一种北芪菇中黄芪苷的提取纯化与检验方法与流程
本发明涉及北芪菇中活性物质的提取纯化与检验领域,具体为一种北芪菇中黄芪苷的提取纯化与检验方法。
背景技术:
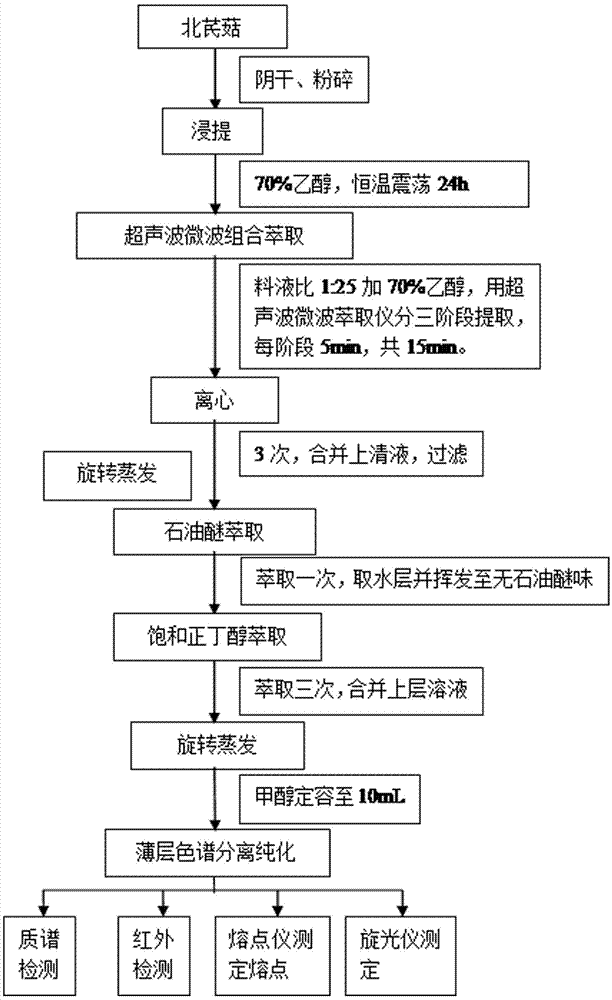
北芪菇,担子菌门下伞菌目侧耳属平菇,将正北芪、多种中草药、农作物副产品作培养料,采用科学的配方和特殊的工艺多年培育而成的新型侧耳属平菇,是山西浑源富硒地区开发的新型食用菌。根据生物富集作用,北芪菇内含浑源黄芪的药理成分,在生长发育过程中对黄芪苷有一定程度的富集。北芪菇中含有多种人体必需的氨基酸、多糖蛋白、抗生素、牛黄酸、菌糖、多种酶、维生素以及硒元素,是全能营养型的食用菌,其营养成份远超于普通平菇,含有较高的食用价值和药用价值。北芪菇具有较强的生物转化作用,其重要的代谢产物,如蛋白多糖、抗生素、微量牛黄酸有抗肿瘤、抗菌消炎、降低血压和防治脑血管障碍等作用,菌多糖和甘露糖有益于调理胃肠作用,以及含有帮助消化的各种酶等。在补气固表、润肤、脱毒排脓、有强身健体、保护肝脏等有较为显著的作用,尤其是在治愈银霄病和其它瘙痒性皮肤病有特殊的功效。另外,北芪菇是一种能产生新型氨基酸的微生物,其丰富的赖氨酸(1.19g/100g)和硒(0.54ppm/100g)可能与吡咯赖氨酸和l-硒代半胱氨酸或l-硒-甲基硒代半胱氨酸的生物合成存在着密切的关系,有望在抑制肿瘤、抗氧化、辅助治疗心血管疾病、解毒排毒等方面进行开发。近年我国对不同植物中的黄芪苷进行了各方面的研究,相继报道了黄芪苷有免疫调节、抗炎,降压,镇静镇痛,保肝护心,清除多种自由基等多方面的药理作用。关于黄芪中黄芪苷的提取及含量测定早有报道,但对产于山西浑源县北芪菇中黄芪苷的含量少有报道,如今北芪菇中黄芪苷的研究备受人们重视,有关黄芪苷的研究变成了科学界的热点。随着社会经济和科学技术的发展,黄芪苷的开发研究和应用范围会更广,开发前景会更好,从而促进医疗事业的发展。北芪菇中的黄芪苷以黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ和黄芪甲苷为主。黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ在抗氧化、调节免疫功能、保肝、抗病毒和对心脑血管等多方面具有显著作用。黄芪甲苷为环阿尔廷型三萜皂苷类化合物,白色至淡黄色粉末,mp29510~29610e,cas编号:83207-58-3,分子式为c41h68o14,分子量为784.9702,熔点295.0~296.0℃,易溶于甲醇、乙醇、丙酮,难溶于氯仿、乙酸乙酯等弱极性有机溶剂,生物活性高,是黄芪的主要药理成份,是黄芪及含黄芪制剂的重要鉴定指标。黄芪甲苷对人体拥有非常高的药理作用,其在器官保护(尤其是肝脏、肺、心肌、肾脏、脑等器官)、免疫调节、抗细胞凋亡、抗炎抗病毒、抗糖尿病、清除自由基、抗衰老等方面具有广泛的药理作用。黄芪苷的含量测定与检测曾尝试采用高效液相紫外检测器检测,但由于黄芪苷在紫外区仅有微弱的吸收,溶剂噪音对结果有较大影响,灵敏度低,测定误差大。索氏回流提取需要碱洗及大孔树脂富集过程,洗脱富集复杂,不易操作。此外,还有采用了水浸提法、碱水提取法、回流法、co2超临界流体萃取法等多种方法,但是这些方法均存在分离纯化量少,精度低,误差大的缺点,无法实现高精度的大量制备。为解决北芪菇中黄芪苷的提取和黄芪皂苷ⅰ、ⅱ、ⅲ和黄芪甲苷提取纯化的操作繁琐、结果不可靠的问题,本发明提供了一种易于操作、试验耗时短且提取效率高、实现从微克量级的分离分析到克量级的分离制备的北芪菇中黄芪苷提取纯化与检测的方法。技术实现要素:本发明的目的在于提供一种测定北芪菇中黄芪苷提取纯化和检验的方法,用以克服上述技术缺陷。本发明是通过如下技术方案来实现的:一种测定北芪菇中黄芪苷提取纯化和检验的方法,以北芪菇为原料,微波超声波萃取仪辅助处理,采用萃取法对黄芪苷进行粗提取,经薄层层析法分离纯化后,利用质谱和红外质谱检测标准品和供试品,测定旋光度和比旋光度,结晶后测定其熔点,其中,以70%的乙醇为提取剂,料液比为1:25,经微波超声波辅助萃取仪辅助处理后获得黄芪苷粗提取液,以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2为展开剂,经薄层层析展开分离纯化黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ,用色谱甲醇和超纯水作为流动相,用质谱仪测定样品分子质量信息(分子离子或碎片离子的质荷比),用红外检测其官能团,并分析其分子结构,利用旋光仪测定旋光度和比旋光度,待溶液蒸发结晶后测定熔点。进一步地,具体过程为:步骤a,以北芪菇为原料,加入70%乙醇,料液比为1:25,振荡浸提,超声波微波萃取仪分三阶段提取,每阶段5min,共15min,超声波频率为50khz,电机转速900r/min,提取液于4000r/min下离心10min,上清液过滤、浓缩,石油醚去杂后,用饱和正丁醇萃取,合并上层溶液、浓缩至无液体溢出,采用甲醇定容备用;步骤b,将黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ标准品分别加于色谱甲醇中,超声条件下充分溶解后制得2mg/ml的标准品溶液,置于2~4℃的冰箱中保存备用;步骤c,将层析板加热活化,用微量进样器吸取黄芪苷粗提液及标准品溶液25μl,依次点供试液20μl,标准品溶液2μl、4μl、6μl,以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,密封后,预饱和20min,放入层析缸中上行展开,待展开前沿距层析板顶端1cm处时,取出放入通风口处,均匀喷10%的硫酸乙醇溶液,加热5分钟后取出,用玻璃板覆盖,四周胶布固定,紫外365nm下检视,薄层色谱成像系统拍照,并标志;步骤d,用色谱甲醇和超纯水作为流动相,用微量进样器吸取10μl的标准品溶液,进样,对谱图进行分析、保存;将供试品层析板置于tlc-质谱接口上,使激光对准圈出的待测斑点中心处,取样,对谱图进行分析、保存;步骤e,打开傅里叶红外光谱仪,测试背景,用毛细管吸取标准品溶液于点样中心处,测试样品,对红外谱图进行处理分析,轻刮层析板上的待测斑点置于烧杯中,加入色谱甲醇,过滤取滤液,制得待测液,待测液的红外检测方法与标准品的检测相同;步骤f,校正旋光仪,分别倒入标准品、待测液,调节模式,测定旋光度和比旋光度,记录数据;步骤g,将待测液蒸发结晶后测定其熔点。与现有技术相比较本发明的有益效果在于:微波联合超声波提取比单一的超声波、微波提取效率更高,该方法不仅能使细胞内的目标物质充分溶解于提取剂中,不造成目标物质的损失,而且操作简单、用时短,大大的提高了提取效率,广泛的适用于各种物质的提取。薄层层析能实现对黄芪苷的分离纯化,使得黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ准确分离,ms和ir联用对其质荷比和所含官能团进行表征与分析,准确度高。tlc-ms-ir联用能实现从微克量级的分离分析到克量级的分离制备,可用于天然产物粗提物的去除杂质和单个产物的精制,适用于从北芪菇中进行物质的制备分离。附图说明图1为本发明的含量测定的流程图。图2为黄芪甲苷的结构式。图3为黄芪皂苷ⅰ标准品的结构式。图4为黄芪皂苷ⅱ标准品的结构式。图5为黄芪皂苷ⅲ标准品的结构式。图6为本发明的北芪菇供试品与标准品tlc成像图,图中1为黄芪皂苷ⅰ标准品,2为黄芪皂苷ⅱ标准品,3为黄芪皂苷ⅲ标准品,4为黄芪甲苷标准品。图7为本发明的正北芪供试品与标准品tlc成像图,图中5为黄芪皂苷ⅰ标准品,6为黄芪皂苷ⅱ标准品,7为黄芪皂苷ⅲ标准品,8为黄芪甲苷标准品。图8为黄芪甲苷标准品的质谱波谱图。图9为北芪菇中黄芪甲苷的质谱波谱图。图10为正北芪中黄芪甲苷的质谱波谱图。图11为黄芪甲苷标准品的红外图谱。图12为北芪菇中黄芪甲苷的红外波谱图。图13为正北芪中黄芪甲苷的红外波谱图。图14为黄芪皂苷ⅰ标准品的质谱波谱图。图15为北芪菇中黄芪皂苷ⅰ的质谱波谱图。图16为正北芪中黄芪皂苷ⅰ的质谱波谱图。图17为黄芪皂苷ⅱ标准品的质谱波谱图。图18为北芪菇中黄芪皂苷ⅱ的质谱波谱图。图19为正北芪中黄芪皂苷ⅱ的质谱波谱图。图20为黄芪皂苷ⅲ标准品的质谱波谱图。图21为北芪菇中黄芪皂苷ⅲ的质谱波谱图。图22为正北芪中黄芪皂苷ⅲ的质谱波谱图。图23黄芪皂苷ⅰ标准品的红外图谱。图24北芪菇中黄芪皂苷ⅰ的红外波谱图。图25正北芪中黄芪皂苷ⅰ的红外波谱图。图26黄芪皂苷ⅱ标准品的红外图谱。图27北芪菇中黄芪皂苷ⅱ的红外波谱图。图28正北芪中黄芪皂苷ⅱ的红外波谱图。图29为黄芪皂苷ⅲ标准品的红外图谱。图30为北芪菇中黄芪皂苷ⅲ的红外波谱图。图31为正北芪中黄芪皂苷ⅲ的红外波谱图。具体实施方式以下结合附图,对本发明上述的和另外的技术特征和优点作更详细的说明。本发明以北芪菇为原料,微波超声波萃取仪辅助处理,采用萃取法对黄芪苷进行粗提取,经薄层层析法分离纯化后,利用质谱和红外质谱检测标准品和供试品,测定旋光度和比旋光度,结晶后测定其熔点,其中,以70%的乙醇为提取剂,料液比为1:25,经微波超声波辅助萃取仪辅助处理后获得黄芪苷粗提取液,以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2为展开剂,经薄层层析展开分离纯化黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ,用色谱甲醇和超纯水作为流动相,用质谱仪测定样品分子质量信息(分子离子或碎片离子的质荷比),用红外检测其官能团,并分析其分子结构,利用旋光仪测定旋光度和比旋光度,待溶液蒸发结晶后测定熔点。请参阅图1所示,具体过程为:步骤a,以北芪菇为原料,加入70%乙醇,料液比为1:25,振荡浸提,超声波微波萃取仪分三阶段提取,每阶段5min,共15min,超声波频率为50khz,电机转速900r/min,提取液于4000r/min下离心10min,上清液过滤、浓缩,石油醚去杂后,用饱和正丁醇萃取,合并上层溶液、浓缩至无液体溢出,采用甲醇定容备用;步骤b,将黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ标准品分别加于色谱甲醇中,超声条件下充分溶解后制得2mg/ml的标准品溶液,置于2~4℃的冰箱中保存备用;步骤c,将层析板加热活化,用微量进样器吸取黄芪苷粗提液及标准品溶液25μl,依次点供试液20μl,标准品溶液2μl、4μl、6μl,以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,密封后,预饱和20min,放入层析缸中上行展开,待展开前沿距层析板顶端1cm处时,取出放入通风口处,均匀喷10%的硫酸乙醇溶液,加热5分钟后取出,用玻璃板覆盖,四周胶布固定,紫外365nm下检视,薄层色谱成像系统拍照,并标志;步骤d,用色谱甲醇和超纯水作为流动相,用微量进样器吸取10μl的标准品溶液,进样,对谱图进行分析、保存;将供试品层析板置于tlc-质谱接口上,使激光对准圈出的待测斑点中心处,取样,对谱图进行分析、保存;步骤e,打开傅里叶红外光谱仪,测试背景,用毛细管吸取标准品溶液于点样中心处,测试样品,对红外谱图进行处理分析,轻刮层析板上的待测斑点置于烧杯中,加入色谱甲醇,过滤取滤液,制得待测液,待测液的红外检测方法与标准品的检测相同;步骤f,校正旋光仪,分别倒入标准品、待测液,调节模式,测定旋光度和比旋光度,记录数据;步骤g,将待测液蒸发结晶后测定其熔点。下面通过实施例进行说明。实施例1步骤a,黄芪苷的粗提取:将北芪菇于通风处阴干至材料发脆,恒温箱内(40~50℃)烘干,用高速多功能粉碎机粉碎,过80目筛筛选,装入玻璃器皿中保存,准确称取10.0g于500ml的锥形瓶中分别加入250ml的70%乙醇,50℃下气浴恒温振荡器中振荡浸提24h,充分浸泡溶解形成粗提浸提液;超声波微波萃取仪分三阶段提取,每阶段5min,共15min,根据表1选取最佳微波超声波条件,在超声波恒定的条件下萃取,设置温度为45℃、50℃、55℃,最终测得超声波功率为800、900、1000w,微波功率为250、300、350w为佳,超声波频率为50khz,模式15:10,电机转速900r/min,提取液于4000r/min下离心10min,反复提取3次,合并上清液过滤、浓缩至体积的1/10;表1微波及超声波参数用2倍体积的石油醚萃取水提取液,保留水相,去杂后,待水相挥发至无石油醚味后,用等体积的饱和正丁醇萃取3次,合并上层正丁醇相溶液,添加纯水使其形成共沸物后于旋转蒸发仪减压浓缩干燥,设置水浴温度为80℃,回收正丁醇,浓缩至无液体溢出时用色谱纯甲醇将其定容至10ml,作为北芪菇供试品溶液备用;步骤b,黄芪甲苷标准品的制备:精确称量20mg黄芪甲苷标准品加色谱纯甲醇定容于用超声洗净的10ml容量瓶中,超声条件下充分溶解后得到2mg/ml的黄芪甲苷标准品溶液,并将其置于2~4℃的冰箱中保存备用;步骤c,薄层层析分离纯化黄芪甲苷:点样:用平头微量进样器分别吸取北芪菇、正北芪供试品溶液及黄芪甲苷标准品溶液25μl,用全自动点样机依次点供试液20μl,黄芪甲苷标准品溶液2μl、4μl、6μl,点样参数为点样数为4、点样宽度8mm、点样间距10mm、点样速度5s/μl);展开:以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,以氯仿:甲醇:水=13:7:2静置过夜的下相为正北芪供试品展开剂,密封后,预饱和20min使展开剂蒸汽在层析缸中均匀分布,将点样后的聚酰胺层析板以约75°倾角放入展开剂液面高约2-3mm高的平卧式层析缸内,上行展开,展开剂不浸没点样斑点,以防点样物质在展开剂中溶解,待展开前沿距层析板顶端1cm处时,取出层析板;显色:将层析板晾干后,用显色喷雾瓶向层析板均匀喷10%的硫酸乙醇溶液,使之显色,于105-110℃电热鼓风干燥箱加热至显色清晰为止。用同样大小的玻璃板覆盖,周围用胶布固定,减慢氧化速度;检视:将显色后的层析板放入薄层色谱成像系统,于紫外波长365nm下对层析板检视,观察对比标准品位置,rf值相同,呈橙黄色荧光斑点的为北芪菇和正北芪样品中的黄芪甲苷,薄层色谱成像系统拍照,并标志;步骤d,质谱检测:黄芪甲苷标准品的质谱检测:用色谱甲醇和超纯水作为流动相,将expressions质谱仪和软件打开使其处于工作状态,用微量进样器吸取10μl的黄芪甲苷标准品溶液插入到进样口处,打到load档开始进样,进完样后打回到inject档。点开谱图界面对谱图进行分析并保存。待测液的质谱检测:用色谱甲醇和超纯水作为流动相,使高分辨质谱仪和软件处于工作状态,将供试品层析板置于tlc-质谱接口上,使激光对准圈出的黄芪甲苷斑点中心处,用软件操作进行取样,点开谱图界面对谱图进行分析并保存。步骤e,红外检测:黄芪甲苷标准品的红外检测:打开傅里叶红外光谱仪选择相应的工作界面,用擦镜纸擦干净点样中心处,,测试背景,用毛细管吸取少量的黄芪甲苷标准品溶液于点样中心处,测试样品,对谱图界面出现的红外谱图进行处理分析。待测液的红外检测:用小刀轻刮层析板上的画出的待测黄芪甲苷斑点置于烧杯中,画出的待测黄芪甲苷斑点的rf值与黄芪甲苷标准品相同。在烧杯中加入10ml色谱甲醇,过滤取滤液,制得待测液,打开傅里叶红外光谱仪选择相应的工作界面,用擦镜纸擦干净点样中心处,测试背景,用毛细管吸取少量的待测液于点样中心处,测试样品,对谱图界面出现的红外谱图进行处理分析。步骤f,旋光仪进行物理表征检测:用甲醇校正旋光仪,分别倒入标准品、待测液于旋光仪,调节模式,测定旋光度和比旋光度,记录数据。步骤g,熔点的测定:将待测液蒸发结晶,利用测定其熔点。结果分析:1.黄芪甲苷薄层色谱分析:以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,以氯仿:甲醇:水=13:7:2静置过夜的下相为正北芪供试品展开剂,供试品及标准品溶液经点样展开并显色后,日光下北芪菇供试品、正北芪供试品在与标准品在相应的位置上显相同的棕褐色斑点,在紫外(365nm)检视下,图6、7可见,北芪菇供试品、正北芪供试品色谱在与黄芪甲苷标准品图谱相应的位置上都呈现橙黄色荧光斑点,且荧光斑点位置一致,其rf值均为0.31。由此可以判断,北芪菇和正北芪中均含有黄芪甲苷。2.黄芪甲苷质谱检测分析:分子离子峰的识别首先是该离子峰范围内最大的峰值(m/z),其次需要判断该物质是否符合氮律,不含n或含偶数n的有机分子,其分子离子峰的m/z(即分子量)为偶数。含奇数n的有机分子,其分子离子峰的m/z(即分子量)为奇数,这两点是识别离子峰的关键。参照图2,黄芪甲苷为环阿尔廷型三萜皂苷类化合物,分子式为c41h68o14,分子量为784.97,且结构式中不含n。由图8-10可知,可以观察到该离子峰范围内的最大峰值为807.6,与黄芪甲苷的分子量相差约等于23,表示该物质在电子的轰击下增加了一个na离子[m+na+=807.97],这为合理性的增加,所以可以确定黄芪甲苷的离子峰为807.6。由图8-10可知,有相同的离子峰,这就可以确定从薄层板中刮出来的物质可能是黄芪甲苷。3.黄芪甲苷红外质谱检测分析:观察图11-13可知,可以看到三者在1600-1700cm-1之间有相似的峰,说明都含有苯环;在3000-3200cm-1之间三者有相似的峰,说明都含有羟基;在2900-3000cm-1之间有相似的峰,说明都含有-c-h单键;在1380cm-1附近有相似的峰,说明都含有-ch3键;在1000-1500cm-1之间有相似的峰,说明都含有c-o单键;在700-1000cm-1之间有相似的峰,说明三者都具有=c-h弯曲振动吸收峰,参照图2,这些峰值基本符合黄芪甲苷结构的特征峰,所以可以确定北芪菇中可能含有黄芪甲苷。4.黄芪甲苷的旋光度、比旋光度分析参考表2,测得的黄芪甲苷标准品与北芪菇待测液基本一致,由此进一步确认北芪菇中分离纯化得到的待测液为黄芪甲苷。表2黄芪甲苷的旋光度、比旋光度旋光度测定值旋光度平均值比旋光度黄芪甲苷标准品+55.7/+55.2/+55.4/+56.2/+54.2/+56.1+55.5+24.1北芪菇待测液+56.9/+55.8/+56.4/+55.2/+54.9/+55.7+55.8+24.45.黄芪甲苷的熔点分析参考表3,北芪菇样品与黄芪甲苷标准品的平均熔点均为295℃,在熔点295.0~296.0℃范围内,而且易溶于甲醇、乙醇、丙酮等极性溶剂,进一步确认根据本发明的方法从北芪菇中提取纯化的物质为黄芪甲苷。表3黄芪甲苷的熔点熔点测定值℃熔点平均值℃黄芪甲苷标准品295/296/294/295北芪菇待测液296/295/294295结论:以北芪菇为原料,70%乙醇为提取剂,微波超声波萃取仪分三阶段提取,每阶段5min,共15min。在超声波恒定的条件下,设置温度为45℃、50℃、55℃,超声波功率为800、900、1000w,微波功率为250、300、350w,超声波频率为50khz,模式15:10,电机转速900r/min,经多级萃取后获得黄芪甲苷粗提取,以氯仿:甲醇:乙酸乙酯:水=13:7:2:2(v/v)为展开剂,薄层层析获得的黄芪甲苷,荧光斑点显色清晰,与黄芪甲苷标准品对比荧光斑点位置一致,其rf值为0.31。质谱图谱显示:北芪菇样品所测的m/z与黄芪甲苷标准品基本保持一致,m/z均为807.6,与黄芪甲苷的分子量相差约23,表示该物质在电子的轰击下增加了一个na离子[m+na+=807.97],为合理性的增加。将层析板上荧光斑点刮下,制得溶液,进行红外质谱分析,红外谱图显示:北芪菇所测样品所含官能团与黄芪甲苷标准品基本相同。北芪菇样品与黄芪甲苷标准品熔点均为295℃,在熔点295.0~296.0℃范围内,而且易溶于甲醇、乙醇、丙酮等极性溶剂。黄芪甲苷的旋光度:-56.6(c,0.13indmf),与北芪菇所测样品基本一致。实施例2步骤a,黄芪苷的粗提取:将北芪菇于通风处阴干至材料发脆,恒温箱内(40~50℃)烘干,用高速多功能粉碎机粉碎,过80目筛筛选,装入玻璃器皿中保存,准确称取10.0g于500ml的锥形瓶中分别加入250ml的70%乙醇,50℃下气浴恒温振荡器中振荡浸提24h,充分浸泡溶解形成粗提浸提液;超声波微波萃取仪分三阶段提取,每阶段5min,共15min,根据表4选取最佳微波超声波条件,在超声波恒定的条件下萃取,设置温度为45℃、50℃、55℃,最终测得超声波功率为800、900、1000w,微波功率为250、300、350w为佳,超声波频率为50khz,模式15:10,电机转速900r/min,提取液于4000r/min下离心10min,反复提取3次,合并上清液过滤、浓缩至体积的1/10;表4微波及超声波参数用2倍体积的石油醚萃取水提取液,保留水相,去杂后,待水相挥发至无石油醚味后,用等体积的饱和正丁醇萃取3次,合并上层正丁醇相溶液,添加纯水使其形成共沸物后于旋转蒸发仪减压浓缩干燥,设置水浴温度为80℃,回收正丁醇,浓缩至无液体溢出时用色谱纯甲醇将其定容至10ml,作为北芪菇供试品溶液备用;步骤b,黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ标准品的制备:精确称量20mg黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ标准品分别加色谱纯甲醇定容于用超声洗净的10ml容量瓶中,超声条件下充分溶解后得到2mg/ml的黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ标准品溶液,并将其置于2~4℃的冰箱中保存备用;步骤c,薄层层析分离纯化黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ:活化:将100x100mm的硅胶gh254层析板,于110~115℃的电热鼓风干燥箱中加热活化20~30min,除去薄层板吸附的水分子,增强其吸附能力;点样:用平头微量进样器分别吸取北芪菇、正北芪供试品溶液及黄芪皂苷ⅰ标准品溶液25μl,用全自动点样机依次点供试液20μl,黄芪皂苷ⅰ标准品溶液2μl、4μl、6μl,点样参数为点样数为4、点样宽度8mm、点样间距10mm、点样速度5s/μl);黄芪皂苷ⅱ、黄芪皂苷ⅲ点样方法同上。展开:以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,以氯仿:甲醇:水=13:7:2静置过夜的下相为正北芪供试品展开剂,密封后,预饱和20min使展开剂蒸汽在层析缸中均匀分布,将点样后的硅胶g层析板以约75°倾角放入展开剂液面高约2-3mm高的平卧式层析缸内,上行展开,展开剂不浸没点样斑点,以防点样物质在展开剂中溶解,待展开前沿距层析板顶端1cm处时,取出层析板;显色:将层析板晾干后,用显色喷雾瓶向硅胶板均匀喷10%的硫酸乙醇溶液,使之显色,于105-110℃电热鼓风干燥箱加热至显色清晰为止。用同样大小的玻璃板覆盖,周围用胶布固定,减慢氧化速度;检视:将显色后的层析板放入薄层色谱成像系统,于紫外波长365nm下对层析板检视,观察对比黄芪皂苷ⅰ、ⅱ、ⅲ标准品位置,rf值相同,呈橙黄色荧光斑点的为北芪菇和正北芪样品中的黄芪皂苷ⅰ、黄芪皂苷ⅱ、黄芪皂苷ⅲ,薄层色谱成像系统拍照,并标志;步骤d,质谱检测:黄芪皂苷ⅰ标准品的质谱检测:用色谱甲醇和超纯水作为流动相,将expressions质谱仪和软件打开使其处于工作状态,用微量进样器吸取10μl的黄芪皂苷ⅰ标准品溶液插入到进样口处,打到load档开始进样,进完样后打回到inject档。点开谱图界面对谱图进行分析并保存。待测液的质谱检测:用色谱甲醇和超纯水作为流动相,使高分辨质谱仪和软件处于工作状态,将供试品层析板置于tlc-质谱接口上,使激光对准圈出的黄芪皂苷ⅰ斑点中心处,用软件操作进行取样,点开谱图界面对谱图进行分析并保存。黄芪皂苷ⅱ和黄芪皂苷ⅲ质谱检验方法同上。步骤e,红外检测:黄芪皂苷ⅰ标准品的红外检测:打开傅里叶红外光谱仪选择相应的工作界面,用擦镜纸擦干净点样中心处,测试背景,用毛细管吸取少量的黄芪皂苷ⅰ标准品溶液于点样中心处,测试样品,对谱图界面出现的红外谱图进行处理分析。待测液的红外检测:用小刀轻刮层析板上的画出的待测黄芪皂苷ⅰ斑点置于烧杯中,画出的待测黄芪皂苷ⅰ斑点的rf值与黄芪皂苷ⅰ标准品相同。在烧杯中加入10ml色谱甲醇,过滤取滤液,制得待测液,打开傅里叶红外光谱仪选择相应的工作界面,用擦镜纸擦干净点样中心处,测试背景,用毛细管吸取少量的待测液于点样中心处,测试样品,对谱图界面出现的红外谱图进行处理分析。黄芪皂苷ⅱ和黄芪皂苷ⅲ红外检验方法同上。步骤f,旋光仪进行物理表征检测:用甲醇校正旋光仪,分别倒入标准品、待测液于旋光仪,调节模式,测定旋光度和比旋光度,记录数据。步骤g,熔点的测定:将待测液蒸发结晶,利用测定其熔点。结果分析:1.黄芪皂苷ⅰ、ⅱ、ⅲ薄层色谱分析:以氯仿:甲醇:乙酸乙酯:水的体积比13:7:2:2静置过夜的下相为北芪菇供试品展开剂,以氯仿:甲醇:水=13:7:2静置过夜的下相为正北芪供试品展开剂,供试品及标准品溶液经点样展开并显色后,日光下北芪菇供试品、正北芪供试品在与标准品在相应的位置上显相同的棕褐色斑点,在紫外(365nm)检视下,图6、7可见,北芪菇供试品、正北芪供试品色谱在与黄芪皂苷ⅰ、ⅱ、ⅲ标准品图谱相应的位置上都呈现橙黄色荧光斑点,且荧光斑点位置一致,黄芪皂苷ⅰ、ⅱ、ⅲ的rf值分别为0.71、0.54、0.34。由此可以判断,北芪菇和正北芪中均含有黄芪皂苷ⅰ、ⅱ、ⅲ。2.质谱检测分析:黄芪皂苷ⅰ的质谱检测分析:黄芪皂苷ⅰ的分子离子峰大致在m/z=784.9,根据图14-16对比分析,可知m/z=742.3的离子峰为该图谱最高质量的离子峰,且质荷比为偶数,为奇电子离子,参考图3,由于黄芪皂苷ⅰ不包含氮原子,故其满足氮规则;假设此峰为分子离子峰,那其与碎片离子峰之间质荷比的差值δm=784.9-742.3≈43就表示该物质在离子源轰击下碎裂丢失了一个-ch3co,即产生了合理的碎片丢失,则该假设成立。因此可推断出m/z=742.3可能为黄芪皂苷ⅰ的分子离子峰。由图14-16可知,有相同的离子峰,这就可以确定从层析板中刮出来的物质是黄芪皂苷ⅰ。黄芪皂苷ⅱ的质谱检测分析:黄芪皂苷ⅱ的分子离子峰大致范围在m/z=827.0,对比分析图17-19,可知m/z=825.8的离子峰为该图谱最高质量的离子峰,且为偶数,参考图4,由于黄芪皂苷ⅱ不包含氮原子,故其满足氮规则;假设此峰为分子离子峰,那其与碎片离子峰之间质荷比的差值△m=827.0-825.8≈1就表示该物质在离子源中碎裂丢失了一个h+,即产生了合理的碎片丢失,该假设成立。因此可推断出m/z=825.8可能为黄芪皂苷ⅱ的分子离子峰。由图17-19可知,有相同的离子峰,这就可以确定从层析板中刮出来的物质是黄芪皂苷ⅱ。黄芪皂苷ⅲ的质谱检测分析:黄芪皂苷ⅲ的分子离子峰大致范围在m/z=785.0左右,对比分析图20-22,可知m/z=768.1的离子峰为该图谱最高质量的离子峰,且为偶数,参考图5由于黄芪皂苷ⅲ不包含氮原子,故其满足氮规则;假设此峰为分子离子峰,那其与碎片离子峰之间质荷比的差值△m=785.0-768.1≈17就表示该物质在离子源轰击下碎裂丢失了一个-oh,即产生了合理的碎片丢失,则该假设成立。因此可推断出m/z=768.1可能为黄芪皂苷ⅲ的分子离子峰。由图20-22可知,有相同的离子峰,这就可以确定从层析板中刮出来的物质是黄芪皂苷ⅲ。3.红外质谱检测分析;黄芪皂苷ⅰ红外质谱检测分析:对比分析图23-25,三者分别在3100-3000cm-1之间出现相似波峰,为不饱和c-h伸缩振动吸收,有可能为烯、炔、芳香化合物:在c=c骨架振动区1600-1450cm-1内出现强烈波峰,证明其均含苯环;在3391处都出现清晰波峰,证明其均含羟基;在1657附近都呈现出明显波峰,证明其均含c=o键;在1000-1300之内出现波峰,证明其均含芳香醚。根据图3分析显示,北芪菇和正北芪中均含有黄芪皂苷ⅰ的官能团,符合黄芪皂苷ⅰ结构的特征峰,证明均分离纯化出了黄芪皂苷ⅰ。黄芪皂苷ⅱ红外质谱检测分析:对比分析图26-28,三者分别在3100-3000cm-1之间出现相似波峰,为不饱和c-h伸缩振动吸收,有可能为烯、炔、芳香化合物:在c=c骨架振动区1600-1450cm-1内出现强烈波峰,证明其均含苯环;在3391处都出现清晰波峰,证明其均含羟基;在1657附近都呈现出明显波峰,证明其均含c=o键;在1000-1300之内出现波峰,证明其均含芳香醚。根据图4分析显示,北芪菇和正北芪中均含有黄芪皂苷ⅱ的官能团,符合黄芪皂苷ⅱ结构的特征峰,证明均可能分离纯化出了黄芪皂苷ⅱ。黄芪皂苷ⅲ红外质谱检测分析:对比分析图29-31,三者分别在3100-3000cm-1之间出现相似波峰,为不饱和c-h伸缩振动吸收,有可能为烯、炔、芳香化合物:在c=c骨架振动区1600-1450cm-1内出现强烈波峰,证明其均含苯环;在3391处都出现清晰波峰,证明其均含羟基;在1657附近都呈现出明显波峰,证明其均含c=o键;在1000-1300之内出现波峰,证明其均含芳香醚。根据图5分析显示,北芪菇和正北芪中均含有黄芪皂苷ⅲ的官能团,符合黄芪皂苷ⅲ结构的特征峰,证明均可能分离纯化出了黄芪皂苷ⅲ。4.北芪菇中黄芪皂苷的旋光度、比旋光度分析:测得的北芪菇中黄芪皂苷ⅰ、ⅱ、ⅲ旋光度如下表所示,进一步确认北芪菇中分离纯化得到的待测液为黄芪皂苷。表5黄芪皂苷ⅰ、ⅱ、ⅲ的旋光度、比旋光度旋光度测定值旋光度平均值比旋光度黄芪皂苷ⅰ+24.6/+24.2/+24.4/+24.5/+24.3/+24.4+24.4+10.6黄芪皂苷ⅱ+30.4/+30.3/+30.4/+30.5/+30.4/+30.4+30.4+13.2黄芪皂苷ⅲ+21.2/+21.4/+21.2/+21.6/+21.4/+21.6+21.4+9.35.黄芪皂苷ⅰ、ⅱ、ⅲ的熔点分析测得的北芪菇中黄芪皂苷ⅰ、ⅱ、ⅲ熔点如下表所示,进一步确认北芪菇中分离纯化得到的待测液为黄芪皂苷ⅰ、ⅱ、ⅲ。表6黄芪皂苷ⅰ、ⅱ、ⅲ的熔点熔点测定值℃熔点平均值℃黄芪皂苷ⅰ228/230/229229黄芪皂苷ⅱ246/248/247248黄芪皂苷ⅲ248/244/246246结论:以北芪菇为原料,70%乙醇为提取剂,微波超声波萃取仪分三阶段提取,每阶段5min,共15min。在超声波恒定的条件下,设置温度为45℃/50℃/55℃,超声波功率为800/900/1000w,微波功率为250/300/350w,超声波频率为50khz,模式15:10,电机转速900r/min,经多级萃取后获得黄芪皂苷粗提取,以氯仿:甲醇:乙酸乙酯:水=13:7:2:2(v/v)为展开剂,薄层层析获得的黄芪皂苷ⅰ、ⅱ、ⅲ,荧光斑点显色清晰,与黄芪皂苷ⅰ、ⅱ、ⅲ标准品对比荧光斑点位置一致,在硅胶板上展开时黄芪皂苷ⅰ距离点样原点最远,跑的最快,说明其极性最小,黄芪皂苷ⅱ比黄芪皂苷ⅲ极性小,但比黄芪皂苷ⅰ极性大,黄芪皂苷ⅰ、ⅱ、ⅲ的rf值分别为0.71、0.54、0.34。经由红外及高分辨质谱仪化学表征检测,观察其与黄芪皂苷ⅰ、ⅱ、ⅲ所含官能团保持一致,且均为其特征官能团,由此进一步分析可能纯化获得黄芪皂苷ⅰ、ⅱ、ⅲ单体,实现从微克量级的分离分析到克量级的分离制备,利用熔点测定器测定其熔点,自动旋光仪检测其旋光度与比旋光度,黄芪皂苷样品ⅰ、ⅱ、ⅲ的熔点分别为229℃、248℃、246℃,旋光度分别为+24.4、+30.4、+21.4,比旋光度分别为+10.6、+13.2、+9.3。本发明选定以乙醇作为本实验的提取溶剂,微波联合超声波提取比单一的超声波或单一的微波提取效率更高,该方法不仅能使细胞内的目标生物活性物质充分溶解于提取剂中,不造成待提取的目标物质的损失,而且操作简单、用时短,能有效提高提取效率,广泛的适用于各种生物活性物质的提取。经次优化试验条件,采取tlc-ms-ir联用分离纯化、检测北芪菇中黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ,其分离效果较好,实现从微克量级的分离分析到克量级的分离制备,是一种有效的提取北芪菇中黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ的方法,为黄芪甲苷、黄芪皂苷ⅰ、ⅱ、ⅲ的标准品的制备检验提供了一种新的快速有效的方法。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1