一种中药组合物的指纹图谱的测定方法与流程
本发明涉及到一种中药组合物的指纹图谱的测定方法。
背景技术:
中药指纹图谱是指某些中药材或中药制剂经适当处理后,采用一定的分析手段,得到的能够标示其化学特征的色谱图或光谱图。中药指纹图谱是一种综合的,可量化的鉴定手段,它是建立在中药化学成分系统研究的基础上,主要用于评价中药材以及中药制剂半成品质量的真实性、优良性和稳定性。“整体性”和“模糊性”为其显著特点。以指纹图谱作为中药(天然药物)提取物及其制剂的质量控制方法,已成为目前国际共识,各种符合中药(天然药物)特色的指纹图谱控制技术体系正在研究和建立。美国食品药品管理局(fda)允许草药保健品申报资料中提供色谱指纹图谱;世界卫生组织(who)在1996年草药评价指导原则中也规定,如果草药的活性成分不明确,可以提供色谱指纹图谱以证明产品质量的一致;欧共体在草药质量指南中亦称,单靠测定某种有效成分考查质量的稳定性是不够的,因为草药及其制剂是以整体为活性物质。色谱指纹图谱尤其是薄层色谱的鲜明的指纹图谱是很有用的。国外指纹图谱的应用,目的在于解决成分复杂,有效成分不明确的植物药质量检测和产品批次间质量差异的问题。
高效液相色谱(hplc)指纹图谱,具有很高的分离度,可把复杂的化学成分进行分离而形成高低不同的峰组成一张色谱图,这些色谱峰的高度和峰面积分别代表了各种不同化学成分和其含量。由此可见,中药指纹图谱比dna指纹图谱更进一步的发展在于:不但有特征的体现(各种化学成分的个数和相对位置——保留时间)可作定性鉴别使用,还体现了量的概念。峰的高度和峰面积表示了某个化学成分的含量,而各峰的峰高(或峰面积)的比值体现了各种化学成分间的相对含量;量的概念的引入、定性和定量的结合赋予中药指纹图谱更大的功效;中药指纹图谱不仅可以进行个体、某物种的“唯一性”的鉴定,还可以将其“量”的特征和其他体系挂钩。因此,中药指纹图谱不仅是一种中药质量控制模式和技术,更可以发展成为一种采用各种指纹图谱来进行中药理论(复杂系统)和新药开发的研究体系和研究模式。指纹图谱应该具备指纹性,即:(1)专属性强。指所制订的指纹图谱应该是该中药所独有的、能与其它中药相区别的,其反映的化学信息是具有高度的选择性的;(2)稳定性好。即中药的指纹图谱应该是从某中药的多批次中归纳出的共性,图谱中的共有峰或特征峰应相对稳定;(3)重现性好。所制订的指纹图谱在规定条件下应能再现指纹特征(如共有峰数目、大小、位置等),其误差应在允许的范围内。只有这样,所制订的指纹图谱才具有实用价值,才可以有效控制药品的质量。采用uplc指纹图谱方法控制药品质量具有灵敏、快速、简便、准确等特点,通过优选色谱条件测定药品制剂的指纹图谱,可以很好控制药品质量。
中药指纹图谱可用于中药制剂研究、生产过程的各个阶段,按应用对象来分类,可分为中药材(原料药材)指纹图谱、中药原料药(包括饮片、配伍颗粒)指纹图谱和中药制剂指纹图谱。如分得更细,还可包括用于工艺生产过程中间产物的指纹图谱。值得注意的是建立中药材化学指纹图谱必须考虑诸多因素的影响。中药品种混杂,历史上形成的同名异物、异名同物,以及同一物种受产地环境、不同有效部位所含成分的波动大等因素,给质量控制带来了很多困扰。因此对于某一种中药材的指纹图谱,需要有大量的数据积累,才能制定合理的相似性标准。中药及其制剂均为多组分复杂体系,因此评价其质量应采用与之相适应的,能提供丰富鉴别信息的检测方法,但现行的显微鉴别、理化鉴别和含量测定等方法都不足以解决这一问题,建立中药指纹图谱将能较为全面地反映中药及其制剂中所含化学成分的种类与数量,进而对药品质量进行整体描述和评价。这也正好符合中医药整体学说。在此基础上,如果进一步开展谱效学研究,可使中药质量与其药效真正结合起来,有助于阐明中药作用机理。总之,中药指纹图谱的研究和建立,对于提高中药质量,促进中药现代化具有重要意义。
专利申请号为201210450660.0,发明名称为:一种抗病毒中药组合物的制备工艺及指纹图谱的测定方法,公开了连翘、大黄、麻黄、鱼腥草超声逆流提取的提取液的指纹图谱测定方法,标定了27个共有峰,指认了其中4个共有峰,分别为连翘酯苷a、连翘苷、松脂素-4-o-glc、大黄酸,该申请没有公开本发明的技术内容。本发明要求保护一种中药组合物的高效液相色谱指纹图谱测定方法,该中药组合物由以下药材组成:连翘、金银花、板蓝根、苦杏仁、薄荷脑、鱼腥草、大黄、广藿香、绵马贯众、红景天、麻黄、甘草、石膏,该指纹图谱测定方法,标定了15个主要色谱峰,指认了其中9个,分别为新绿原酸,绿原酸,隐绿原酸,异连翘酯苷a,连翘酯苷a,连翘苷,槲皮苷,4,5-二-o-咖啡酰奎宁酸,连翘苷,甘草酸,而现有技术中并没有公开该技术内容。
技术实现要素:
本发明提供一种中药组合物的指纹图谱测定方法。
该中药组合物由如下重量份的原料药制成:连翘200-300、麻黄60-100、大黄40-60、鱼腥草200-300、金银花200-300、板蓝根200-300、广藿香60-100、绵马贯众200-300、红景天60-100、薄荷脑5-9、苦杏仁60-100、甘草60-100、石膏200-300,指纹图谱测定方法如下:
供试品溶液制备:取所述中药组合物0.1-0.5g,精密称定,置具塞锥形瓶中,精密加入40-100%甲醇10-50ml,称定重量,超声处理20-40分钟,放冷,再称定重量,用40-100%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为c18色谱柱;检测波长239nm,流速0.1-0.5ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各1-10µl,注入超高液相色谱仪,测定,记录色谱图,即得。
本发明所述的中药组合物优选的重量份原料药为:连翘200、金银花300、板蓝根200、大黄40、广藿香60、绵马贯众300、红景天100、薄荷脑9、麻黄60、苦杏仁100、鱼腥草200、甘草100、石膏200。
本发明所述的中药组合物优选的重量份原料药为:连翘300、金银花200、板蓝根300、大黄60、广藿香100、绵马贯众200、红景天60、薄荷脑5、麻黄100、苦杏仁60、鱼腥草300、甘草60、石膏300。
本发明所述的中药组合物优选的重量份原料药为:连翘278、金银花294、板蓝根285、大黄55、广藿香95、绵马贯众290、红景天87、薄荷脑8.5、麻黄88、苦杏仁80、鱼腥草284、甘草95、石膏277。
本发明所述的中药组合物优选的重量份原料药为:连翘255、金银花255、板蓝根255、大黄51、广藿香85、绵马贯众255、红景天85、薄荷脑7.5、麻黄85、苦杏仁85、鱼腥草255、甘草85、石膏255。
本发明所述的中药组合物制剂的制备方法为:
(1)按照原料药重量比例称取中药材,净选,酌情碎断;
(2)广藿香碎断,加10倍量水提取挥发油,提油时间8小时,收集挥发油,备用;提取液过滤后,残渣弃去,滤液备用;
(3)连翘、麻黄、鱼腥草、大黄,用12倍量70%的乙醇提取3次,每次2.5小时,提取液合并过滤,回收乙醇,滤液备用;
(4)金银花、石膏、板蓝根、绵马贯众、甘草、红景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小时,提取液合并过滤,所得滤液与步骤(2)广藿香提油后的滤液合并,浓缩成在60℃时测定相对密度为1.10-1.15的清膏,加入乙醇,调节至醇浓度为70%,冷藏放置,过滤,回收乙醇至无醇味,得清膏备用;
(5)将步骤(4)所得清膏与步骤(3)所得醇提液合并,浓缩至在60℃时测定相对密度为1.15-1.20的清膏,干燥,得干膏粉,备用;
(6)将步骤(5)所得干膏粉加入适当药学上可接受的辅料制粒;
(7)将薄荷脑、步骤(2)所得挥发油加入乙醇溶解,喷入步骤(6)所得颗粒,密闭,混匀,压片或者装胶囊、或者装袋。
本发明所述指纹图谱测定方法优选为:
供试品溶液制备:取所述中药组合物0.25g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,称定重量,超声处理(功率500w,频率40khz)30分钟,放冷,再称定重量,用50%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersacquityuplchsst3色谱柱,柱长为100mm,内径为2.1mm,粒径为1.8µm;检测波长239nm,流速0.3ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各2µl,注入超高液相色谱仪,测定,记录色谱图,即得。
本发明所述指纹图谱测定方法还优选为:
供试品溶液制备:取所述中药组合物0.1g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理40分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersacquityuplchsst3色谱柱;检测波长239nm,流速0.1ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各1µl,注入超高液相色谱仪,测定,记录色谱图,即得。
本发明所述指纹图谱测定方法还优选为:
供试品溶液制备:取所述中药组合物0.5g,精密称定,置具塞锥形瓶中,精密加入40%甲醇10ml,称定重量,超声处理20分钟,放冷,再称定重量,用40%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersxbridgetmshieldrp18色谱柱;检测波长239nm,流速0.5ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各1µl,注入超高液相色谱仪,测定,记录色谱图,即得。
本发明所述指纹图谱测定方法还优选为:
供试品溶液制备:取所述中药组合物0.2g,精密称定,置具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声处理40分钟,放冷,再称定重量,用90%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为phenomenexkinetxxb-c18;检测波长239nm,流速0.4ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各6µl,注入超高液相色谱仪,测定,记录色谱图,即得。
用实施例1制备的样品,对本发明中药组合物的指纹图谱测定方法,从多个方面评价其可行性,评价方法如下:
1.仪器、试剂与试药
1.1仪器
美国watersuplch-class超高效液相色谱仪(pda检测器,empower3.0色谱工作站),循环式多用真空泵(郑州长城科工贸有限公司),kq250db型超声波清洗器(昆山市超声仪器有限公司),瑞士mettlertoledoal204型电子分析天平,瑞士mettlertoledoab135-s型电子分析天平,上海精科ja2603b电子天平。
1.2试剂
连翘苷(中国食品药品检定研究院,批号110821-201213,纯度95.3%),连翘酯苷a(中国食品药品检定研究院,批号111810-201304,纯度94.3%),新绿原酸(成都普瑞法科技开发有限公司,批号:13112712,纯度≥98%),绿原酸(中国食品药品检定研究院,批号:110753-201314,纯度,96.6%),隐绿原酸(上海源叶生物科技有限公司,批号:zf0226ba14,纯度≥98%),异连翘酯苷a(上海源叶生物科技有限公司,批号:ya0817hb14,纯度≥98%)、4,5-二-o-咖啡酰奎宁酸(中国食品药品检定研究院,批号:111894-201102,纯度,94.1%),槲皮苷(hwianalytikgmbhpharmasolutions,批号:hwi01112,纯度,91.7%),甘草酸铵(councilofeurope-edqm,批号:batch:1.1,纯度≥98%),甲醇(分析纯,天津市光复科技发展有限公司,批号2014年1月13日),磷酸(分析纯,天津市光复科技发展有限公司,批号2014年3月26日),乙腈(色谱纯,美国fisher公司,lot106246)。
1.3试药
本发明中药组合物(石家庄以岭药业股份有限公司)
2.色谱条件优化
2.1.检测波长的选择
在210nm~400nm考察供试品溶液不同检测波长下的色谱图,以239nm下的信息量较丰富,因此检测波长最优选择239nm,具体信息见下附图1-4。
2.2流动相系统的选择
本研究比较了甲醇-0.1%磷酸、乙腈-0.1%磷酸和甲醇-乙腈-0.1%磷酸三个流动相系统。结果显示甲醇-乙腈-0.1%磷酸系统色谱峰的分离度最好,具体信息见附图5-7,因此,确定流动相系统为甲醇-乙腈-0.1%磷酸,梯度洗脱表见表1,流速0.3ml/min。
表1流动相梯度洗脱表
3.供试品溶液和对照品溶液的制备
3.1提取溶剂的考察
分别比较了甲醇,90%甲醇,70%甲醇,50%甲醇的提取效果,结果表明50%甲醇提取所得指纹图谱中色谱峰最多,具体信息见附图8-11,因此选择50%甲醇作为提取溶剂。
3.2提取方法的考察
分别比较了超声和回流两种提取方法,超声和回流提取两种方法所得的指纹图谱色谱峰的数目和强度没有明显区别,具体信息见附图12-13,由于超声提取方法简便,因此选择超声提取。
3.3超声时间的考察
分别比较了超声20分钟、30分钟、40分钟,结果表明超声30分钟与40分钟所得指纹图谱中色谱峰信息量大,无明显区别,具体信息见附图14-16,因此最终确定超声时间为30分钟。
3.4供试品溶液制备方法的确定:取本发明中药组合物内容物适量,研细,取0.25g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,称定重量,超声处理(功率500w,频率40khz)30分钟,放冷,再称定重量,用50%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
3.5对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得。
3.6参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
主要色谱峰的光谱特征和辨认
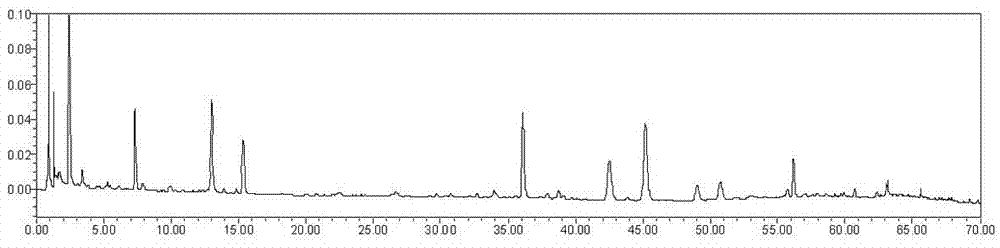
在已确定的色谱条件下,分别对对照品溶液和供试品溶液进样,供试品溶液的色谱图中确定16个主要色谱峰。根据色谱峰的保留时间和光谱特征,确定指纹图谱中2号峰为新绿原酸,3号峰为绿原酸,5号峰为隐绿原酸,8号峰为异连翘酯苷a,11号峰为连翘酯苷a,13号峰为槲皮苷,14号峰为4,5-二-o-咖啡酰奎宁酸,15号峰为连翘苷,16号峰为甘草酸,以上对照品与供试品色谱图及光谱图见附图17-28。
精密度
取供试品溶液,按确定的色谱条件连续进样6次,各主要色谱峰峰面积的rsd小于5%,表明进样精密度良好,结果见表2。
重复性
取本发明中药组合物内容物6份,按已确定的“供试品溶液制备方法”制备,注入液相色谱仪检测,测定各主要色谱峰峰面积的rsd均小于5%,表明方法重复性较好,结果见表3。
稳定性
取供试品溶液,分别在0h,2h,4h,8h,12h,24h进样,测定各主要色谱峰峰面积的rsd均小于10%,表明供试品溶液在24h内稳定,结果见表4。
指纹图谱的建立及相似度分析
8.1指纹图谱的建立:取10批本发明中药组合物样品,按3.4项下要求制备供试品溶液,在上述色谱条件下进样分析,得到10批本发明中药组合物的指纹图谱,并采用国家药典委员会开发的中药色谱指纹图谱相似度评价系统2004a版软件对10批样品的指纹图谱进行分析,采用中位数法对各指纹图谱色谱峰进行了多点校正和自动匹配,计算相似度并生成对照指纹图谱,见附图29-30,相似度结果见表5,指纹图谱中共有色谱峰16个,用标准品指认了其中9个共有峰:新绿原酸(2号峰),绿原酸(3号峰),隐绿原酸(5号峰),异连翘酯苷a(8号峰),连翘酯苷a(11号峰),槲皮苷(13号峰),4,5-二-o-咖啡酰奎宁酸(14号峰),连翘苷(15号峰),甘草酸(16号峰)。其中以出峰稳定,峰面积较大的11号色谱峰连翘酯苷a为参照峰:
本发明建立了本发明中药组合物的液相指纹图谱方法,共有色谱峰16个,用标准品指认了其中9个共有峰:新绿原酸(2号峰),绿原酸(3号峰),隐绿原酸(5号峰),异连翘酯苷a(8号峰),连翘酯苷a(11号峰),槲皮苷(13号峰),4,5-二-o-咖啡酰奎宁酸(14号峰),连翘苷(15号峰),甘草酸(16号峰)。其中以出峰稳定,峰面积较大的11号色谱峰连翘酯苷a为参照峰,10批本发明中药组合物指纹图谱的相似度较高。由以上实验结果可知,本方法具有良好的精密度和稳定性,以及重复性,为提高该中药组合物的质量控制提供一种新方法。
附图说明
图1:检测波长320nm下供试品溶液色谱图
图2:检测波长290nm下供试品溶液色谱图
图3:检测波长254nm下供试品溶液色谱图
图4:检测波长230nm下供试品溶液色谱图
图5:甲醇-0.1%磷酸供试品溶液色谱图
图6:乙腈-0.1%磷酸供试品溶液色谱图
图7:甲醇-乙腈-0.1%磷酸供试品溶液色谱图
图8:50%甲醇提取供试品溶液色谱图
图9:70%甲醇提取供试品溶液色谱图
图10:90%甲醇提取供试品溶液色谱图
图11:甲醇提取供试品溶液色谱图
图12:超声30分钟供试品溶液色谱图
图13:回流1小时供试品溶液色谱图
图14:超声20分钟供试品溶液色谱图
图15:超声30分钟供试品溶液色谱图
图16:超声40分钟供试品溶液色谱图
图17:对照品溶液的色谱图
图18:连翘酯苷a参照物溶液的色谱图
图19:供试品溶液的色谱图
图20:对照品溶液与供试品溶液中新绿原酸光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图21:对照品溶液与供试品溶液中绿原酸光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图22:对照品溶液与供试品溶液中隐绿原酸光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图23:对照品溶液与供试品溶液中异连翘酯苷a光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图24:对照品溶液与供试品溶液中连翘酯苷a光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图25:对照品溶液与供试品溶液中槲皮苷光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图26:对照品溶液与供试品溶液中4,5-二-o-咖啡酰奎宁酸光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图27:对照品溶液与供试品溶液中连翘苷光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图28:对照品溶液与供试品溶液中甘草酸光谱图比较,虚线为供试品光谱图,实线为对照品光谱图
图29:10批本发明中药组合物色谱指纹图谱
图30:本发明中药组合物对照指纹图谱
图31:实施例1指纹图谱,其中a图中峰1为新绿原酸,2为绿原酸,3为隐绿原酸,4为异连翘酯苷a,5为连翘酯苷a,6为槲皮苷,7为4,5-二-o-咖啡酰奎宁酸,8为连翘苷,9为甘草酸;b图为连翘酯苷a参照物溶液的色谱图;c为供试品溶液指纹色谱图
图32:实施例2指纹图谱,其中a图中峰1为新绿原酸,2为绿原酸,3为隐绿原酸,4为异连翘酯苷a,5为连翘酯苷a,6为槲皮苷,7为4,5-二-o-咖啡酰奎宁酸,8为连翘苷,9为甘草酸;b图为连翘酯苷a参照物溶液的色谱图;c为供试品溶液指纹色谱图
图33:实施例3指纹图谱,其中a图中峰1为新绿原酸,2为绿原酸,3为隐绿原酸,4为异连翘酯苷a,5为连翘酯苷a,6为槲皮苷,7为4,5-二-o-咖啡酰奎宁酸,8为连翘苷,9为甘草酸;b图为连翘酯苷a参照物溶液的色谱图;c为供试品溶液指纹色谱图
图34:实施例4指纹图谱,其中a图中峰1为新绿原酸,2为绿原酸,3为隐绿原酸,4为异连翘酯苷a,5为连翘酯苷a,6为槲皮苷,7为4,5-二-o-咖啡酰奎宁酸,8为连翘苷,9为甘草酸;b图为连翘酯苷a参照物溶液的色谱图;c为供试品溶液指纹色谱图。
具体实施方式
实施例1
按比例称取:连翘200g、金银花300g、板蓝根200g、大黄40g、广藿香60g、绵马贯众300g、红景天100g、薄荷脑9g、麻黄60g、苦杏仁100g、鱼腥草200g、甘草100g、石膏200g,按照以下工艺提取:
(1)按照原料药重量比例称取中药材,净选,酌情碎断;
(2)广藿香碎断,加10倍量水提取挥发油,提油时间8小时,收集挥发油,备用;提取液过滤后,残渣弃去,滤液备用;
(3)连翘、麻黄、鱼腥草、大黄,用12倍量70%的乙醇提取3次,每次2.5小时,提取液合并过滤,回收乙醇,滤液备用;
(4)金银花、石膏、板蓝根、绵马贯众、甘草、红景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小时,提取液合并过滤,所得滤液与步骤(2)广藿香提油后的滤液合并,浓缩成在60℃时测定相对密度为1.10-1.15的清膏,加入乙醇,调节至醇浓度为70%,冷藏放置,过滤,回收乙醇至无醇味,得清膏备用;
(5)将步骤(4)所得清膏与步骤(3)所得醇提液合并,浓缩至在60℃时测定相对密度为1.15-1.20的清膏,干燥,得干膏粉,备用;
(6)将步骤(5)所得干膏粉加入适当药学上可接受的辅料制粒;
(7)将薄荷脑、步骤(2)所得挥发油加入乙醇溶解,喷入步骤(6)所得颗粒,装胶囊,即得。
指纹图谱测定方法:
供试品溶液制备:取所述中药组合物0.25g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,称定重量,超声功率500w,频率40khz,超声30分钟,放冷,再称定重量,用50%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersacquityuplchsst3色谱柱,柱长为100mm,内径为2.1mm,粒径为1.8µm;检测波长239nm,流速0.3ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各2µl,注入超高液相色谱仪,测定,记录色谱图,即得。
结论:指纹图谱见附图31,结果令人满意可以用于控制该中药组合物的质量。
实施例2
按比例称取:连翘300g、金银花200g、板蓝根300g、大黄60g、广藿香100g、绵马贯众200g、红景天60g、薄荷脑5g、麻黄100g、苦杏仁60g、鱼腥草300g、甘草60g、石膏300g,按照以下工艺提取:
(1)按照原料药重量比例称取中药材,净选,酌情碎断;
(2)广藿香碎断,加10倍量水提取挥发油,提油时间8小时,收集挥发油,备用;提取液过滤后,残渣弃去,滤液备用;
(3)连翘、麻黄、鱼腥草、大黄,用12倍量70%的乙醇提取3次,每次2.5小时,提取液合并过滤,回收乙醇,滤液备用;
(4)金银花、石膏、板蓝根、绵马贯众、甘草、红景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小时,提取液合并过滤,所得滤液与步骤(2)广藿香提油后的滤液合并,浓缩成在60℃时测定相对密度为1.10-1.15的清膏,加入乙醇,调节至醇浓度为70%,冷藏放置,过滤,回收乙醇至无醇味,得清膏备用;
(5)将步骤(4)所得清膏与步骤(3)所得醇提液合并,浓缩至在60℃时测定相对密度为1.15-1.20的清膏,干燥,得干膏粉,备用;
(6)将步骤(5)所得干膏粉加入适当药学上可接受的辅料制粒;
(7)将薄荷脑、步骤(2)所得挥发油加入乙醇溶解,喷入步骤(6)所得颗粒,压片,即得。
指纹图谱测定方法:
供试品溶液制备:取所述中药组合物0.1g,精密称定,置具塞锥形瓶中,精密加入100%甲醇50ml,称定重量,超声处理40分钟,放冷,再称定重量,用100%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersacquityuplchsst3(柱长为100mm,内径为2.1mm,粒径为1.8µm)色谱柱;检测波长239nm,流速0.1ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各1µl,注入超高液相色谱仪,测定,记录色谱图,即得。
结论:指纹图谱见附图32,结果令人满意可以用于控制该中药组合物的质量。
实施例3
原料药配方为:连翘278g、金银花294g、板蓝根285g、大黄55g、广藿香95g、绵马贯众290g、红景天87g、薄荷脑8.5g、麻黄88g、苦杏仁80g、鱼腥草284g、甘草95g、石膏277g,按照以下工艺提取:
(1)按照原料药重量比例称取中药材,净选,酌情碎断;
(2)广藿香碎断,加10倍量水提取挥发油,提油时间8小时,收集挥发油,备用;提取液过滤后,残渣弃去,滤液备用;
(3)连翘、麻黄、鱼腥草、大黄,用12倍量70%的乙醇提取3次,每次2.5小时,提取液合并过滤,回收乙醇,滤液备用;
(4)金银花、石膏、板蓝根、绵马贯众、甘草、红景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小时,提取液合并过滤,所得滤液与步骤(2)广藿香提油后的滤液合并,浓缩成在60℃时测定相对密度为1.10-1.15的清膏,加入乙醇,调节至醇浓度为70%,冷藏放置,过滤,回收乙醇至无醇味,得清膏备用;
(5)将步骤(4)所得清膏与步骤(3)所得醇提液合并,浓缩至在60℃时测定相对密度为1.15-1.20的清膏,干燥,得干膏粉,备用;
(6)将步骤(5)所得干膏粉加入适当药学上可接受的辅料制粒;
(7)将薄荷脑、步骤(2)所得挥发油加入乙醇溶解,喷入步骤(6)所得颗粒,装袋,即得。
指纹图谱测定方法:
供试品溶液制备:取所述中药组合物0.5g,精密称定,置具塞锥形瓶中,精密加入40%甲醇10ml,称定重量,超声处理20分钟,放冷,再称定重量,用40%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为watersxbridgetmshieldrp18(5µm,4.6mm×250mm)色谱柱;检测波长239nm,流速0.5ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各1µl,注入超高液相色谱仪,测定,记录色谱图,即得。
结果:指纹图谱见附图33,结果令人满意可以用于控制该中药组合物的质量。
实施例4
原料药配方为:按比例称取:连翘255g、金银花255g、板蓝根255g、大黄51g、广藿香85g、绵马贯众255g、红景天85g、薄荷脑7.5g、麻黄85g、苦杏仁85g、鱼腥草255g、甘草85g、石膏255g,按照以下工艺提取:
(1)按照原料药重量比例称取中药材,净选,酌情碎断;
(2)广藿香碎断,加10倍量水提取挥发油,提油时间8小时,收集挥发油,备用;提取液过滤后,残渣弃去,滤液备用;
(3)连翘、麻黄、鱼腥草、大黄,用12倍量70%的乙醇提取3次,每次2.5小时,提取液合并过滤,回收乙醇,滤液备用;
(4)金银花、石膏、板蓝根、绵马贯众、甘草、红景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小时,提取液合并过滤,所得滤液与步骤(2)广藿香提油后的滤液合并,浓缩成在60℃时测定相对密度为1.10-1.15的清膏,加入乙醇,调节至醇浓度为70%,冷藏放置,过滤,回收乙醇至无醇味,得清膏备用;
(5)将步骤(4)所得清膏与步骤(3)所得醇提液合并,浓缩至在60℃时测定相对密度为1.15-1.20的清膏,干燥,得干膏粉,备用;
(6)将步骤(5)所得干膏粉加入适当药学上可接受的辅料制粒;
(7)将薄荷脑、步骤(2)所得挥发油加入乙醇溶解,喷入步骤(6)所得颗粒,装胶囊,即得。
指纹图谱测定方法:
供试品溶液制备:取所述中药组合物0.2g,精密称定,置具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声处理40分钟,放冷,再称定重量,用90%的甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:分别称取新绿原酸、绿原酸、隐绿原酸、异连翘酯苷a、连翘酯苷a、连翘苷、4,5-二-o-咖啡酰奎宁酸、槲皮苷、甘草酸铵对照品适量,制成分别含18.4µg/ml新绿原酸、27.2µg/ml绿原酸、21.2µg/ml隐绿原酸、27.2µg/ml异连翘酯苷a、21.6µg/ml连翘酯苷a、16.0µg/ml连翘苷、14.4µg/ml4,5-二-o-咖啡酰奎宁酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸铵混合对照品溶液即得;
参照物溶液的制备:取连翘酯苷a对照品适量,精密称定,加50%甲醇制成每1ml含101.2µg的溶液,即得。
色谱条件:流动相为甲醇-乙腈-0.1%磷酸,梯度洗脱,洗脱比例如下表:
色谱柱为phenomenexkinetxxb-c18色谱柱;检测波长239nm,流速0.4ml/min;
测定法:分别精密吸取对照品溶液与供试品溶液各6µl,注入超高液相色谱仪,测定,记录色谱图,即得。
结论:指纹图谱见附图34,结果令人满意可以用于控制该中药组合物的质量。
- 还没有人留言评论。精彩留言会获得点赞!