一种以24‑脱氢胆固醇还原酶(DHCR24)为靶点进行降胆固醇药物的虚拟筛选方法与流程
本发明涉及药物筛选方法,尤其是以24-脱氢胆固醇还原酶(3β-脱氢胆固醇-Δ24还原酶,3β-hydroxysterolΔ-24-reductase,DHCR24)为靶点,利用分子对接软件,对小分子数据库进行降胆固醇药物的虚拟筛选方法。
背景技术:
胆固醇是人体组织细胞不可或缺的重要物质,但胆固醇含量过高却会诱发产生疾病,尤其是心血管疾病。2016年5月是我国首个“胆固醇月”。根据《中国心血管病报告2014》最新数据显示,我国心血管病患者约为2.9亿,且患病率仍在持续升高。我国心血管病死亡率的上升趋势,主要是由于胆固醇引起的缺血性心脏病死亡上升所致。根据研究数据表明,中国人冠心病死亡率增加77%,主要是由于胆固醇升高所引起。而在众多危险因素当中,胆固醇指标的异常是导致心肌梗死、冠心病等疾病的重要危险因素。在人体内胆固醇来源主要通过两条途径:内源性的胆固醇的生物合成和肠道对摄入食物中胆固醇的摄取与吸收,其中内源性合成占80%左右。在食物摄入方面,可以通过人为控制,来达到一定程度的降低胆固醇的目的。但胆固醇的内源性合成是由遗传的因素来决定的。因而从内源性合成方面控制胆固醇的生成,是降低胆固醇的有效思路。例如目前临床普遍使用的他汀类降胆固醇药物,就是控制内源性胆固醇的生物合成来实现药效的。但是,他汀类药物的作用机理是通过竞争性抑制胆固醇生物合成过程初始阶段的酶羟甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂来实现。然而需要长期服用的他汀类药物,却存在合并高血糖、高血压、肌肉疾病以及肝酶升高等副作用,并且存在一部分高血脂病人对他汀类药物不敏感。3β-脱氢胆固醇-Δ24还原酶(DHCR24)是催化胆固醇合成最后一步的酶,催化链甾醇合成胆固醇。因此以DHCR24为靶点,通过开发DHCR24抑制剂进行新型降胆固醇药物的研发,意义重大。
传统的药物筛选方法。例如无论是高通量细胞平台还是动物水平筛选,从确认有效成分,到分离单体,到最终成药上市,需要投入大量的人力,物力。因而寻找到一种高效的药物筛选方法就变得尤为重要。伴随着计算机的广泛发展,利用计算机技术进行虚拟筛选的方法,逐步被重视并得到应用。虚拟筛选是利用对接软件模拟分子间的相互作用,从而计算出分子间的亲和能大小,初步预测潜在的药物分子。目前的研究表明,这种虚拟筛选的阳性率可以高达20%左右,远远大于传统的新药筛选方法,从而可以节约大量的时间和研发成本,大大缩短了研发的周期和资金投入。因此虚拟筛选这种近年来不断发展完善的新型方法,在药物研发中受到了越来越广泛的关注和重视。
目前被广泛使用的小分子数据库很多,例如DrugBank,TCM。这些数据库已经被频繁使用于新药的虚拟筛选研发与设计,其中包括以现有临床正在使用的药物和处于研发阶段的小分子药物数据库,如DrugBank(http://www.drugbank.ca/)可以被用来针对“老药”进行筛选加以“新用”,也可以大大提高开发的效率。“老药新用”是指已上市药物的新适应证或者新的用途开发。全新药物的研发一般从确立项目到上市需要10多年时间,成本也极高,而成功率却不足10%。“老药新用”则可以大大减少研发的风险性。
因而利用小分子数据库,尤其是以“老药新用”的小分子药物数据库进行虚拟筛选研究,建立一种高效的DHCR24为靶点的新型降胆固醇药物筛选体系意义重大。
技术实现要素:
本发明的目的是建立一种创新性的,高效性的以DHCR24为靶点,利用现有小分子药物数据库或者其他小分子数据库进行新型降胆固醇药物的虚拟筛选方法。
本发明利用目前较为先进的计算机虚拟筛选技术,结合已知小分子数据库,特别是小分子药物数据库,旨在从现有小分子数据库中,发现能竞争性抑制链甾醇与DHCR24(即以链甾醇与DHCR24结合口袋为靶点)结合从而降低胆固醇合成的新型小分子药物,以达到老药新用,提高药物筛选效率,节约时间,降低成本的目的。
DHCR24基因的长度约为46.4kb,包含有9个外显子以及8个内含子,位于染色体的lp31.1-p33,DHCR24序列中还包含有保守结构域,能与FAD等非共价结合,表现出FAD依赖型氧化还原酶类家族的特征。因此在对接时也要同时将FAD对于链甾醇和DHCR24结合所产生的影响考虑在内。
人源DHCR24蛋白由516个氨基酸组成,其末端具有FAD(黄素腺嘌呤二核苷酸)结合域及位于N端的信号肽。在人体中DHCR24基因突变(如E191K、N294T、K306N、Y471S和两点突变N294T/K306N),导致DHCR24酶活性降低,引起胆甾醇血症,多为多发性的先天性畸形及发育异常,病因为与发育相关的能与胆固醇共价结合的Hedgehog蛋白信号通路发生异常导致。其表现特征是链甾醇在组织和血浆中的积累及胆固醇的缺失。低胆固醇血症导致的先天性畸形主要原因也是由于胎儿不能有效地从母体摄入足够的外源性胆固醇,而内源性合成又因为DHCR24的活性异常低下而造成。成人则有相对稳定的胆固醇调节机制,通过调节胆固醇的生物合成和外源供给而实现细胞内胆固醇维持在较为稳定的水平。目前已知存在小分子抑制剂DMHCA(N,N-dimethyl-3β-hydroxycholenamide),可以通过竞争性机制抑制DHCR24活性(Thomas Pfeifer and et al.,Curr Pharm Biotechnol.2011);以及U18666A可以通过非竞争性抑制机制抑制其活性,从而降低胆固醇的含量(Bae SH and et al.Biochem J.,1997)。这两种抑制剂均可以通过抑制DHCR24活性发挥降低胆固醇作用,证明DHCR24的突变或者活性抑制可以影响胆固醇的含量,为通过抑制其酶活性来开发降胆固醇药物提供了理论基础。
基于上述机理,本发明构思如下:由于目前DHCR24的晶体结构目前还未被测得,首先需要在UniProt上下载DHCR24蛋白的氨基酸序列,用同源建模的方法获得其三维结构,并确定其结构的合理性。如果DHCR24结构被测得,可以直接从共享的蛋白质数据库中获得其晶体结构数据。在chimspider上下载黄素腺嘌呤二核苷酸(flavin adenosine dinucleotide FAD)与链甾醇(desmosterol)的结构数据。在此基础上,利用对接软件Autodock vina1.1.2,将DHCR24蛋白分子先与FAD按照全局搜索策略对接,根据DHCR24蛋白结构域的基本信息,确定DHCR24-FAD复合物最佳构象;之后以此为基础,对接底物链甾醇,进行全局搜索对接,根据打分结果,确定几种组分的最佳结合构象,以此确定DHCR24酶的底物催化活性口袋。选用小分子数据库(如Drugbank数据库)作为药物小分子来源,并对其小分子结构进行适合用于筛选的处理。从drugbank几千种小分子药物中筛选出与活性口袋亲和性较好的小分子药物,并将链甾醇与DHCR24的对接结果作为对照,初步确定出能竞争性抑制底物链甾醇与DHCR24结合的候选药物小分子。最后挑选得到的候选药物小分子,进行细胞水平的降胆固醇效果的验证。本方法提供了一种全新的利用DHCR24为靶点的新型降胆固醇药物的虚拟筛选方法和策略。
本发明采用的技术方案是:一种以24-脱氢胆固醇还原酶(DHCR24)为靶点进行降胆固醇药物的虚拟筛选方法,包括以下步骤:
1)确定DHCR24的分子结构;
所述的确定DHCR24的分子结构,包括:利用同源建模方法构建DHCR24的三维结构模型,利用打分软件进行打分,确定打分较高的结构模型作为DHCR24蛋白的三维结构模型;或者直接从网上蛋白质数据库下载DHCR24蛋白的晶体结构数据。
2)对DHCR24进行前期处理和优化;
所述的对DHCR24进行前期处理和优化,包括:加非极性氢,去除水分子,加电荷;
3)采用分子对接软件,依据链甾醇与DHCR24对接后的结合构象,确定药物小分子对接的活性中心,设定活性口袋;优选的,所述的活性口袋为:中心坐标为x=72.194、y=51.111、z=24.361,网格大小为8×10×18,步长设置为1;具体方法为:
3.1)将DHCR24与FAD进行对接;然后以此结构为基础,采用全局性搜索,对接链甾醇,根据链甾醇与DHCR24的结合构象的autodock vina打分结果,初步挑选打分高的结合构象;
3.2)以初步挑选的打分高的结合构象为基础,采用autodock4半柔性对接方式,对结合构象进行再次精确对接,确定虚拟筛选用的活性口袋。具体为:以步骤3.1)初步挑选的打分高的结合构象为基础,采用autodock4半柔性对接方式,对结合构象进行再次精确对接,对对接后产生的不同结合构象的成簇情况进行综合分析,选择能量最低簇并且结合构象最为集中,且形成一个氢键的第一个构象作为最佳结合构象及活性中心,从而确定药物小分子的活性口袋。
4)整理对接用小分子配体数据库,并进行筛选,筛选项目为毒性筛选、类药性筛选;
5)根据设定的活性口袋,利用分子对接软件,将对接用小分子配体数据库中的小分子配体与活性口袋依次进行对接;
6)对初步对接筛选出的结果,进行精确对接,对药物小分子配体与活性口袋的相互作用方式,进行综合性评估,初步确定具有降胆固醇作用效果的药物。
上述的一种以24-脱氢胆固醇还原酶为靶点进行降胆固醇药物的虚拟筛选方法,步骤4)置于步骤1)至步骤5)任何一个步骤之前。
上述的一种以24-脱氢胆固醇还原酶为靶点进行降胆固醇药物的虚拟筛选方法,所述的小分子配体数据库包括药物小分子配体数据库、天然产物数据库或其他化学小分子数据库。
本发明的有益效果是:本发明通过对现有小分子数据库进行虚拟筛选,能在短时间内得到现有小分子数据库中,与DHCR24和其底物链甾醇的结合口袋结合能力较强的药物小分子,将目标从成千上万个小分子快速且有效地集中到几十个,再利用高通量的细胞筛选以及后续的动物水平筛选,得到降胆固醇的药物,从而可以大大提高效率,缩短研发周期。
附图说明
图1是DHCR24一级结构序列。
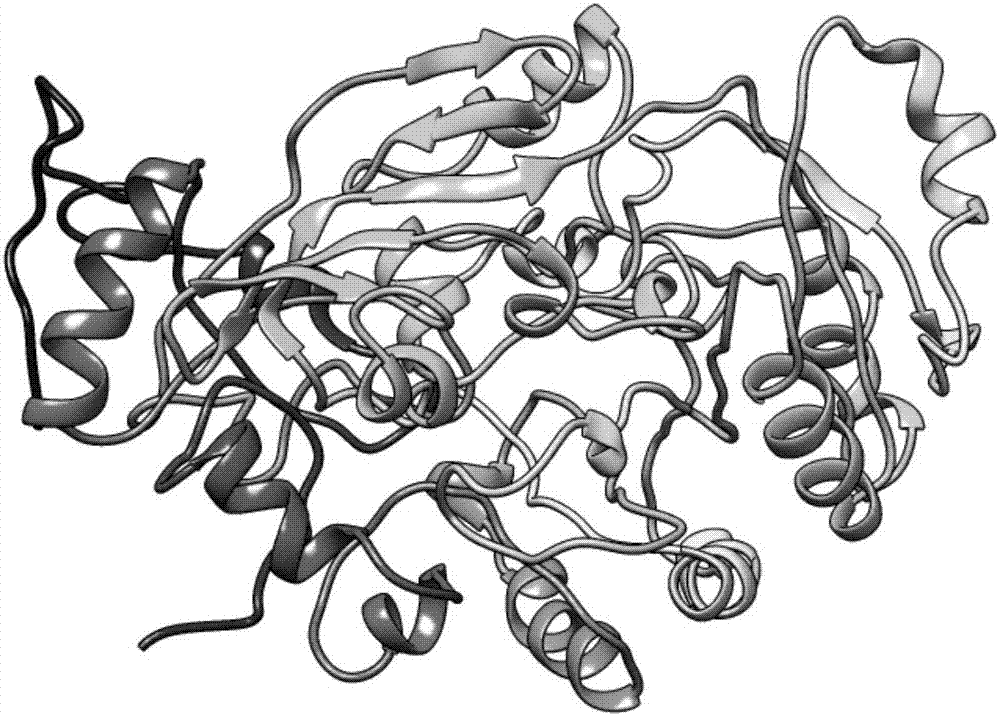
图2是同源建模所获DHCR24三维结构。
图3是黄素腺嘌呤二核苷酸(FAD)的结构式。
图4是化合物链甾醇(desmosterol)的结构式。
图5是DHCR24与链甾醇的结合位点。
图6是筛选用autodock vina配置文件内容。
图7是药物小分子配体对接中使用的SHELL脚本。
图8A药物小分子Conivaptan结构式。
图8B药物小分子Risperidone结构式。
图9 autodock vina精确对接得到的desmosterol与2种药物小分子binding energy的对比图。
图10A对照链甾醇与活性中心的成簇分析及相互作用图。
图10B Conivaptan与活性中心的成簇分析及相互作用图。
图10C Risperidone与活性中心的成簇分析及相互作用图。
图11 desmosterl和cholesterol标准曲线图。
图12各样品中每10*E06个细胞中胆固醇的含量。
具体实施方式
以下实施例将有助于本领域的普通技术人员进一步理解本发明,但不以任何形式限制本发明。
实施例1一种以24-脱氢胆固醇还原酶为靶点进行降胆固醇药物的虚拟筛选方法
步骤如下:
(一)DHCR24酶催化活性口袋的确立
1、在UniProt上下载DHCR24蛋白的氨基酸序列(图1),用同源建模的方法获得其三维结构。在I-TASSER Server在线服务器上预测蛋白结构。利用打分软件LGscore(>1.5correct model)打分结果为2.53,Maxsub(>0.1correct model)打分结果为0.17,VERIFY3D(>80%of the residues had an averaged 3D-1D score>=0.2)打分结果为82.79%,Ramachandran plot(favoured and allowed region>90%)打分结果为95%,最终确定打分较高的结构模型作为DHCR24蛋白的三维结构模型(图2)。
2、大分子受体DHCR24的前期处理。将步骤1中挑选出的打分较高的DHCR24在MGLTools中进行处理。包括加非极性氢,去除水分子,加电荷Kollman Charges。最后保存为DHCR24.pdbqt。
3、活性口袋的确定。
3.1)依据已知的DHCR24的FAD(图3)结合的结构域及其氧化还原功能域的预测结构信息(Xiuli Lu等,Endocrinology,2008),对DHCR24与FAD进行对接,得到dh_fad.pdbqt。然后以此结构为基础,采用全局性搜索的策略,对接desmosterol(图4),并根据desmosterol与DHCR24的结合构象的autodock vina打分结果(binding energy为-8.4Kcal/mol),挑选其最佳结合构象。
3.2)以该结合构象为基础,采用autodock4半柔性对接方式,对该结构进行再次精确对接,以确定虚拟筛选用的最佳结合口袋。对对接后产生的不同结合构象的成簇情况进行综合分析,可以看到在desmosterol结合到DHCR24-FAD复合体上的红色簇为能量最低簇并且构象最为集中,在其中选择形成一个氢键的第一个构象(附图10A),确定其为最佳构象及活性中心,从而确定药物小分子的结合口袋,进而进行虚拟筛选。根据对接结果,经过反复验证,最终确定了活性口袋的设置条件如下,以desmosterol与DHCR24结合口袋(图5虚线圈出)为活性中心的口袋:中心坐标为x=72.194、y=51.111、z=24.361,网格大小为8×10×18,步长(spacing)设置为1。对接每个配体产生30个对接构象,对接时采用半柔性对接,即蛋白质分子固定,小分子配体可以自由转动,配置文件如图6。
(二)以DHCR24酶为受体大分子对小分子数据库(本实施例中为DrugBank药物数据库)进行对接
4、药物小分子配体文件的准备。由于DrugBank(http://www.drugbank.ca/)下载下来的小分子数据库格式为sdf是二维结构模型,Autodock vina软件不予识别,应先转化格式,利用OpenBabel进行批量文件格式转化将sdf格式转化为pdbqt格式,并利用批量改名软件,将DrugBank批量改名称为n.pdbqt(n=1、2、3……n)。
5、利用脚本实现虚拟筛选。利用Autodock vina1.1.2分子对接软件,进行虚拟筛选的程序设置。这需要运用SHELL脚本来完成(图7)。在终端运行脚本文件virtual_screen.sh。循环运行Autodock vina将DHCR24与每一个药物小分子配体逐一对接,并将每一个药物小分子对接最优的前9个构象结果输出到log文件中。挑选其中结合能较强的药物小分子,作为初步筛选结果。
6、经过以上筛选,初步确定和desmosterol与DHCR24结合位点结合能力较强的药物小分子50余个,选择其中2种药物小分子是已经成药的小分子,分别是Conivaptan(DB00872)和Risperidone(DB00734),进行进一步的分子对接验证和细胞平台降胆固醇药效学验证(图8A和图8B)。
7、对2种药物小分子进行精确对接(用autodock vina1.1.2),设置对接次数为5000次,其余对接参数不变。通过观察可以得到这2种药物小分子在desmosterol与DHCR24结合位点的binding energy值均较高,并比desmosterol与DHCR24的binding energy值高,说明其亲和力较之于desmosterol更强(图9)。并同时使用autodock 4进行对接,得到的成簇分析图和结构图如图10B和图10C所示,显示了这2种药物小分子分别与DHCR24酶活性口袋的具体的相互作用方式。
通过以上筛选,初步确定了以链甾醇与DHCR24结合位点为靶点,结合能在-9kJ/mol左右具有较强结合能的小分子药物50余种,选择其中的2种药物小分子,进行精确对接,结果显示这2种药物可以作为DHCR24的有效的竞争性酶抑制剂,并进一步开发为新型以DHCR24为靶点的降胆固醇药物。这为进一步筛选降胆固醇药物极大的节约时间与成本,缩小了范围。
实施例2候选药物的细胞水平降胆固醇药效的初步确认。
为了进一步确认虚拟筛选所得的候选药物小分子的降胆固醇的药效,对Conivaptan(DB00872)和Risperidone(DB00734)这两种药物进行细胞水平的验证。首先培养hepG2细胞(人肝癌细胞),根据文献和实验观察得到各个药物的最适给药浓度,设置完全培养基(S+),无血清无抗生素培养基(S(-)),空白对照组),U18666a(已知的DHCR24抑制剂,阳性对照组,浓度为2μM)和两种药物组(Conivaptan为5μM,Risperidone为1μM),给药24小时后,收集细胞沉淀提取总脂质,进行高效液相的测定。首先利用胆固醇(cholesterol)和胆甾醇(desmosterol)标准品测定标准曲线(如图11),然后进行各实验组的总脂质的样品检测。结果如图12所示,可以看到,与空白对照组相比,在给定的给药浓度下,两个候选药物给药组中均未检测到胆固醇,有力证明了这两种候选药物有显著的抑制胆固醇合成的效果。这一细胞水平的实验结果,进一步确认了计算机虚拟筛选所得候选药物小分子在细胞水平上具有降胆固醇的效果,充分证明了本专利中所发明的以DHCR24为靶点进行降胆固醇药物虚拟筛选策略的合理性。
- 还没有人留言评论。精彩留言会获得点赞!