一种环状RNA的高通量芯片数据处理及分析流程控制方法与流程
本发明设计医学基因组学和计算生物学领域,具体涉及一种环状RNA的高通量芯片数据处理及分析流程控制方法。
背景技术:
环状RNA(circular RNA,circRNA)是最近发现的一类特殊的非编码RNA,它大量存在于真核细胞胞质内,主要由pre-mRNA通过可变剪切加工产生。具有闭合环状结构,大部分的环状RNA是由外显子序列构成,在不同的物种中具有保守性,同时存在组织及不同发育阶段的表达特异性。由于环状RNA对核酸酶不敏感,所以比线性RNA更为稳定,这使得环状RNA在作为新型临床诊断标记物的开发应用上具有明显优势。然而使用环状RNA作为标记物也存在诸多问题,例如大量的环状RNA被鉴定出来,但是其功能机制都了解的太少,给科研工作者带来了挑战和困难,尤其是面对高通量大数据的时候。如何分析环状RNA数据,研究其潜在功能成为该领域目前急需解决的问题。
技术实现要素:
本发明的目的是提供一种环状RNA的高通量芯片数据处理及分析流程控制方法,以解决现有的技术对circRNA高通量芯片数据处理中的不准确性、以及不懂如何分析circRNA等问题。
为实现上述目的,本发明采用的技术方案为:
一种环状RNA的高通量芯片处理及分析流程控制方法,包括如下步骤:
步骤1,自定义参数配置文件的生成:导入环状RNA高通量原始芯片数据,经过信号值筛选和标准化得到理论上有效的环状RNA,在此基础上进行生物信息学参数分析;
步骤2,输入步骤:用户根据需要,输入设定的各参数配置文件;
步骤3,分析步骤:根据上述步骤输入设定的参数配置文件,通过环状RNA高通量数据处理流程模块生成对应的自动化分析流程;
步骤4,执行及输出步骤:执行上述步骤所生成的自动化分析流程,获得并输出环状RNA分析结果报告。
优选的,所述的步骤1具体包括如下步骤:
步骤1.1,导入环状RNA高通量芯片原始信号值文件;
步骤1.2,对所述的环状RNA高通量芯片原始信号文件进行质量分析并剔除低质量信号数据,获得经过筛选的信号数据;
步骤1.3,将所述的经过筛选的数据进行前景值和背景值校正,得到消除噪音污染的环状RNA信号数据;
步骤1.4,将校正过的信号数据进行标准化,并去除极值,得到理论上有效的环状RNA表达值。
优选的,所述的步骤1.2中,所述低质量信号数据是指扫描微阵列芯片荧光强度作为RNA表达信号值且荧光强度小于30的数据,同一探针的重复信号数据采用中位数计算法取中位值作为该探针的表达值。
优选的,所述的步骤1.3中,使用针对Affymetrix芯片原理设计的Affy软件包中的MAS5或者RMA方法根据不同的芯片类型进行芯片数据预处理,其中不同的芯片类型是指单、双色通道;MAS5得到的数据是原始信号强度,RMA得到的是经过对数变换的信号值。
优选的,所述的步骤1.4中,使用limma软件包进行芯片间归一化,得到标准化的环状RNA表达谱数据。
优选的,所述的步骤1中,生物信息学参数分析包括差异表达环状RNA的筛选,环状RNA的功能性分析和对环状RNA的调控机制分析。
优选的,所述的差异表达环状RNA的筛选包括输入指令选取1.5倍或者2倍的差异倍数,选用三个标准Benjamini–Hochberg方法、FDR方法或者Bonforroni方法校正P-value得到差异表达的环状RNA。
优选的,所述的环状RNA的功能性分析包括环状RNA和基因数据的共表达分析,基因本体分析,代谢通路分析,化学反应分析和调控网络的构建;
其中,所述的环状RNA和基因数据的共表达分析采用Pearson相关系数法或Spearman相关系数法;
所述的基因本体分析采用g:Profiler法从生物过程、分子功能和细胞组分三个成分进行注释和富集分析;
所述的代谢通路分析和化学反应采用g:Profiler法通过KEGG和Reactive数据库信息进行分析。
优选的,所述的对环状RNA的调控机制分析包括miRNA结合位点预测,靶基因预测和竞争性内源RNA调控网络的构建;
其中,所述的microRNA结合位点预测采用TargetScan算法和Miranda算法;
所述的靶基因预测采用miRWalk和TargetScan数据库信息。
有益效果:利用本发明,将环状RNA各分析步骤模块分和流程分,能够单独运行一个模块或流程中的局部分析模块,并进行模块内规定数据分析流程的快速执行。从而通过不同模块的选取,帮助科研人员迅速完成一套高通量数据的前期数据质控、功能分析和结果报告。该工具能够优化生物信息分析人员和科研人员的工作时间,显著提高工作效率,降低科研成本,本发明的分析流程思路清晰,其实现方法简单,可广泛应用于生物学研究工作中,也可用于临床相关应用。
本发明的方法首先由系统生成自定义参数配置文件,再根据用户设定参数后的自定义参数文件和高通量数据处理流程模块生成与数据流程对应的批处理可执行文件;由系统执行批处理可执行文件,实现数据流程自动化,最终生成结果报告文件。从而能高效的帮助生物信息分析人员完成一套标准化的高通量数据分析流程,甚至可以让不懂高通量数据分析的科研人员自己完成高通量数据分析。从而可以达到优化科研人员的工作效率,降低科研成本的目的。本发明不仅仅可以用于环状RNA高通量数据分析流程,也可用于其他非编码RNA例如lncRNA等高通量芯片分析流程,甚至在任何物种中通用,其实现方法简单,应用范围较为广泛。
附图说明
图1是环状RNA自动化分析流程;
图2是环状RNA生物信息学分析步骤;
图3是环状RNA-共表达基因网络示意图;
图4是环状RNA生物通路富集调控示意图;
图5是环状RNA的竞争性内源RNA调控网络示意图。
具体实施方式
下面结合附图对本发明作更进一步的说明。
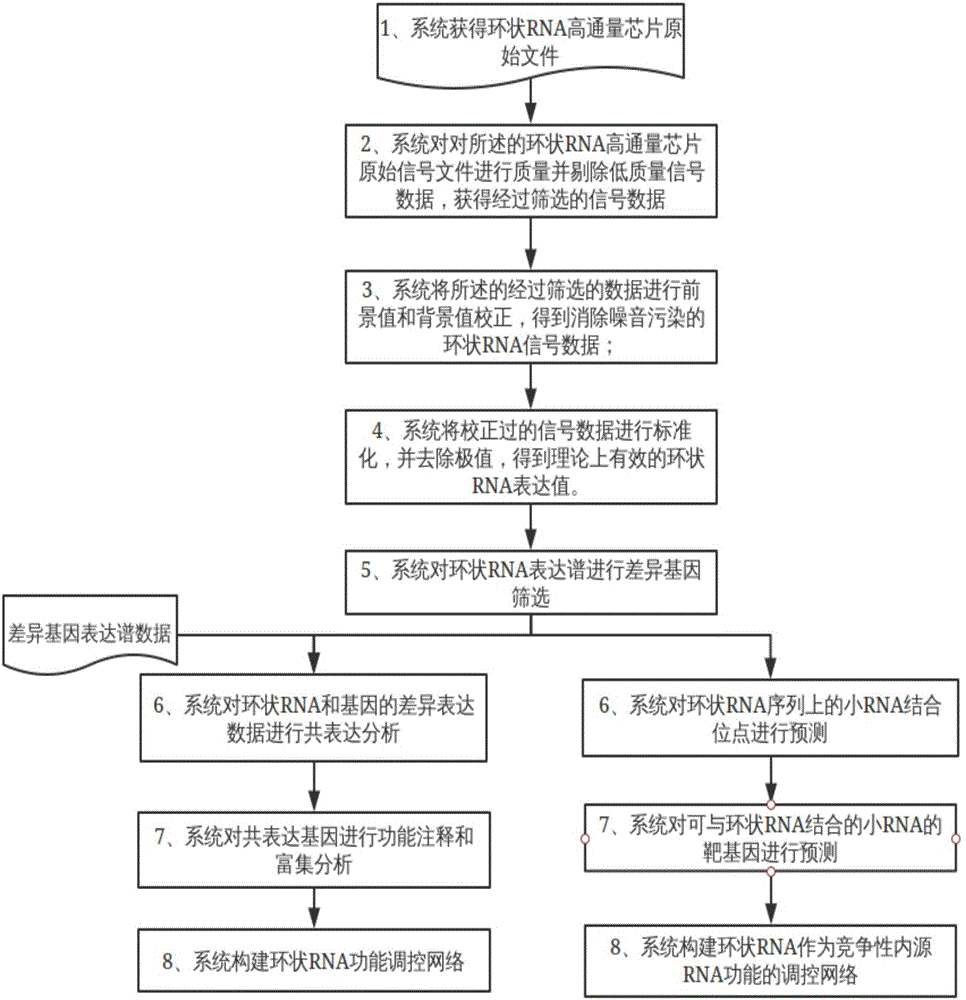
如图1所示,本发明的一种环状RNA的高通量芯片处理及分析流程控制方法,包括如下步骤:
步骤1,自定义参数配置文件的生成:导入环状RNA高通量原始芯片数据,经过信号值筛选和标准化得到理论上有效的环状RNA,在此基础上进行生物信息学参数分析;
步骤2,输入步骤:用户根据需要,输入设定的各参数配置文件;
步骤3,分析步骤:根据上述步骤输入设定的参数配置文件,通过环状RNA高通量数据处理流程模块生成对应的自动化分析流程;
步骤4,执行及输出步骤:执行上述步骤所生成的自动化分析流程,获得并输出环状RNA分析结果报告。
优选的,所述的步骤1具体包括如下步骤:
步骤1.1,导入环状RNA高通量芯片原始信号值文件;
步骤1.2,对所述的环状RNA高通量芯片原始信号文件进行质量分析并剔除低质量信号数据,获得经过筛选的信号数据;其中,低质量信号数据是指扫描微阵列芯片荧光强度作为RNA表达信号值且荧光强度小于30的数据,同一探针的重复信号数据采用中位数计算法取中位值作为该探针的表达值。
步骤1.3,将所述的经过筛选的数据进行前景值和背景值校正,得到消除噪音污染的环状RNA信号数据;其中,使用针对全球销量第一的Affymetrix芯片原理设计的Affy软件包中的MAS5或者RMA方法根据不同的芯片类型进行芯片数据预处理,其中不同的芯片类型是指单、双色通道;MAS5得到的数据是原始信号强度,RMA得到的是经过对数变换的信号值。
步骤1.4,将校正过的信号数据进行标准化,并去除极值,得到理论上有效的环状RNA表达值;其中,使用目前芯片处理最通用的limma软件包进行芯片间归一化,得到标准化的环状RNA表达谱数据。
如图2所示,步骤1中,生物信息学参数分析包括差异表达环状RNA的筛选,环状RNA的功能性分析和对环状RNA的调控机制分析。
其中,差异表达环状RNA的筛选包括输入指令选取1.5倍或者2倍的差异倍数(Fold change),选用三个标准Benjamini–Hochberg方法、FDR方法或者Bonforroni方法校正P-value得到差异表达的环状RNA。
本发明的优选技术方案中,所述的步骤1中,对环状RNA的功能性分析包括环状RNA和基因数据的共表达分析,基因本体分析,代谢通路分析,化学反应分析和调控网络的构建。
本发明的优选技术方案中,所述的步骤1中,对环状RNA的调控机制分析,包括miRNA结合位点预测,靶基因预测和竞争性内源RNA调控网络的构建。
在本发明的一个实施方案中,在R平台,使用limma软件包的linear model线性拟合数据,通过经验Bayes t test得到差异表达的环状RNA结果。
linear model是limma软件的线性模型算法,用来分析实验以及评估差异表达。
E[yj]=Xαj
上式中,Yj表示gene J的表达值;X是实验设计矩阵;Αj是系数向量。
经验Bayes t test检验是检验样本平均数与总体平均数的离差统计量。
上式中,为样本平均数;μ为总体平均数;N为样本容量;σx为样本标准差。
在本发明的一个实施方案中,在R平台,对差异环状RNA的结果进行错误发现率矫正。可以采用Benjamini–Hochberg,FDR和Bonferroni方法。
Benjamini–Hochberg方法
上式中,α是给定的显著性阀值;K代表样本容量;M代表从小到大的排列顺序。
FDR方法
上式中,M0代表零假设是真的时候的样本总数;M代表样本容量;Q显著性阀值。
Bonferroni方法
P=α/k
上式中,α是给定的显著性阀值;K是样本容量。
在本发明的一个实施方案中,在R平台,对环状RNA和基因表达谱数据进行共表达分析,可以使用Pearson和Spearman两种算法,将样品进行计算。
Pearson相关系数是用来衡量两个数据集合是否在一条线上面,它用来衡量定距变量间的线性关系。
上式中,Z:代表正态分布中,数据偏离中心点的距离;等于变量减掉平均数再除以标准差;N为样本容量。
Spearman相关系数对原始变量分布不作要求,属于非参数统计方法。
rs=1-6∑(Xi-Yi)2/n(n2-1)
上式中,Xi和Yi分别为两个变量按大小排位的等级;n为样本容量。
在本发明的一个实施方案中,在R平台,与环状RNA显著共表达的基因采用g:Profiler法从生物过程、分子功能和细胞组分三个成分进行基因本体注释和富集分析,差异显著可以用Benjamini–Hochberg和Bonferroni。
在本发明的一个实施方案中,在R平台,采用g:Profiler法整合KEGG和Reactive数据库信息对与环状RNA显著共表达的基因进行代谢通路和化学反应分析,差异显著可以用Benjamini–Hochberg和Bonferroni方法。
在本发明的一个实施方案中,因为环状RNA和共表达的基因具有相近的功能,在得到基因的基因本体、代谢或者反应信息确定其功能后,将两者结合,生成含有这些信息的网络文件。可以用Cytoscape软件打开,图形化展示环状RNA潜在功能调控网络。
在本发明的一个实施方案中,对环状RNA序列的miRNA结合位点预测采用miRanda和TargetScan方法。
miRanda方法
miRanda是Enright等人于2003年开发一种miRNA靶标预测软件。miRanda的核心思想主要是基于碱基互补,近似于Smith-Waterman算法,但对碱基配对的原则作出了改进,允许G-U间的错配。考虑到miRNA与靶标位点结合时存在对5’端匹配程度要求较高的特性,软件使用scale参数对5’端的11个碱基的得分作出矫正。而对结合能计算方面,miRanda基于ViennaRNA软件包中RNAlib程序来计算miRNA-靶序列间的结合能。对于多个miRNA靶向同一位点的情况,miRanda采用贪婪算法选取得分最高结合能最低的结果。
TargetScan方法
Stark等人于2005年根据实验结果分析miRNA靶标位点序列的结构需求,提出了miRNA具有一个7bp左右的核心序列,也就是种子序列。这段序列只允许Watson-Crick配对。作为靶标位点核心的种子序列通常在物种间高度保守。TargetScan基于这一原则对脊椎动物的miRNA靶标位点进行预测。首先根据miRNA在各物种间的保守情况将其划分为广泛保守、保守和弱保守的miRNA及家族,并考虑了靶标位点在多个物种间的保守性,并根据保守性的得分高低区分为保守靶标位点和弱保守的靶标位点。
在本发明的一个实施方案中,对小RNA靶基因采用miRWalk和TargetScan数据库信息进行预测,并取其交集。
miRWalk数据库
miRWalk是一个综合性数据库,提供来自人类、小鼠和大鼠的miRNA的预测信息和经过验证的位于其靶基因上的结位点,共整合了13个公共数据库资源。
TargetScan数据库
TargetScan是由microRNA领域大牛Bartel实验室开发的数据库。基于靶mRNA序列的进化保守等特征搜寻动物的microRNA靶基因。是预测microRNA靶标假阳性率较低的数据库。
在本发明的一个实施方案中,得到环状RNA、环状RNA通过结合位点吸附的小RNA以及小RNA调控的靶基因数据,构建环状RNA作为竞争性内源RNA的调控网络,生成含有所有信息的文件。可以用Cytoscape软件打开,图形化展示环状RNA的竞争性内源RNA调控网络。
以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不是限制本发明的范围。实施例中采用的实施条件可以根据具体应用要求的条件做进一步调整,未注明的实施条件通常为常规实验中的条件。
实施例
首先对原始数据进行过滤处理,然后去除低质量信号和噪音污染的数据,经过标准化后得到有效的环状RNA表达值。基于环状RNA分析结果,可以结合共表达的基因表达谱对其进行功能预测;也可以基于其序列特征,进行竞争性内源RNA调控分析。环状RNA-共表达基因网络示意图如图3所示。在上述分析的基础上,可进行一系列的统计学和可视化分析。
1.环状RNA原始信号文件如表1所示
分析平台:R平台
分析软件:Affy,limma
表1
列名解释:
2.环状RNA芯片表达结果如表2所示
分析平台:R平台
分析软件:limma,sva
表2
列名解释:
3.差异表达的环状RNA结果如表3所示
分析平台:R平台
分析软件:limma,openxlsx
表3
列名解释:
4.环状RNA和基因的相关系数如表4所示
分析平台:R平台
分析方法:Pearson,Spearman
表4
列名解释:
5.基因功能分析
为了得到与环状RNA显著共表达的基因的功能,通过David数据库对其从生物过程、分子功能和细胞组成进行基因本体分析,代谢通路分析和化学反应分析。
分析平台:R平台
分析软件:g:Profiler
结果如表5-9所示,环状RNA生物通路富集调控示意图如图4所示。
表5生物通路富集分析
列名解释
表6分子功能富集分析
列名解释:
表7细胞组分富集分析
列名解释:
表8KEGG代谢通路富集分析
列名解释:
表9Reactive化学反应富集分析
列名解释:
6.环状RNA潜在功能调控网络的构建
分析平台:R平台
图形化软件:Cytoscape
7.环状RNA的miRNA结合位点预测
分析方法:miRanda和TargetScan
miRanda算法是基于位点结合自由能和序列互补配对得分的方法。默认参数使用strict种子序列互补配对法,score得分大于140分,最小自由能为-15KJ/mol。
分析平台:linux平台
结果如表10所示:
表10miRanda结果
列名解释:
TargetScan算法是在多重比对序列基础上通过寻找保守的种子序列来识别其靶基因的方法。
分析平台:perl平台
结果如表11所示:
表11TargetScan结果
列名解释:
8.对miRNA靶基因采用miRWalk和TargetScan数据库信息进行预测,并取其交集。得到环状RNA、环状RNA通过结合位点吸附的miRNA以及miRNA调控的靶基因数据,构建环状RNA作为竞争性内源RNA的调控网络,生成含有所有信息的文件。
图形化软件:Cytoscape,环状RNA的竞争性内源RNA调控网络示意图如图5所示。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实例的限制,上述实例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等同物界定。
- 还没有人留言评论。精彩留言会获得点赞!