包含决奈达隆的药物组合物和剂型,及其制备方法与流程
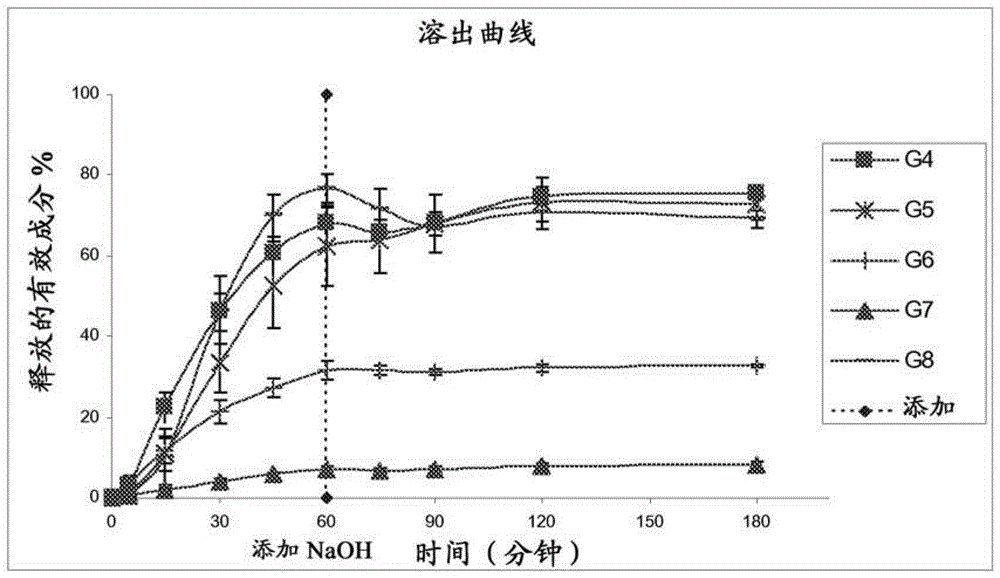
包含决奈达隆的药物组合物和剂型,及其制备方法本发明总体上涉及药物组合物,其用于口服给药的包含具有抗心律失常活性的有效成分。更精确地,本发明涉及既定有利地在胶囊类型剂型中使用的半固体或液体药物组合物,所述组合物包含至少一种苯并呋喃衍生物作为具有抗心律失常活性的有效成分和至少一种脂质赋形剂。本发明还涉及制备这种基于所述药物组合物的剂型的方法,还涉及所述组合物或所述剂型的治疗应用。"具有抗心律失常活性的苯并呋喃衍生物"在本发明的上下文中表示选自专利US3248401,US5223510和EP338746以及专利申请WO88/07996,WO89/02892,WO90/02743和WO94/29289中所述的那些苯并呋喃化合物。在这些化合物中,我们可提及2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃或决奈达隆及其在专利EP1315709中所述的药学上可接受的盐以及2-正丁基-3-(3,5-二碘代-4-二乙基氨基乙氧基苯甲酰基)苯并呋喃或胺碘酮及其在专利US3248401中所述的药学上可接受的盐。有利地,该具有抗心律失常活性的苯并呋喃衍生物选自决奈达隆或下文所示的式(D)的游离碱形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃及其衍生物,如下文所述的药学上可接受的盐。"药学上可接受的盐"是指当以标准剂量给药时对其给药的对象没有毒性的盐。因此,作为决奈达隆的药学上可接受的盐,我们可提及例如2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃盐酸盐,2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃富马酸盐和2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃草酸盐。在本文中使用的抗心律失常化合物,特别是碱形式的决奈达隆和胺碘酮,或其盐,特别是其盐酸盐,其特征在于在水性介质中具有低溶解度,这构成使所述有效成分可用于口服途径给药的主要障碍。因此,这些抗心律失常化合物在模拟胃介质(3mg/ml,在pH=1.5)中具有低溶解度且在模拟肠介质(1μg/ml,在pH=6.5)中具有非常低的溶解度。例如,盐酸决奈达隆在室温作为pH的函数的溶解度曲线显示在pH为大约3至5时最大溶解度为约1至2mg/ml,但在pH为约6至7时显示非常低的溶解度,因为在pH=7其不大于10μg/ml。对于胺碘酮盐酸盐,其溶解度在室温在pH为3至4时为0.3至0.9mg/ml,在pH=7时为几个μg/ml。因此,可将400mg盐酸决奈达隆溶于200ml在pH=4缓冲的水性介质液(0.1MNaH2PO4水溶液)。相反,在用pH=7缓冲的水溶液(0.1MNa2HPO4水溶液)稀释至1/10的该介质中,盐酸决奈达隆发生沉淀(该介质的最终pH=6.7)。因为这些溶解度条件类似于胃肠道中的条件,可以设想,在胃中,盐酸决奈达隆将经受对其溶出有利的酸性条件,但一旦其进入肠中,其将遇到pH为6至7的介质,即不溶解介质,其中它将有发生沉淀的风险。目前,主要在肠中发生有效成分的吸收,且目前众所周知的是通过口服途径给药需要将有效成分有利地保持在溶液中,以期望获得沿着胃肠道的充分渗透,和因此获得可接受的暴露,以达到显著的治疗效果。考虑到溶解度和生物利用度的问题,已开发出一种剂型且目前在市场上销售,其为426mg盐酸决奈达隆的膜包衣片剂,相当于400mg决奈达隆,以商标名销售,成人的推荐剂量为每日两次,每次一片,且其必须随餐服用以保证所述有效成分的最佳作用。事实上,从药代动力学的观点看,在随餐口服给药后,决奈达隆吸收良好(至少70%)。然而,由于系统前首过代谢,该药品(随食品服用)的绝对生物利用度不大于15%。相对于没有同时摄取食物而服用药物,食物的同时消耗将产品的生物利用度扩大为2至4倍。在随餐口服给药后,决奈达隆及其主要循环活性代谢物(N-脱丁基代谢物)的血浆峰值浓度在3至6小时内达到。决奈达隆及其N-脱丁基代谢物的药代动力学适度地偏离与剂量成比例的原则:剂量加倍导致Cmax和AUC增加至约2.5至3.0倍。目前,对于患者当然优选能够得到治疗而不用随餐或不随餐服药的限制,特别是在治疗心节律,尤其是心律失常疾病的领域。因此,能产生可接受的生物利用度的用于口服给药具有抗心律失常活性的有效成分的药物组合物(无论是否同时摄取食物)的开发,即涉及有限的用餐效果而生效的组合物的开发,仍然是相当感兴趣的。目前已发现了新的药物组合物,非常惊人和出乎意料地,其允许口服给药至少一种抗心律失常的有效成分,有利地为2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃或其衍生物,例如其盐,而没有上述缺点。该组合物(其包含掺入在基质中的至少一种有效成分,该基质由所述组合物的其它成分,特别是其它赋形剂构成)证明为充分稳定的且具有合适的溶解度以在胃肠道中幸存直到其达到吸收部位。而且,该组合物可空腹服用或随小吃或甚至低脂食物以单或多剂量服用。本发明因此涉及用于口服给药具有抗心律失常活性的有效成分的药物组合物,该有效成分例如为(i)碱形式,(ii)药学上可接受的盐形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃,其特征在于除了所述有效成分,其包含至少一种HLB值介于2至20的两亲性脂质赋形剂。本发明因此涉及用于口服给药具有抗心律失常活性的有效成分的药物组合物,该有效成分例如为(i)碱形式,(ii)药学上可接受的盐形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃,其特征在于除了所述有效成分,其包含至少一种HLB值介于1至20的两亲性脂质赋形剂。所述本发明的组合物可在胶囊类型或半固体或液体胶囊的剂型中。事实上,其可有利地包装在胶囊类型剂型中,还更有利地在硬胶囊类型的剂型中。在本发明的上下文中使用以下术语:胶囊,具有硬或软壳的剂型;硬胶囊,具有硬壳的胶囊,其具有两部分:一部分称为囊体,一部分称为囊帽;生物利用度,用于描述药品的药代动力学性质的术语,即到达血流的剂量的比例。其评价到达血流的吸收药品的量和所述药品的吸收率;有效成分,任何具有治疗效果,例如抗心律失常效果的物质。在本发明的上下文中,其具体为下文限定的任何具有抗心律失常活性的苯并呋喃衍生物,特别是碱形式的决奈达隆,与有机酸或无机酸加成的药学上可接受的盐形式的决奈达隆。所述盐可用药学上可接受的酸制备,但可用于例如纯化或分离式(I)的化合物的其它酸的盐也构成本发明的一部分。赋形剂,任何对有生命的有机体无活性的或惰性的物质,与有效成分相对,且其促进药品的制备和给药;脂质赋形剂,本领域技术人员已知的作为脂质溶剂的任何赋形剂,有利地为两亲性的,其具有的HLB值(下文定义的术语)根据本发明为小于20且大于1;基质,除了本发明的组合物的一种或多种有效成分的所有成分,特别是赋形剂;HLB值,亲水-亲脂平衡值,根据Griffin开发的分类法,其为本领域技术人员众所周知的;表面活性剂,一种赋形剂,其通过其两亲性质促进粉末的润湿,改善溶解度/溶出和/或减缓再沉淀;共溶剂,改善本发明的组合物的制备方法的可行性的任何溶剂,其基于所述组合物的基质的粘度和熔点的关键参数以及所述基质中有效成分的溶出或分散;稀释剂,用于获得足够体积的组合物的赋形剂,以制备所需尺寸的剂型,例如硬胶囊,且对硬胶囊所选的制备方法具有合适的物理特性;崩解剂,通过增加脆性和减少剂型的硬度而在胃中提供该剂型令人满意的崩解且因此提供有效成分的崩解的赋形剂;抗粘附剂,在制备剂型过程中,例如当填充胶囊时用于防止颗粒彼此粘附和粘附至生产设备的赋形剂。润滑剂,用于促进制备剂型的步骤中的赋形剂,归因于它们在滑动中的作用,即增加机器管道中的颗粒流动性;增塑剂,用于允许有效成分从剂型中恒定释放的赋形剂,通过插入在聚合物链之间且通过使它们相对彼此滑动。其根据其溶解度来选择。在本发明的组合物中,我们可提及第一组药物组合物,其包含:·1-60wt%的本发明的至少一种有效成分,有利地为1至50%,还更有利地为10至45%,还更优选为20%至40%;·40-99wt%的本发明的至少一种脂质赋形剂,有利地为45至80%,还更有利地为50%至60%,·0-30wt%的至少一种选自以下的化合物:表面活性剂、共溶剂、稀释剂、崩解剂、润滑剂、有机或无机碱和增塑剂,有利地为1至20%,还更优选为1至10%,该百分比以相对于所述组合物总重量的重量表示。在本发明的组合物中,我们可提及第二组药物组合物,其包含:·1-60wt%的本发明的至少一种有效成分,有利地为1至50%,还更有利地为10至45%,还更优选为20%至40%;·37-99wt%的本发明的至少一种脂质赋形剂,有利地为45至80%,还更有利地为45%至55%,·0-30wt%的至少一种选自以下的化合物:表面活性剂、共溶剂、稀释剂、崩解剂、润滑剂、有机或无机碱和增塑剂,有利地为1至20%,还更优选为1至10%,该百分比以相对于所述组合物总重量的重量表示。在本发明的组合物中,我们可提及第三组药物组合物,其包含:·1-60wt%的本发明的至少一种有效成分,有利地为1至50%,还更有利地为10至45%,还更优选为20%至40%;·40-99wt%的本发明的至少一种脂质赋形剂,有利地为45至80%,还更有利地为50%至60%,·0-30wt%的至少一种表面活性剂,有利地为1%至20%,还更有利地为5%至15%,和·0-29wt%的至少一种共溶剂,有利地为1至20%,还更有利地为2至15%;该百分比以相对于所述组合物总重量的重量表示。组合物总计为100wt%。在本发明的组合物中,我们可提及第四组药物组合物,其包含:·1-60wt%的本发明的至少一种有效成分,有利地为1至50%,还更有利地为10至45%,还更优选为20%至40%;·37-99wt%的本发明的至少一种脂质赋形剂,有利地为45至80%,还更有利地为50%至60%,·0-30wt%的至少一种表面活性剂,有利地为1%至20%,还更有利地为5%至10%,和·0-29wt%的至少一种共溶剂,有利地为1至20%,还更有利地为2至15%;该百分比以相对于所述组合物总重量的重量表示。组合物总计为100wt%。在本发明的组合物中,我们可提及第五组药物组合物,其包含:·60-200wt%的本发明的至少一种脂质赋形剂,有利地为120至180%,还更有利地为180%,·0-30wt%的至少一种表面活性剂,有利地为5%至30%,还更有利地为10%至30%,·0-30wt%的至少一种共溶剂,有利地为1至20%;该百分比以相对于所述有效成分总重量的重量表示。本发明的药物组合物包含至少一种具有抗心律失常活性的有效成分和至少一种脂质赋形剂。在本发明的具有抗心律失常活性的有效成分中,我们可提及碱形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃或决奈达隆及其在专利EP1315709中所述的药学上可接受的盐。作为药学上可接受的盐,我们可提及例如2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃盐酸盐,2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃富马酸盐和2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃草酸盐。有利地,本发明的组合物包含2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃或2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃盐酸盐作为有效成分。根据一个变化方案,碱形式的有效成分的溶出可通过以下获得:由如上所述的决奈达隆的药学上可接受的盐起始,且通过用有机或无机碱改变pH而原位复原为碱形式的有效成分。包含至少一种决奈达隆的药学上可接受的盐,且进一步包含至少一种有机或无机碱(有利地相对碱形式的有效成分以化学计算摩尔量)的本发明的组合物构成本发明的一部分。作为指导,可用于组合物中的碱的性质可为有机的,例如乙醇胺,或无机的,例如碳酸钠或碳酸钾。其有利地为碳酸钠。本发明的一种或多种有效成分在本发明的组合物中存在的比例为1-60wt%,有利地为1至50%,还更有利地为10至45%,还更优选为20%至40wt%,基于组合物的总重量计。该脂质赋形剂为HLB值介于1至20且熔点低于50°C的两亲性脂质赋形剂。该脂质赋形剂为HLB值介于2至20且熔点低于50°C的两亲性脂质赋形剂。在室温为半固体的两亲性赋形剂和室温为液体的两亲性赋形剂不同。本发明的脂质赋形剂可选自:·半固体的取代的甘油酯(substitutedglycerides),·液体的取代的甘油酯,·半固体的取代的聚乙二醇甘油酯(substitutedpolyoxylglycerides),·液体的取代的聚乙二醇甘油酯,·及其混合物。我们可提及,例如以下的组,其中脂质赋形剂选自:·作为本发明的半固体的取代的甘油酯,例如以商标名33/01,39/01,43/01和PeceolTM销售的Gelucires,·作为本发明的液体的取代的甘油酯,例如以商标名LabrafacWL1349销售的那些,·作为本发明的半固体的取代的聚乙二醇甘油酯,例如以商标名44/14,50/13销售的Gelucire,·作为本发明的液体的取代的聚乙二醇甘油酯,例如以商标名M1944CS,M2125CS,M2130CS和销售的那些。我们可提及,例如另一组,其中所述脂质赋形剂选自本发明的半固体的取代的聚乙二醇甘油酯(可商购的Gelucire),更具体地为以商标名44/14销售的月桂酰聚乙二醇甘油酯。当有效成分为盐形式时,该组合物可为所述固体有效成分在室温下在固体基质中的分散体的形式(当半固体脂质赋形剂的使用量充足)或为固体在室温下在油中的分散体的形式(当液体脂质赋形剂的使用量充足),而且所述有效成分在组合物中的溶解度为包含该组合物的介质的pH的函数。本发明的组合物中的在室温为半固体的两亲性脂质赋形剂具有以下优点:使得固态分散或热溶解有效成分于所述组合物的基质中,和当基质溶解于胃和/或肠的水性环境中时促进有效成分的溶解。本发明的组合物中的在室温为液体的两亲性脂质赋形剂具有以下优点:促进有效成分溶解于胃和/或肠的水性环境中。有利地,本发明的组合物包含至少一种HLB值介于5至18的两亲性脂质赋形剂。本发明的HLB值介于5至18的两亲性脂质赋形剂可选自:·中链单甘油酯和二甘油酯,例如Capmul(HLB值介于5.5至6),Abitec公司销售,·丙二醇单月桂酸酯,例如90(HLB值等于5)和Capmul分别由Gattefosse和Abitec公司销售,·辛酰己酰聚乙二醇-8甘油酯,例如(HLB值等于14),由Gattefosse公司销售,·月桂酰聚乙二醇甘油酯,例如44/14(HLB值等于14)和50/13(HLB值等于13),由Gattefosse公司销售,·丙二醇辛酸单酯,例如PG-8(HLB值等于6),由Abitec公司销售,·及其混合物。更具体地,HLB值介于5至18的两亲性脂质赋形剂选自Capmul90,Capmul44/14,50/13,PG-8,及其混合物。根据一个实施方案,本发明的脂质赋形剂选自HLB值介于12至18的两亲性脂质赋形剂。本发明的一种或多种脂质赋形剂在本发明的组合物中存在的比例为40-99wt%,有利地为45至80%,还更有利地为50%至60wt%,基于组合物的总重量。本发明的一种或多种脂质赋形剂在本发明的组合物中存在的比例为37-99wt%,有利地为45至80%,还更有利地为50%至60wt%,基于组合物的总重量。本发明的一种或多种脂质赋形剂在本发明的组合物中存在的比例为100-200wt%,有利地为110至180%,还更有利地为50%至60wt%,基于有效成分的总重量。本发明的药物组合物可进一步包含至少一种表面活性剂和/或至少一种共溶剂。该表面活性剂有利地为亲水性和/或非离子性。其可选自:·环氧乙烷/环氧丙烷共聚物,下文称为泊洛沙姆,如以商标名SYNPERONICPE/L44销售的泊洛沙姆124;以商标名PLURONICF68或SYNPERONICPE/F68销售的泊洛沙姆188;以商标名PLURONICF87或SYNPERONICPE/F87销售的泊洛沙姆237;以商标名SYNPERONICPE/F108销售的泊洛沙姆338或以商标名PLURONICF127,SYNPERONICPE/F127或LUTROLF127销售的泊洛沙姆407;·聚乙氧基蓖麻油,如以商标名CREMOPHORRH40销售的那些;·乙氧基化聚山梨酯,如分别以商标名TWEEN20,TWEEN40,TWEEN60和TWEEN80销售的聚山梨酯20,聚山梨酯40,聚山梨酯60和聚山梨酯80;和·聚乙二醇硬脂酸酯(polyethylenehydroxystearate),如以商标名SOLUTOLHS15销售的聚乙二醇硬脂酸酯660。更具体地,该表面活性剂可选自:·环氧乙烷/环氧丙烷共聚物,下文称为泊洛沙姆,如以商标名SYNPERONICPE/L44销售的泊洛沙姆124;以商标名PLURONICF68或SYNPERONICPE/F68销售的泊洛沙姆188;或以商标名PLURONICF127,SYNPERONICPE/F127或LUTROLF127销售的泊洛沙姆407;·聚乙氧基蓖麻油,如以商标名CREMOPHORRH40销售的那些;·乙氧基化聚山梨酯,如以商标名TWEEN60销售的聚山梨酯60;和·聚乙二醇硬脂酸酯,如以商标名SOLUTOLHS15销售的聚乙二醇硬脂酸酯660。有利地,本发明的一种或多种表面活性剂选自称为泊洛沙姆的环氧乙烷/环氧丙烷共聚物,还更有利地其为泊洛沙姆407。所述表面活性剂可存在于本发明的组合物中的比例为0%至30wt%,基于所述组合物的总重量,有利地为1%至20wt%,还更有利地为5%至15wt%的表面活性剂。所述表面活性剂可以存在于本发明的组合物中的比例为至少一种表面活性剂的0-30wt%,有利地为5%至20%,还更有利地为10%至20%,基于有效成分的总重量。本发明的共溶剂可选自醇性有机溶剂或二醇衍生物。我们可提及以下作为共溶剂:·例如醇如乙醇和异丙醇;·丙二醇及其衍生物,其任选被取代,如以商标名PG,LauroglycolTM90,LauroglycolTMFCC,CapryolTM90,CapryolTMPGMC销售的那些。所述共溶剂可以以所述组合物总重量的0%至29wt%的比例存在于本发明的药物组合物中,有利地为1%至20wt%,还更有利地为2%至15wt%的共溶剂。所述共溶剂可以以有效成分的总重量的0-30wt%的比例存在于本发明的药物组合物中的至少一种共溶剂(有利地为1至20%)。根据一个实施方案,该共溶剂为取代的二醇衍生物和/或含量低于29wt%,基于所述组合物的总重量,有利地该共溶剂为丙二醇和/或含量为约20wt%。根据一个实施方案,该共溶剂为取代的二醇衍生物和/或含量低于有效成分的总重量的30wt%,有利地该共溶剂为丙二醇和/或含量为有效成分的总重量的约20wt%。根据一个实施方案,本发明的组合物包含:·碱形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃或2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃盐酸盐,作为有效成分,和/或·至少一种HLB为1至20的半固体脂质赋形剂,有利地HLB为5至18,还更有利地为12至18,有利地选自半固体的取代的甘油酯和半固体的取代的聚乙二醇甘油酯,有利地其为44/14,和/或·至少一种表面活性剂,有利地选自称为泊洛沙姆的环氧乙烷/环氧丙烷共聚物,还更有利地其为泊洛沙姆407,和任选至少一种如上定义的共溶剂。在其它脂质赋形剂中,该药物组合物在室温为液体或半固体形式(即具有糊状稠度),取决于使用的一种或多种赋形剂的稠度和性质。在室温为液体或半固体的赋形剂将形成半固体基质且因此产生糊状稠度的本发明的组合物,而在室温为液体的脂质赋形剂将形成液体基质且因此产生液体稠度的本发明的组合物。因此,当本发明的脂质赋形剂选自液体或半固体赋形剂时,本发明的组合物可通过以下制备:使用溶解或冷固态分散或热固态分散于脂质赋形剂中形成脂质基质的已知方法。组合物的制备包括,例如,在约30°C至60°C(例如约44°C)的温度将本发明的有效成分和任选本发明的其它赋形剂溶解或分散于所述脂质赋形剂中,所述温度作为使用的所述脂质赋形剂的熔点的函数进行选择。根据一个特别有利的实施方案,制备本发明的组合物的方法包括在约44°C将有效成分(有利地为碱形式的2-正丁基-3-[4-(3-二-正丁基氨基丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃)溶解于脂质赋形剂(有利地为月桂基聚乙二醇甘油酯,例如44/14)中。当本发明的脂质赋形剂选自液体脂质赋形剂时,本发明的组合物可通过以下制备:使用将有效成分溶解或分散于脂质赋形剂中形成在室温为液体的脂质基质的已知方法。制备本发明的药物组合物的方法通过本领域技术人员已知的常规技术进行。然后由此获得的本发明的液体或半固体药物组合物可原样掺入硬胶囊中。包囊根据常规包囊方法进行,并考虑所述组合物和所述方法的物理化学限制。所述组合物可任选转化为粉末,其可制粒或任选掺入胶囊中或原样使用。因此,本发明也涉及包含本发明的药物组合物的剂型。该剂型可为包含本发明的组合物的胶囊形式或任选为可提供在多剂量容器中的粉末或颗粒形式或为单位剂量,如小包或囊剂形式。胶囊为由可变形状和容量的硬或软壳构成的固体制剂,通常包含单位剂量的有效成分。所述壳基于明胶或其它天然或合成的物质,其稠度可通过添加例如甘油或山梨醇而调节。其它赋形剂如表面活性剂,遮光剂,抗微生物防腐剂,增甜剂,着色剂和/或香料也可添加至胶囊壳的组成中。我们可提及以下胶囊:硬胶囊,软壳胶囊,肠溶胶囊和改良释放胶囊。有利地,本发明的剂型为硬胶囊。制备包含体和帽的硬胶囊的方法由以下组成:(i)通过混合如上定义的成分而制备本发明的组合物,然后(ii)通过适合粉末(压缩/计量装置,平整方法,具有交替的平整和装填或夯实的方法,蜗杆方法或小室计量方法)或适合半固体(铸造熔融或液体产物)的方法通过体积分配而填充胶囊的帽和/或体部分,和最后通过将形成所述胶囊的帽和体的部分连接在一起而封闭胶囊。在软胶囊的情况下,在胶囊根据常规制备方法形成于模中的同时倒入液体制剂。作为非限制性指导,有效成分的量可为每剂量单位50至500mg,例如(i)胶囊,有利地硬胶囊,或(ii)粉末囊剂,或颗粒,且脂质赋形剂的量为0.5至100mg。优选地,本发明的剂型(例如硬胶囊)可包含200至400mg有效成分。本发明的药物组合物和包含所述组合物的剂型目标是在口服给予人后限制用餐影响。该脂质赋形剂一方面使得可增溶本发明的有效成分且另一方面保护其对抗肠道中pH的不利作用,从而允许明显地避免用餐影响。表面活性剂(例如泊洛沙姆)在所述组合物中的存在使得可限制有效成分在胃肠道中时出现再沉淀和聚集。图1显示本发明的硬胶囊(硬胶囊G1至硬胶囊G6)、对比参考硬胶囊和具有10%泊洛沙姆的对比片剂的溶出曲线,所有这些制剂包含本发明的有效成分。图2显示本发明的硬胶囊(胶囊G4至胶囊G8)的溶出曲线,所有这些制剂包含本发明的有效成分。图3显示本发明的硬胶囊(胶囊G1,G3,G9)的溶出曲线,所有这些制剂包含本发明的有效成分。图4显示本发明的硬胶囊(胶囊G5,G10,G13)的溶出曲线,所有这些制剂包含本发明的有效成分。图5显示本发明的硬胶囊(胶囊G1,G9,G11,G12)的溶出曲线,所有这些制剂包含本发明的有效成分。图6显示本发明的硬胶囊(胶囊G1,G22,G24,G26,G28)的溶出曲线,所有这些制剂包含本发明的有效成分。图7显示本发明的硬胶囊(胶囊G5,G23,G25,G27,G29)的溶出曲线,所有这些制剂包含本发明的有效成分。这些曲线表示为释放的有效成分的重量百分比与时间(以分钟表示)的函数。在60分钟粗体显示的垂直线标记了NaOH碱性溶液添加至模拟胃介质以模拟肠介质中的时刻。实施例下表A显示盐酸决奈达隆和碱形式的决奈达隆在本发明的脂质赋形剂中的溶解度。表A本发明的组合物按照下表1,3和4详细所示的组成制备。对比组合物(并非本发明的组合物)以下表2所示的组成制备。用于制备所述组合物的化合物的量以mg表示于所述表1和2中。"-"表示不存在的组分。"QS"表示足量。"HCl盐"表示盐酸决奈达隆。"碱"表示决奈达隆碱形式。"Pol."表示"泊洛沙姆"。"Geluc."表示"Gelucire"。"Crem.RH40"表示"CREMOPHORRH40)"表1*对应于含27%氢氧化钠的水溶液将制剂G1至G6和本发明的其它制剂稀释至百分之一的组成示于下表3和4。表3表4然后使用以下给出的实施例的组合物和使用对比组合物2制备不透明的0号尺寸的白色胶囊,以获得本发明的硬胶囊G1-G29和不是本发明的硬胶囊(即参比硬胶囊)。对比组合物1(非本发明)用于制备非本发明的片剂。表2制备所述组合物和硬胶囊(该方法如下所述)所需的设备如下:磁力搅拌器,烧杯,调节至称重量的精密天平,筛0.315mm,水浴,活塞式GilsonPipette1000μL,胶囊制造机。而且,用于制备所述组合物的Gelucire44/14在制备的前一天晚上在55°C烘烤。通过翻转锅而手动混匀。硬胶囊G1的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛选盐酸盐形式的决奈达隆,然后称重,-熔化和混合44/14和泊洛沙姆407,在水浴温度为55-60°C时以200rpm的搅拌速度缓慢搅拌约10分钟,-在水浴温度为55-60°C时以200rpm的搅拌速度添加丙二醇,-缓慢添加预先筛分的盐酸决奈达隆且在添加过程中以300-650rpm的速度剧烈搅拌而分散。添加后,以500rpm的混合速度和在水浴温度为55-60°C时混合30分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。硬胶囊G2的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛选盐酸盐形式的决奈达隆,然后称重,-熔化和混合44/14和泊洛沙姆407,在水浴温度为55-60°C时以200rpm的搅拌速度缓慢搅拌约10分钟,-缓慢添加预先筛分的盐酸决奈达隆且在添加过程中以300-650rpm的速度剧烈搅拌而分散。添加后,在水浴温度为55-60°C以500rpm的混合速度混合10分钟,-以500rpm的搅拌速度添加27%氢氧化钠溶液。添加后,在水浴温度为55-60°C混合30分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。硬胶囊G3的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛选盐酸盐形式的决奈达隆,然后称重,-缓慢添加预先筛分的盐酸决奈达隆且在添加过程中以300-650rpm的速度剧烈搅拌而分散。添加后,以350rpm的混合速度且在水浴温度为55-60°C混合30分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。硬胶囊G4的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛分决奈达隆,然后称重,-熔化和混合44/14和泊洛沙姆407,在水浴温度为55-60°C时以200rpm的搅拌速度缓慢搅拌约10分钟,-逐渐添加和溶解预先筛分的碱形式的决奈达隆,在添加过程中以300-650rpm的速度剧烈搅拌。添加后,以500rpm的混合速度和在水浴温度为55-60°C时混合30分钟,-逐渐添加水,以500rpm的搅拌速度且在水浴温度为55-60°C搅拌约10分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。硬胶囊G5的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛分决奈达隆,然后称重,-熔化和混合44/14和泊洛沙姆407,在水浴温度为55-60°C时以200rpm的搅拌速度缓慢搅拌约10分钟,-在水浴温度为55-60°C时以200rpm的搅拌速度添加丙二醇,-逐渐添加和溶解预先筛分的碱形式的决奈达隆,在添加过程中以300-650rpm的速度剧烈搅拌。添加后,以500rpm的混合速度和在水浴温度为55-60°C时混合30分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。硬胶囊G6的制备:操作:-将预先在用于制备的烧杯中熔化的44/14称重,-以0.315mm筛网筛分决奈达隆,然后称重,-熔化和混合44/14,在水浴温度为55-60°C时以200rpm的搅拌速度缓慢搅拌约10分钟,-逐渐添加和溶解预先筛分的碱形式的决奈达隆,在添加过程中以300-650rpm的速度剧烈搅拌。添加后,以500rpm的混合速度和在水浴温度为55-60°C时混合30分钟,-使用自动移液器,填充不透明的0号尺寸的白色硬胶囊。用单位重量填充胶囊。分配过程中的搅拌速度为500rpm且水浴温度为55-60°C,-密封后,将胶囊以垂直位置放置在胶囊制造机上以在室温进行固化。参比硬胶囊根据如胶囊G6相同的方案进行制备且具有以上表1和2所述的比例和成分。由如表2所述的对比组合物1构成的片剂根据制备该种剂型的通常技术进行制备。硬胶囊G7至G29根据如胶囊G6相同的方案进行制备且具有以上表3和4所述的比例和成分。评价当穿过胃肠道时药物组合物的有效成分在水性介质中的溶出为再现pH对有效成分的影响且特别是对其在穿过胃肠道过程中的溶出的影响,通过再现胃部的pH然后使用pH骤变再现肠的pH来模拟胃肠环境。溶出动力学使用简单的体外溶出测试通过pH骤变来研究。000原理:该原理由以下构成:通过研究其在37°C的溶出动力学(首先在pH4的模拟胃介质中,然后在pH6.5的模拟肠介质中,其时间间隔与胃肠道一致)确定配制的有效成分的溶出。000设备和方法000设备:精密天平(MettlerAE200或AT261),pH计(Knick或Schott或Inolab),恒温溶出浴6或7溶出杯(thermostatedDissolutest6or7bowls)(SotaxAT6或AT7),具有注射器(Térumo)的5μm滤器(PALLVersapore25mm),UV分光光度计(GilfordResponseII或PerkinElmer)或HPLC(Merck或Agilent)。溶出介质:代表胃肠道的模拟生理介质获自USP推荐的模拟胃液和模拟肠液,但在没有胃蛋白酶或胰酶的情况下使用。·模拟的胃液-USP(不含胃蛋白酶):-每900ml蒸馏水2g氯化钠,-使用浓盐酸(37%)将pH调节至1.2,-使用蒸馏水定量至1000ml。·模拟的肠液-USP(不含胰酶):-每900ml蒸馏水6.8g磷酸二氢钾,-使用浓氢氧化钠(10M)将pH调节至7.5,-使用蒸馏水定量至1000ml。通过以不同比例混合两种模拟液体而得到pH4的模拟胃介质,使用pH电极监测。肠类型的溶出介质使用几滴浓碳酸钠(10M)调节为pH6.5以模拟有效成分排入肠的起点,而不导致稀释。方法:·校准:有效成分的标准溶液在溶剂介质中制备,优选在流动相或乙醇或甲醇中制备,然后在有效成分的特征波长进行分析。然后测定校准直线,其显示了作为光密度(通过UV分光光度分析法)的函数或作为峰下面积(HPLC分析)的函数的浓度。所得直线的方程式可用于根据光密度或峰下面积的测量值确定溶解的有效成分的浓度。·000溶出动力学:使用的浓度对应于250ml中的剂量(即4个胶囊在500-ml杯中)。溶出动力学首先在溶出测试中在37°C恒温的杯中于pH4的模拟胃介质中进行研究,其中桨片在75rpm搅拌1小时,在5,15,30,45和60分钟时取样,然后在肠介质中进行研究(通过添加少量的浓碳酸钠(例如:500mlpH4的介质使用0.400ml)将pH升高至6.5,使用pH电极监测。监测动力学3小时且在75,90,120和180分钟时取样,各样品以5μm过滤,然后分析。·分析:可根据所需的灵敏度和研究的剂型使用两种分析方法:UV分光光度法(仅用于有效成分)或HPLC(仅用于有效成分或其制剂)。在时间t的浓度基于之前建立的校准进行测定。在通过UV分光光度法分析的过程中,对整个吸收光谱进行监测,至少在动力学研究结束时进行。此外,介质的pH在动力学研究结束时监测。000结果:绘制了有效成分作为时间的函数的溶出动力学(表示为释放的产物的百分比),首先在pH4(模拟胃介质)、然后在pH6.5(模拟肠介质),连续绘制在一个相同的曲线上,如图1所示。该溶出动力学以与硬胶囊G1-G7和参比硬胶囊相同的条件测量,这些硬胶囊具有如上限定的组成。相对参比硬胶囊,该片剂在有效成分的释放百分比方面显示有意义的、显著的改善,但仅在模拟的肠pH条件下改善。相比参比硬胶囊或相比片剂,在模拟的胃部pH的条件下在脂质赋形剂基质中包含盐酸决奈达隆而不含表面活性剂的硬胶囊G3在有效成分的释放百分比方面显示显著的改善。相比硬胶囊G3、相比参比硬胶囊和相比片剂,在表面活性剂的存在下在脂质赋形剂基质中包含盐酸决奈达隆的硬胶囊G1在有效成分的释放百分比方面显示非常显著的改善。相比参比硬胶囊,相比片剂和相比硬胶囊G3,在脂质赋形剂基质中包含从盐酸决奈达隆原位形成的决奈达隆碱的硬胶囊G2在有效成分的释放百分比方面显示非常显著的改善。相比参比硬胶囊,相比片剂和相比硬胶囊G3,在脂质赋形剂基质中包含游离碱形式的决奈达隆的硬胶囊G4和G5在有效成分的释放百分比方面也显示非常显著的改善,而且在溶出过程中的性能与硬胶囊G2相当。相比参比硬胶囊和片剂(其中不存在脂质赋形剂),在包含碱形式或盐酸盐形式的决奈达隆的制剂中脂质赋形剂的存在的有益效果可从图1所示的曲线看出。而且,可以看出相对于不含表面活性剂的硬胶囊,在组成中进一步包含表面活性剂的硬胶囊在有效成分的释放百分比方面有非常显著的改善(G1的曲线对比G3的曲线,以及G5的曲线对比G6的曲线)。而且,在基质中包含碱形式的决奈达隆的制剂的情况下,在模拟的胃部pH的条件下有效成分的释放百分比的曲线从最开始就很快,这与包含盐酸决奈达隆的制剂相反,该包含盐酸决奈达隆的制剂的释放在肠pH的条件下观察到最佳情况。Gelucire44/14的比例对碱形式的决奈达隆的影响可从图2所示的曲线看出增加Gelucire的比例的正影响。Gelucire44/14的比例对盐酸决奈达隆的影响可从图3所示的曲线看出增加Gelucire的比例的正影响。表面活性剂的比例对碱形式的决奈达隆的影响可从图4所示的曲线看出在根据本发明组合物中的表面活性剂的比例对于有效成分的释放没有影响。表面活性剂的比例对盐酸决奈达隆的影响可从图5所示的曲线看出在根据本发明组合物中的表面活性剂的比例对有效成分的释放具有有益效果,其中最佳比例为10至30%。表面活性剂的性质对碱形式的决奈达隆和盐酸决奈达隆的影响可从图6所示的曲线看出碱形式的释放动力学相当,无论使用哪种非离子表面活性剂。可从图7所示的曲线看出盐酸决奈达隆的释放动力学在HLB介于15至25时具有更好的曲线,尤其为约22。生物利用度的评价生物利用度是指药品吸收的量。其与给予的药品达到全身循环的剂量的比例相关,且与其达到该目标的速率相关。口服给药的生物利用度尤其取决于消化吸收和肠与肝中的首过代谢。方案:12名健康状况良好的年轻受试者,以空腹或在高脂肪食物过程中,通过摄取具有以上定义组成的硬胶囊G1、G2或G3而接受单剂量的400mgBID决奈达隆。血样在48小时内定期收集且收集的血浆通过LC-UV方法进行测试以确定作为时间的函数的决奈达隆的血浆浓度。基于由此获得的曲线测量Cmax、Tmax和AUC。所得结果示于下表4和5中。Cmax对应于决奈达隆的血浆浓度峰值。tmax对应于达到Cmax的时间。AUC对应于作为时间t的函数的血浆浓度的曲线下面积或积分。表5*Cmax(禁食)对应于禁食患者摄取本发明的硬胶囊所测量的Cmax与该相同禁食患者摄取参比硬胶囊所测量的Cmax的比例。**AUC(禁食)对应于禁食患者摄取本发明的硬胶囊所测量的AUC与该相同禁食患者摄取参比硬胶囊所测量的AUC的比例。***Cmax(随餐)对应于在用餐过程中患者摄取本发明的硬胶囊所测量的Cmax与在用餐过程中该相同患者摄取参比硬胶囊所测量的Cmax的比例。****AUC(随餐)对应于在用餐过程中患者摄取本发明的硬胶囊所测量的AUC与在用餐过程中该相同患者摄取参比硬胶囊所测量的AUC的比例。表6¤Cmax对应于在用餐过程中患者摄取给定硬胶囊所测量的Cmax与禁食患者摄取相同的给定硬胶囊所测量的Cmax的比例。¤¤用餐影响对应于在用餐过程中患者摄取给定硬胶囊所测量的AUC与禁食患者摄取相同的给定硬胶囊所测量的AUC的比例。结果:结果表明在禁食条件,相比参比硬胶囊,本发明的硬胶囊G1、G2、G3的生物利用度显著增加,胶囊G2最有效。而且,可以看出相对参比硬胶囊,本发明的硬胶囊用餐影响显著降低,胶囊G2具有最低用餐影响,大约为1.46。生物利用度的评价方案治疗和给药使用的剂量为60mg/动物,无论周期/条件,相当于6mg/kg(假设狗的重量为10kg)且相当于人使用的剂量为400mg(即对于体重70kg的人为约6mg/kg)。给药条件如下:·禁食周期:动物在给药前的晚上不进食。分别在给药1小时和4小时后给予水以及常规食物(SSNIFFhdH)。·进食周期:动物在给药10分钟前接受50g高脂肪食物(高脂肪SSNIFFEFDogFDAModel)(该食物具有的能量值为100kcal且由15%蛋白质,25%碳水化合物和50-60%脂肪构成)。然后分别在给药1小时和4小时后给予水和常规狗粮(SSNIFFhdH)。在给药0.5小时前用五肽胃泌素预处理。五肽胃泌素(6μg/kg,0.25mL/kg)肌内给药且使得可保持动物的胃部pH为2-3,从而模拟人的条件。给药所述胶囊,然后通过管饲法给予30ml水,其大概相当于在临床试验中给予人受试者240mL的体积。所述治疗为:治疗1:60mg盐酸决奈达隆胶囊,禁食条件,口服途径(参比硬胶囊)(ref2)治疗2:60mg盐酸决奈达隆以及Gelucire和泊洛沙姆407的胶囊,禁食条件,口服途径(=G1)。治疗3:从盐酸决奈达隆原位重构的60mg决奈达隆碱形式以及Gelucire和泊洛沙姆407的胶囊,禁食条件,口服途径(=G2)。治疗4:60mg决奈达隆碱形式以及Gelucire和泊洛沙姆407的胶囊,禁食条件,口服途径(=G5)。治疗5:60mg决奈达隆碱形式以及Gelucire和泊洛沙姆407的胶囊,进食条件,口服途径(=G5)。治疗6:60mg决奈达隆碱形式以及Gelucire但不含泊洛沙姆407的胶囊,禁食条件,口服途径(=G8)。治疗7:60mg决奈达隆碱形式以及Gelucire但不含泊洛沙姆407的胶囊,进食条件,口服途径(=G8)。取样和分析在以下样品收集时间将血样收集在包含肝素锂作为抗凝血剂的塑料管中:治疗前和给予各治疗后0.5,1,2,3,4,6,8和24小时。通过液相色谱质谱偶联(LC-MS/MS)使用探索性分析方法测定决奈达隆的血浆浓度。该方法对于测试化合物的检测下限为0.5ng/mL。结果的表示由单独的浓度通过非房室分析使用WinNonLin5.2.1软件(Pharsight,USA)且使用理论取样时间(条件是实际取样时间与理论时间的偏差不超过15%)计算药代动力学参数。对各治疗测量了以下药代动力学参数:-Cmax(ng/mL):对应于观察到的最大血浆浓度,-tmax(h):对应于获得最大浓度的观察时间,-AUClast:对应于作为时间t的函数的血浆浓度的曲线下面积或积分,通过梯形方法从t0计算至对应于最终可以计量的浓度的时间。-AUC:对应于作为外推至无穷大的时间的函数的血浆浓度的曲线下面积或积分。-T1/2z:最终消除半衰期也评估了以下参数:-基于Cmax和AUC的相对生物利用度-基于Cmax和AUC的用餐影响的比例。结果表7-第1组中的决奈达隆的药代动力学参数(平均值±SD(CV%))(每种制剂n=4)*中值(最小值至最大值)表8-第2组中的决奈达隆的药代动力学参数(平均值±SD(CV%))(每种制剂n=4)n=3;*中值(最小值至最大值)表9*中值(最小值至最大值)所有接受参比制剂的狗在禁食条件具有类似的暴露,无论哪一组。表10-禁食条件下具有90%CI的决奈达隆(%)的相对生物利用度(使用胶囊作为参照)对于2B1,n=3所有测试的制剂显示比参比胶囊更高的生物利用度,其在禁食条件下具有的相对生物利用度为271%至500%。含Gelucire、盐酸决奈达隆和原位重构的碱(Frel=301%)的制剂显示比参比胶囊更高的生物利用度,如上文的临床测试所示。含Gelucire和使用天然碱的制剂显示的相对生物利用度类似于使用盐酸决奈达隆和原位重构的碱的Gelucire制剂(当在禁食条件与参照对比时),如置信区间的覆盖区所示。含Gelucire和含或不含泊洛沙姆的制剂显示相似的相对生物利用度,比参比胶囊的生物利用度高3至5倍。表11-对于含Gelucire、决奈达隆碱形式和泊洛沙姆的胶囊,用餐影响的比例治疗CmaxAUClastAUC进食/禁食0.73(0.35-1.53)0.71(0.41-1.23)0.70(0.41-1.21)当含泊洛沙姆的Gelucire胶囊与高脂肪食物一起给予时,Cmax有稍微降低的趋势,不与高脂肪食物一起给予的Cmax为其1.4倍。该降低并不显著,因为CI为90%。表12-对于含Gelucire、决奈达隆碱形式和不含泊洛沙姆的胶囊,用餐影响的比例治疗CmaxAUClastAUC进食/禁食1.93(1.16-3.21)1.39(0.82-2.35)1.32(0.81-2.16)当不含泊洛沙姆的Gelucire胶囊与高脂肪食物一起给予时,存在正向的用餐影响的趋势。事实上Cmax增加为1.9倍,AUClast增加为1.4倍且AUC增加为1.3倍。然而,该增加对AUC并不显著,因为CI为90%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1