血液中未知毒物的气相色谱‑质谱筛查方法与流程
本发明涉及血液中未知毒物的气相色谱-质谱筛查方法,属于化学物质检测技术领域。
背景技术:
常见毒物筛查在法庭科学理化检验中占据了相当大的比重。申请人通过调研山东省近10年来的涉毒案件,发现此类案件物证检验中存在以下问题:
(一)涉案毒物种类明显增多,在侦办某些是否中毒不明确的案件中,需要筛查的毒物不仅仅局限于某几种或某一类,即目标性不强。此种情况下,如果仍沿用老方法仅就几种最常见的毒物进行筛查,极有可能造成漏检。
(二)目前,气质联用法是全省各地市普遍掌握和采用的仪器分析方法,此方法相应的的样品前处理方法不规范,也未经过优化考查。
(三)利用气质联用技术对涉案毒物的检出限进行系统性考查仍属空白,阴性结果评价缺乏科学的数据支撑。
鉴于以上情况,为满足新形势下公安工作的需求,有必要提供一种具有通用性的、操作简便的筛查方法。
技术实现要素:
针对上述现有技术,本发明提供了一种血液中未知毒物的气相色谱-质谱筛查方法。本申请系统开展了毒物(主要包括有机磷类、氨基甲酸酯类、拟除虫菊酯类、有机氯类、除草剂、鼠药等)、药物(主要包括苯并杂氮卓类、巴比妥类、吩噻嗪类、其他安眠镇静类药物等)的检验方法学研究,在最优条件下得到它们的检出限,为阴性结果评价提供数据支撑,继而为相关的临床诊断、抢救及刑事侦查、诉讼提供有力的科学依据。
本发明是通过以下技术方案实现的:
一种血液中未知毒物的气相色谱-质谱筛查方法:取待检测血液,进行前处理,然后进行gc-ms分析,根据分析结果判断待检测血液样品中是否含有毒物成分;所述前处理的方法为液液萃取方法或固相萃取方法。
所述液液萃取方法具体为:(1)当目标检测物为毒物时:取待检测血液2ml,置于15ml尖底离心试管中,加100μg/ml环氧七氯工作液10μl,振荡混匀,经乙酸乙酯:苯(体积比1:1)6ml×2提取,振荡10min,离心10min,分离有机相,合并于试管中,浓缩至近干,加甲醇定容至100μl,供gc-ms分析;(2)当目标检测物为药物时:取待检测血液2ml,置于15ml尖底离心试管中,加200μg/mlskf525a、烯丙异丙巴比妥内标工作液各2μl,用8%氢氧化钠溶液调ph至11~12,经乙酸乙酯:苯(体积比1:1)6ml×2提取,振荡10min,离心10min,分离有机相,合并于试管中,剩余液体用10%盐酸调ph至2~3,经乙酸乙酯:苯(体积比1:1)6ml×2提取,振荡10min,离心10min,分离有机相,合并于试管中;将以上两步所得提取液合并,浓缩至近干,加甲醇定容至100μl,供gc-ms分析。
所述固相萃取方法具体为:(1)取待检测血液2ml,置于15ml尖底塑料离心试管中,加200μg/mlskf525a、烯丙异丙巴比妥内标工作液各2μl,100μg/ml环氧七氯工作液10μl,振荡混匀,加入0.1m磷酸盐缓冲溶液(ph6.0)6ml,振荡10min,离心10min,分离上清液,用于固相萃取上样;(2)固相萃取步骤:①柱活化:分别用5ml甲醇及5ml磷酸盐缓冲溶液(0.1m,ph6.0)活化固相柱,流速1.5ml/min;所用固相柱选自hlb固相萃取柱或pep固相萃取柱;②上样:将样品上清液5ml分别加入到已活化好的固相萃取柱,流速0.5ml/min;③淋洗:分别用3ml水及3ml磷酸盐缓冲液(0.1m,ph6.0)淋洗固相柱,流速5ml/min;之后空气加压5min,流速50ml/min,再通氮气干燥10min;④洗脱:用4ml乙酸乙酯:苯(体积比1:1)洗脱,流速0.5ml/min;收集洗脱液,浓缩近干,甲醇定容至100μl,供gc-ms分析。
上述所涉及的毒物包括:1.灭多威;2.甲胺磷;3.敌敌畏;4.乙酰甲胺磷;5.速灭威;6.异丙威;7.氧化乐果;8.治冥磷;9.久效磷;10.甲拌磷;11.毒鼠强;12.乐果;13.呋喃丹;14.特丁磷;15.抗蚜威;16.乙草胺;17.甲草胺;18.甲基对硫磷;19.马拉硫磷;20.毒死蜱;21.对硫磷;22.甲基异柳磷;23.硫丹内酯;24.环氧七氯;25.杀扑磷;26.丁草胺;27.α-硫丹;28.硫丹醇;29.β-硫丹;30.吡氟甲禾灵;31.胺菊酯;32.氰甲菊酯;33.伏杀磷;34.高效氯氟氰菊酯;35.氯菊酯;36.氯氰菊酯;37.氰戊菊酯;38.溴氰菊酯。
上述所涉及的药物包括:39.巴比妥;40.布洛芬;41.烯丙异丙巴比妥;42.异戊巴比妥;43.戊巴比妥;44.哌替啶;45.眠尔通;46.司可巴比妥;47.咖啡因;48.安替比林;49.氨基比林;50.苯巴比妥;51.扑尔敏;52.阿米替林;53.丙咪嗪;54.多虑平;55.异丙嗪;56.卡马西平;57.skf525a;58.奥沙西泮;59.苯妥英;60.劳拉西泮;61.安定;62.氯丙嗪;63.利眠宁;64.咪达唑仑;65.三氟拉嗪;66.芬太尼;67.氯硝西泮;68.氯氮平;69.艾司唑仑;70.阿普唑仑;71.三唑仑;72.佐匹克隆。
进一步地,进行gc-ms分析时的条件为:色谱柱:db-5ms弹性石英毛细柱(30m×0.25mm×0.25μm)或等效色谱柱;离子源:ei,230℃;升温程序:初始温度80℃保持2min,以10℃/min程序升温至280℃,保持13min;进样口温度:280℃;传输线温度:280℃;载气:高纯氦气,纯度大于等于99.999%;柱流量:1ml/min;扫描方式:scan,质量范围:40~450amu;分流比:10:1。
进一步地,以向空白血液样品中添加检出限浓度的2~5倍量的目标检测物作为阳性对照样品,以空白血液样品作为阴性对照,根据检测结果进行评价:
在相同的实验条件下,待测样品中出现的色谱峰保留时间与添加对照样品的色谱峰保留时间相比较,相对误差在±2%内,且特征碎片离子均出现,所选择的离子相对丰度比与添加对照品的离子相对丰度比之相对误差不超过表1规定的范围,则可判断样品中存在这种化合物。
表1相对离子丰度比的最大允许相对误差(%)
阳性结果评价:如果待测样品中检出毒物成分且空白样品无干扰,则阳性结果可靠;如果待测样品中检出毒物成分且空白样品亦呈阳性,则阳性结果不可靠。
阴性结果评价:如果待测样品中出现与内标环氧七氯、skf525a和烯丙异丙巴比妥rt相同的色谱峰,且信噪比≥3,未检出毒物成分,则阴性结果可靠;如果待测样品中未出现内标环氧七氯、skf525a和烯丙异丙巴比妥的色谱峰,则阴性结果不可靠。
本发明的血液中未知毒物的气相色谱-质谱筛查方法,建立了不同类别毒物的检测方法,检测快速、准确,优化了前处理方法,给出了常见毒物(基本涵盖了法庭科学领域涉及的常见毒物)的检出限(见附录a),确保各类目标物的高灵敏有效检验,为阴性检验结果提供了科学的数据支撑,继而为相关的临床诊断、抢救及刑事侦查、诉讼提供有力的科学依据。
附图说明
图1:38种毒物总离子流色谱图,其中,出峰顺序:1.灭多威;2.甲胺磷;3.敌敌畏;4.乙酰甲胺磷;5.速灭威;6.异丙威;7.氧化乐果;8.治冥磷;9.久效磷;10.甲拌磷;11.毒鼠强;12.乐果;13.呋喃丹;14.特丁磷;15.抗蚜威;16.乙草胺;17.甲草胺;18.甲基对硫磷;19.马拉硫磷;20.毒死蜱;21.对硫磷;22.甲基异柳磷;23.硫丹内酯;24.环氧七氯;25.杀扑磷;26.丁草胺;27.α-硫丹;28.硫丹醇;29.β-硫丹;30.吡氟甲禾灵;31.胺菊酯;32.氰甲菊酯;33.伏杀磷;34.高效氯氟氰菊酯;35.氯菊酯;36.氯氰菊酯;37.氰戊菊酯;38.溴氰菊酯。
图2:34种药物总离子流色谱图,其中,出峰顺序:39.巴比妥;40.布洛芬;41.烯丙异丙巴比妥;42.异戊巴比妥;43.戊巴比妥;44.哌替啶;45.眠尔通;46.司可巴比妥;47.咖啡因;48.安替比林;49.氨基比林;50.苯巴比妥;51.扑尔敏;52.阿米替林;53.丙咪嗪;54.多虑平;55.异丙嗪;56.卡马西平;57.skf525a;58.奥沙西泮;59.苯妥英;60.劳拉西泮;61.安定;62.氯丙嗪;63.利眠宁;64.咪达唑仑;65.三氟拉嗪;66.芬太尼;67.氯硝西泮;68.氯氮平;69.艾司唑仑;70.阿普唑仑;71.三唑仑;72.佐匹克隆。
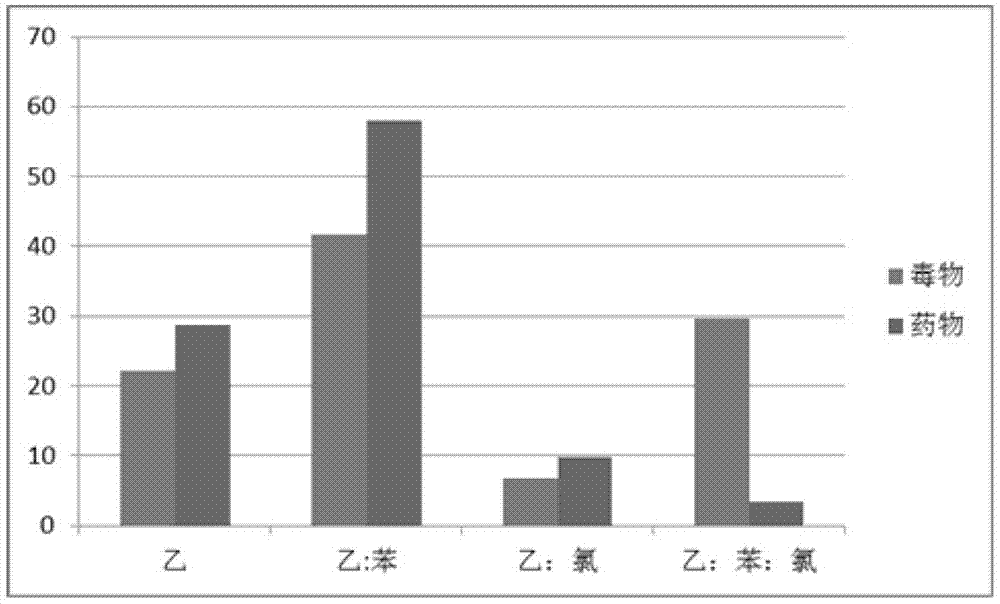
图3:液液萃取四种有机溶剂提取率比较图。
图4:三家实验室不同固相柱目标物最高回收率个数所占比重。
图5:三家实验室不同洗脱体积目标物最高回收率个数所占比重。
具体实施方式
下面结合实施例对本发明作进一步的说明。
下述实施例中所涉及的仪器、试剂、材料等,若无特别说明,均为现有技术中已有的常规仪器、试剂、材料等,可通过正规商业途径获得。下述实施例中所涉及的实验方法,检测方法等,若无特别说明,均为现有技术中已有的常规实验方法,检测方法等。
实验血液中毒物/药物的气相色谱-质谱筛查方法的优化
(一)仪器、试剂与材料
仪器:gc-ms(配有ei源);分析天平(感量0.1mg);高速离心机;电动振荡器;固相萃取仪;移液器。
标准物质:根据目标物前处理提取方式的不同,将标准物质分为两大类:一是毒物,主要包括有机磷类、氨基甲酸酯类、拟除虫菊酯类、有机氯类、除草剂、鼠药等;二是药物,主要包括苯并杂氮卓类、巴比妥类、吩噻嗪类、其他安眠镇静类药物等。标准物质具体名称见表2。
表272种毒(药)物名称、cas号、分子量、化学类别和类型参数表
甲醇,8%氢氧化钠水溶液,10%盐酸水溶液,磷酸盐缓冲溶液(0.1m,ph6.0),所用试剂除另有说明外均为分析纯,实验用水为超纯水。
空白血为健康人血液(血站提供);尖底塑料离心试管;hlb固相萃取柱、c18固相萃取柱、pep固相萃取柱(规格均为6ml、200mg)。
标准物质溶液配制
标准物质溶液:准确称取72种毒(药)物适量,用甲醇配成1.0mg/ml的标准物质储备液,置于冰箱冷冻储存,保存期限为12个月;实验所需浓度的标准物质工作液均由储备液稀释而得。
同时,按照毒物和药物所属类别(有机磷、有机氯、氨基甲酸酯、除草剂、苯并杂氮卓类、巴比妥类、吩噻嗪类,其他类安眠药等类型),配制一批浓度为1mg/ml的混标溶液。
内标物标准溶液:准确称取skf525a和烯丙异丙巴比妥适量,用甲醇配成1mg/ml的混合内标储备溶液,置于冰箱中冷冻储存,保存期限为12个月。将内标储备液用甲醇稀释5倍,得200μg/ml的内标工作液。环氧七氯内标工作液(100μg/ml)购自农业部环境保护监测所。
样品配制
样品配制1(用于提取条件考查):
取空白血液11份,每份2ml,分别向其中添加毒物和药物混标,使目标物浓度均为2.0μg/ml。
样品配制2(用于方法检出限考查):
取空白血液12份,每份2ml,分别向其中添加毒物和药物混标,使目标物浓度分别为0.1、0.2、0.5、1.0、2.0和5.0μg/ml(每个浓度两份)。
(二)gc-ms条件与前处理方法
(1)gc-ms条件
色谱柱:thermoscientifictg-5ms(agilentdb-5ms或其它等效柱)30m×0.25mm×0.25μm弹性石英毛细管柱。
色谱条件:进样口:280℃,柱温:80℃(2min)10℃/min280℃(13min),流量:1ml/min,分流比:10:1,载气:he气。
质谱条件:离子源:ei,质量扫描范围:40~450amu,扫描方式:scan,传输线温度:280℃,离子源温度:230℃。
(2)前处理方法
a.药物
①直接提取方法(液液萃取)
取空白血液2ml(根据实验需要,添加不同浓度的目标分析物),用8%naoh调ph11–12(体积参考值:50μl),经提取溶剂6ml×2提取,振荡10min,离心(8000r/min,10min),分离有机相,合并于尖底玻璃试管中;剩余液体用10%hcl调ph2.0–3.0(加入体积参考值:100μl),经提取溶剂6ml×2提取,振荡10min,离心(8000r/min,10min),分离有机相,合并于尖底玻璃试管中;将以上两步所得提取液合并,浓缩至近干,100μl甲醇定容,供仪器分析。
取空白血液2ml,按上述步骤平行操作,作为空白对照。
②固相萃取方法
固相萃取柱:c18、hlb、pep;
分别采用c18、hlb、pep三种固相萃取柱,平行进行如下操作:
第一步:样品处理:取空白血液2ml(根据实验需要,添加不同浓度的目标分析物),加入0.1m磷酸盐缓冲液(ph6.0)6ml,混匀,振荡10min,离心(8000r/min,10min),分离上清液,用于固相萃取上样。
第二步:固相萃取步骤:
柱活化:分别用5ml甲醇及5ml磷酸盐缓冲溶液(0.1m,ph6.0)活化固相小柱,流速1.5ml/min。
加样:将制备好的样品分别加入到已活化好的c18柱(加样量均为5ml),流速0.5ml/min。
淋洗:分别用3ml水及3ml磷酸盐缓冲溶液(0.1m,ph6.0)淋洗小柱,流速5ml/min;之后空气加压5min(流速50ml/min),再通氮气干燥10min。
洗脱:用6ml洗脱溶剂(直接提取最优溶剂)进行洗脱,流速0.5ml/min。
第三步:收集洗脱液,浓缩至近干,100μl甲醇定容,供仪器分析。
取空白血液2ml,按上述步骤平行操作,作为阴性对照。
(2)毒物
①直接提取方法(液液萃取)
取空白血液2ml(根据实验需要,添加不同浓度的目标分析物),经提取溶剂6ml×2提取,振荡10min,离心(8000r/min,10min),分离有机相,合并于尖底玻璃试管中,常温浓缩至近干,100μl甲醇定容,供仪器分析。
②固相萃取方法
参见上述药物固相萃取方法。收集洗脱液后,常温浓缩至近干,100μl甲醇定容,供仪器分析。
(三)结果与分析
(1)毒(药)物标准物质的gc-ms分析
gc-ms联用技术,既具有gc高分离效能,又具有ms准确定性的特点,可实现多种目标物同时扫描,快速定性和定量分析。大大提高了分析效率。该检测技术已广泛应用于环境、医药、农业、食品安全及法庭科学等领域。
本研究利用gc-ms全扫描采集模式,以保留时间和特征碎片离子来确定目标物。特征离子选择原则遵循以下几点:一是离子丰度相对较高,分子量较大的碎片离子;二是避免来自离子源污染物、柱流失物、基质干扰物等产生的离子,如57、71、73、85、147、207等碎片离子;三是与nist谱图库中目标物的质谱图相匹配。一般以丰度比最高且测定干扰较少的离子作为定量离子(用于计算回收率)。
为摸索72种毒(药)物的气相色谱-质谱仪器分析方法,采用单标确定保留时间和混标测试分离情况的方法,分别取浓度为100μg/ml的单标和混标(分毒物和药物两类),采用前述仪器条件,用气质联用仪进行分析。同时,通过不断稀释混标的浓度,测试目标物对应的仪器检出限。通过分析,72种毒(药)物的保留时间得以确定,每种毒(药)物的仪器检出限浓度(s/n≥3)在0.5μg/ml~40μg/ml范围内。该实验数据证实了采用前述仪器条件,在35min之内,完全能够实现72种毒(药)物地良好分离,确保了检验结果的灵敏度和准确性,具体实验结果如图1、图2、表3所示。
表372种毒(药)物和内标物标准品的保留时间、特征离子和仪器检出限
从以上图表中可以看出,利用前述的仪器分析条件,72种毒(药)物均有着良好的响应值和分离度。表3中特征离子有下划线的,为该物质丰度比最高的离子碎片,作为下步已知标准添加测定回收率的定量离子。
(2)液液萃取条件的优化
物质在不同溶剂中有不同的溶解度。当两种互不混溶的溶剂共存时,溶质在这两种溶剂中能分配不同的溶解量。互不相溶的溶剂通常指水和与水不互溶的有机溶剂,且两者密度具有一定的差距,易于分离。利用这一性质将溶质从一种溶剂转移到另一种溶剂中的过程,称之为液-液萃取。本研究中的72种毒(药)物,均为有机化合物,需要用有机溶剂将其从血液(水相)中萃取出来,经浓缩定容后进行gc-ms分析。不同的有机溶剂对同一种毒(药)物的提取率是不相同的;对于72种毒(药)物,所选用的提取溶剂应能实现对大多数毒(药)物的良好提取,从而达到提高检出灵敏度的目的。因此,提取剂的选择,对血液中未知物的筛查至关重要。
a.液液萃取有机溶剂的选择:根据大量的文献资料和工作实践,选择了乙酸乙酯、乙酸乙酯/苯(1:1)、乙酸乙酯/氯仿(1:1)、乙酸乙酯/苯/氯仿(1:1:2)四种试剂组合,分别按照前述的方法进行提取分析。
为了考查重现性,济宁、潍坊和青岛均按照上述方法平行操作,分别得到72种毒(药)物的液液萃取回收率。回收率计算方法如下:已知空白血添加毒(药)物目标物的峰面积与相应的质量浓度标准溶液的峰面积比值来计算回收率。
在完成每种物质的回收率的计算后,为方便统计和表述,分别统计三家研究单位四种萃取溶剂中回收率最高的毒(药)物种类个数,计算出其在四种溶剂中所占的比重,最后再将三家单位的数据对应四种萃取溶剂,分别求平均值,结果见表4和图3。
表4三家实验室四种溶剂萃取百分比(%)
通过以上图表中的数据可以看出,萃取率最高的毒(药)物所占比重最大的,无论是毒物还是药物,均为乙酸乙酯/苯(1:1)。根据这一实验数据,最终确定液液萃取溶剂和固相萃取中的洗脱溶剂均选用该试剂。
b.液液萃取-气质联用的方法检出限
确定了乙酸乙酯/苯(1:1)作为筛查方法最优萃取溶剂后,我们利用液液萃取-气质联用方法,对72种毒(药)物的检出限进行了系统性考查。济宁、青岛和潍坊三家平行进行操作。按照72种目标物各自的色谱峰(10≥s/n≥3)进行评判,得到三家实验室的72种目标物检出限数据。表5为毒物检出限,表6为药物检出限。根据实验数据分析,在检出限以上浓度(含检出限)已知添加的目标物回收率均在50%~150%之间,完全满足准确定性的需要。
表5液液萃取毒物检出限(μg/ml)
表6液液萃取药物检出限(μg/ml)
由于三家实验室仪器设备新旧不同,人员操作亦存在一定的差异,故液液萃取检出限浓度不尽相同。综合分析三家实验室的数据,最终确定了72种毒(药)物的方法检出限浓度,见表7。
表772种毒(药)物的液液萃取方法检出限表(μg/ml)
(3)固相萃取前处理条件的优化
利用固体吸附剂将液体样品中的目标化合物吸附,与样品的基体和干扰化合物分离,然后再用洗脱液洗脱,达到分离和富集目标化合物的目的。
血液中未知物的筛查,常用到的固相萃取柱依据其萃取填料分为以下三种:(1)烷基键合硅胶基质。主要有c18,常用于中等极性到非极性化合物的分析;(2)亲脂性二乙烯苯和亲水性n-乙烯基吡咯烷酮两种单体按一定比例聚合成的大孔共聚物,如hlb柱,可用于分析酸性、中性和碱性化合物的萃取;(3)官能化聚苯乙烯/二乙烯共聚物,如pep柱,对各种极性、非极性化合物均具有较均衡的吸附作用。
a.不同固相萃取柱的选择
本研究中,我们分别选用了c18、hlb、pep三种小柱比较萃取效果。济宁、潍坊和青岛均按照上述方法平行操作,分别得到72种毒(药)物的固相萃取回收率。为方便统计和表述,分别统计三家单位不同固相萃取柱中回收率最高的毒(药)物种类个数,计算出其在总数中所占的比重,最后再将三家测试单位的数据对应求平均值,结果见表8和图4。
表8三家实验室不同固相柱目标物最高回收率个数所占比重表(%)
从表8和图4中可以看出,对于38种毒物来讲,60%(23种)以上的均为hlb柱回收率最高;对于34种药物来讲,80%(27种)以上均为pep柱回收率最高。综合三家实验室数据,最终选择hlb柱作为毒物的最优固相萃取柱;pep柱作为药物的最优固相萃取柱。
b.洗脱体积的影响
济宁、潍坊和青岛分别对待筛查毒(药)物在不同洗脱体积下的提取率进行了考查。通过测定目标物回收率,计算出不同洗脱体积中回收率最高的个数比,再将三家实验室对应洗脱体积中的个数比求平均值,具体数据见表9和图5。
表9三家实验室不同洗脱体积目标物最高回收率个数所占比重表(%)
从表9和图5中看出,对于毒物而言,随着洗脱溶剂体积的不断增加,大部分毒物的回收率呈下降趋势。对于药物而言,随着洗脱溶剂体积的不断增加,大部分目标物回收率仅以非常小的幅度增加,例如洗脱溶剂体积从4ml增至8ml时,大部分药物回收率提高不到5%。由此证明,提高洗脱溶剂的体积,不但没有大幅提高目标物回收率,反而增加了浓缩时间,造成目标物的损失,从而最终影响检验效果。综合三家实验数据,最终确定固相萃取洗脱体积定为4ml。
c.固相萃取-气质联用方法检出限
在最优条件下,利用固相萃取-气质联用方法对72种毒(药)物的检出限进行了系统性考查。济宁、青岛和潍坊三家平行进行操作。按照72种目标物各自的色谱峰(10≥s/n≥3)进行评判,得到三家实验室的72种目标物固相萃取检出限数据。表10为毒物检出限,表11为药物检出限。根据三家实验数据分析,在检出限以上浓度(含检出限)已知添加的目标物回收率均在50%~150%之间,完全满足准确定性的需要。
表10毒物hlb固相萃取柱检出限比较(μg/ml)
表11药物pep固相萃取柱检出限比较(μg/ml)
同液液萃取检出限情况一样,由于三家实验室仪器设备新旧不同,人员操作亦存在一定的差异,故固相萃取检出限浓度不尽相同。综合分析三家实验室的数据,最终确定了72种毒(药)物的固相萃取方法检出限浓度,见表12。
表1272种毒(药)物的固相萃取方法检出限表(μg/ml)
(4)液液萃取和固相萃取前处理方式的比较
表13液液萃取和固相萃取前处理方式的比较
本研究对液液提取和固相萃取两种前处理方式中影响提取率的重要因素进行了详细考查,并从多个层面对两者进行了比较。从表13可以看出,两种方法各有优缺点。在实际案件检验中,可根据待筛查目标物的性质及实验室前处理设备配置情况,选择最优方法以达到高效、快速、高灵敏筛查的目的。
(5)目标物检验过程中内标的使用
在实际检测工作中,血液中未知毒物筛查的阴性结果所占检案结果的比重更大。阴性结果的判定,现行通用的方法主要有两种。一种是在空白血中做目标物的标准物质添加,添加浓度一般接近该目标物的检出限浓度;另一种是在检血中添加一个或数个内标,通过内标的检出与否来判定阴性结果的可靠性。对于多种目标物的筛查,为保证定性结果准确性,往往需要添加若干种物质,操作起来费时费力。如果选择第二种方法,则可以大大简化操作步骤。运用此方法时,内标物的选择极为关键。按照惯例,内标物应当是一个能得到纯样的化合物,而且是样品中没有且投毒案(事)件中极为罕见,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质、色谱行为和响应特征,且在色谱分析条件下,内标物必须能与样品中各个待测目标物充分分离。
本研究结合实验及数据分析,将烯丙异丙巴比妥、skf525a和环氧七氯三种物质分别确定为酸性药物、中性和碱性药物及毒物的筛查内标。实际案件检验中,可依据前述标准溶液配制方法制备这三种物质的内标工作液,根据所需筛查毒(药)物种类,向检材或样本中添加检出限浓度的对应内标物质,然后按照前述“gc-ms条件与前处理方法”的步骤进行检测分析,即可实现对毒物筛查阴性结果的良好质控。
实验注意事项
本研究中选择了乙酸乙酯:苯(1:1)作为液液萃取的有机溶剂,直接定为6ml。主要是考虑到体积过小,有可能形成乳化,直接影响到目标物回收率;体积过大,又增加了浓缩的时间,从而影响分析效率。但具体的影响情况未做进一步考察。
考虑到毒物中有部分酸性和碱性目标物,故分别在酸性和碱性条件下各提取两次,实验证明目标物的加标回收率均能满足分析要求,所以未做进一步的提取次数考查。
由于三家单位实验室仪器设备型号、状况不同,所用色谱柱型号亦不相同,还有不同操作人员实验操作的差异,造成同一目标物的色谱峰保留时间在不同的实验室间存在细微差异,但72种毒(药)物出峰先后顺序,三家实验室均一致。故筛查目标物时,本研究给出的保留时间可作为参考依据。阳性结果定性时,要注重与标准物质的已知添加进行比对。
在液液萃取前处理方法中,由于血液中基质的干扰,部分目标物被基质峰所覆盖,这种情况往往在目标物浓度较低时出现。在进行谱图分析时,一是通过提取特征离子来寻找目标物;二是在确定了目标物色谱峰位置后,采取扣除背景的方式,能够准确地筛查出目标物,避免漏检。
该研究选择的72种毒(药)物涵盖了农药、鼠药、安眠镇静类药物、镇痛药、消炎药等种类,对毒品、其他人工合成药物等适合用gc-ms法进行分析的毒(药)亦可进行筛查。
结论
本研究对涉案毒物进行了系统调研总结,基本涵盖了法庭科学领域涉及的常见毒物,并且根据不同物质的性质,分别制定了气质联用定性筛查方法,极大程度地避免漏检发生。
针对气质联用法,优化并建立了血液中常见毒物的液液和固相提取方法,提高了目标物回收率,确立了两种定性筛查方法,并提供了两套完整的与不同提取方法对应的常见毒物在血液中的检出限数据。在未知毒物筛查案件中,可以根据每种定性筛查方法的检出限数据对阴性结果进行监控,从而快速、准确地进行定性评价。
附录a72种毒(药)物和内标的gc-ms参考参数
附录a1:毒物(以目标物保留时间先后为序排列)
附录a2:药物(以目标物保留时间先后为序排列)
- 还没有人留言评论。精彩留言会获得点赞!
- 用气相色谱‑正化学源‑质谱联用技术测定食用植物油中生育酚和生育三烯酚含量的方法与流程
- 一种超临界流体色谱‑气相色谱‑质谱测定卷烟主流烟气中苯并[a]芘的方法与流程
- 一种超临界流体色谱‑气相色谱‑质谱测定卷烟主流烟气中酚类化合物的方法与流程
- 一种用于气相催化裂解制备偏二氯乙烯的催化剂的方法与流程
- 一种气相色谱质谱联用检测水域中四溴双酚A的方法与流程
- 用于二恶英在线检测的气相色谱‑质谱间传输线系统的制造方法与工艺
- 一种气相色谱‑质谱法快速测定棉花纤维中4种有机氯农药残留的检测方法与流程
- 一种用于二恶英在线检测的气相色谱‑质谱间传输线系统的制造方法与工艺
- 一种检测纺织品中聚丙烯酸及其酯类聚合物的分析方法与流程
- 血液中未知毒物的气相色谱‑质谱筛查方法与流程