滤。Q-官能化的SAE纤维介质柱用缓冲溶液(25mMTris,pH8)平衡。将100毫升8.2g/L单克隆抗体蛋白质A洗脱(pH8)以1.0mL/分钟的流速经过柱。收集十个10mL馏分。使用1M氯化钠溶液在25mMTris(pH8)中作为洗脱缓冲剂,洗脱结合的HCP。还收集两个10mL洗脱馏分。通过HCP-ELISA和蛋白质AHPLC来分析十个流通馏分和两个洗脱馏分,以分别确定HCP清除水平和单克隆抗体回收率。实施例26.用于病毒的结合/洗脱纯化的SAE纤维介质能力。如以下显示进行噬菌体6的静态结合能力和洗脱回收率实验。根据与在实施例13中描述的那些类似的程序,还可以填充的柱形式实施阴离子交换模式结合/洗脱操作。在5个塑料离心管中加入实施例21的Q-官能化的SAE纤维介质。每一个SAE纤维样品用5mL25mMTris缓冲剂(pH8,具有0.18mg/mLHSA)平衡,搅拌10分钟。将管在室温下在台式顶部离心机中以4000rpm旋转10分钟,使得SAE纤维介质成粒。除去2.5mL上清液,且将2.5mL1.7×107pfu/mL6溶液在25mMTris缓冲剂(pH8,具有0.18mg/mLHSA)中加入到每个管中。将样品在室温下搅拌1小时。随后,将管在室温下在台式顶部离心机中以4000rpm旋转15分钟,使得SAE纤维介质成粒。除去2.5mL上清液,且通过成斑测定,测定这些样品的未结合的6。在离心下,管用25mMTris缓冲剂(pH8,具有0.18mg/mLHSA)的2.5mL洗涤液洗涤3次,使在每次洗涤之间的SAE纤维介质成粒,并且除去2.5mL上清液。在洗涤后,将2.5mL1.0MNaCl溶液在25mMTris缓冲剂(pH8,具有0.18mg/mLHSA)中加入到每一个管(总体积5mL,最终NaCl浓度为0.5M)中。将样品在室温下搅拌10分钟。随后,将管在室温下在台式顶部离心机中以4000rpm旋转10分钟,使得SAE纤维介质成粒。除去2.5mL上清液,且通过成斑测定,测定这些洗脱样品的洗脱的6。Q-官能化的SAE纤维介质可整合成为预填充的设备形式或层析柱,用于流通病毒清除或结合/洗脱病毒纯化应用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1