药物抗体缀合物的制作方法
本发明涉及新型药物缀合物,药物接头化合物,涉及它们的制备方法,包含所述药物缀合物的药物组合物和它们作为抗肿瘤剂的用途。
发明背景
国际公布号WO-A-2007/144423和WO-A-2009/080761公开了新型二氢吡喃-2-酮和四氢吡喃-2-酮衍生物,其呈现非常有希望的抗肿瘤活性。在WO-A-2007/144423中公开的PM060184目前在用于预防和治疗实体瘤的I期临床试验中。
近年来,随着更有效靶向和杀伤癌细胞的药物实体的开发,癌症的治疗进展显著。研究人员利用了由靶细胞如癌细胞选择性地表达的细胞表面受体和抗原,基于在肿瘤的实例中结合肿瘤特异的或肿瘤相关的抗原的抗体,开发了药物实体。为了实现它,已经将细胞毒性分子如化疗药物,细菌和植物毒素以及放射性核素与结合肿瘤特异的或肿瘤相关的细胞表面抗原的单克隆抗体化学连接(参见,例如,国际专利申请WO-A-2004/010957,WO-A-2006/060533和WO-A-2007/024536)。这样的化合物通常被称为药物、毒素和放射性核素“缀合物”。在药物缀合物与肿瘤细胞结合并且释放和/或活化药物部分的细胞毒活性时,发生肿瘤细胞杀伤。由药物缀合物提供的选择性使对正常细胞的毒性最小化,从而提高药物在患者中的耐受性。已经得到上市许可的这种类型的药物抗体缀合物的三个实例是:用于急性骨髓性白血病(acute myelogenous leukemia)的吉姆单抗奥佐米星(Gemtuzumab ozogamicin),用于复发和顽固性霍奇金淋巴瘤(Hodgkin lymphoma)和间变性大细胞淋巴瘤(anaplastic large cell lymphoma)的Brentuximab vedotin,以及用于乳腺癌,特别是HER2+的ado-曲妥珠单抗(Trastuzumab)emtansine。
用于癌症化疗的药物的有效性通常依赖于癌症和正常组织之间生长速度,生化通路和的生理特征的不同。因此,大多数标准的化疗药物是相对非特异的并且呈现剂量限制性毒性,导致非最佳的疗效。一种选择性靶向恶性细胞和不健康组织的途径是使用识别在肿瘤细胞表面上表达的肿瘤相关抗原的特异性单克隆抗体(mAb)[Meyer,D.L.&Senter,P.D.(2003)Recent advances in antibody drug conjugates for cancer therapy(用于癌症治疗的抗体药物缀合物中的近期进展).Annu.Rep.Med.Chem.,38,229-237;Chari,R.V.(2008)Targeted cancer therapy:conferring specificity to cytotoxic drugs(靶向癌症治疗:赋予细胞毒性药物特异性).Acc.Chem.Res.41,98-107]。mAb和衍生物目前是最快速增长的治疗分子类型。在过去25年已经批准了多于30种G型免疫球蛋白(IgG)和相关制剂,主要用于癌症和炎性疾病。在肿瘤学中,mAb常常与细胞毒性药物组合以增强其治疗功效。备选地,小的抗肿瘤分子可以与mAb化学缀合,既用作载体(增加半衰期),也用作靶向剂(选择性)。由于单克隆抗体对肿瘤相关抗原的高选择性,良好的药物代谢动力学,和相对低的固有毒性,已经针对单克隆抗体(mAb)用于靶向药物递送的用途做出了相当大的努力。mAb药物缀合物(ADC)通常经条件稳定的接头体系通过将抗癌药物与mAb共价连接而形成。在与细胞表面抗原结合时,用于多数ADC的mAb被主动转运到溶酶体或其他细胞内隔室,在那里,酶,低pH,或还原剂促进药物释放。
抗原必须具备高的肿瘤细胞选择性,以限制毒性和脱靶(off-target)效应。已经在临床前模型和临床试验中研究了大量肿瘤相关抗原,举几个例子,包括在下列各项中过表达的抗原:B细胞(例如,CD20,CD22,CD40,CD79),T细胞(CD25,CD30),癌细胞(HER2,EGFR,EpCAM,EphB2,PSMA),内皮(endoglin),或基质细胞(成纤维细胞活化的蛋白)[Teicher BA.Antibody-drug conjugate targets(抗体-药物缀合物靶点).Curr Cancer Drug Targets 9(8):982-1004,2009]。ADC靶点的主要和关键性质是其被内在化的能力;这可以是抗原自身的固有特点,或其可以通过抗体与其抗原的结合来引入。实际上,ADC内在化对于降低与药物有效荷载的细胞外递送相关的毒性是关键的。
就缀合的小分子而言,并且与种类繁多的推定的抗原靶点相比,目前在临床试验中积极地研究了在ADC中用作有效荷载的有限数量的家族的细胞毒性药物:刺孢霉素(calicheamycin)(Pfizer),多卡霉素(duocarmycins)(Synthon),吡咯并苯并二氮杂(Spirogen),依立替康(irinotecan)(Immunomedics),美坦生类化合物(maytansinoids)(DM1和DM4;ImmunoGen+Genentech/Roche,Sanofi-Aventis,Biogen Idec,Centocor/Johnson&Johnson,Millennium/Takeda),和阿里他汀(auristatins)(MMAE和MMAF;Seattle Genetics+Genentech/Roche,MedImmune/AstraZeneca,Bayer-Schering,Celldex,Progenics,Genmab)。刺孢霉素,多卡霉素和吡咯并苯并二氮杂是DNA小沟结合剂,依立替康是拓扑异构酶I抑制剂,而美坦生类化合物和阿里他汀是微管解聚剂。它们的共同特征之一是它们与例如,用于第一代ADC的多柔比星(doxorubicin)(10-7M)相比的高游离药物效力(10-9至10-11M)。成功的另一关键要素是对允许释放活性代谢产物的接头连接的“允许”位置的清楚理解(类似于常规的前药)。
有趣的是,这三种细胞毒性来源的ADC的代表已经到达后期临床试验。曲妥珠单抗emtansine(T-DM1),通过稳定接头与美坦生类化合物半合成药物连接的曲妥珠单抗(2013年2月22日FDA批准,用于晚期HER2阳性乳腺癌);伊珠单抗(Inotuzumab)奥佐米星(CMC-544),利用酸不稳定接头(乙酰基苯氧基-丁酸)缀合于刺孢霉素的人源化抗CD22mAb(G5/44,IgG4)(B细胞非霍奇金淋巴瘤);Brentuximab vedotin,经马来酰亚胺己酰基-缬氨酰-瓜氨酸基-对氨基苄基氨基甲酸酯接头连接于一甲基阿里他汀E(MMAE)的人源化抗CD30mAb(2011年8月19日FDA批准,用于间变性大细胞淋巴瘤和霍奇金淋巴瘤)。
接头代表ADC结构的关键组分。已经研究了很多类型的第二代接头,包括酸不稳定腙接头(溶酶体)(例如吉姆单抗和伊珠单抗奥佐米星);基于二硫化物的接头(还原性细胞内环境);不可切割的硫醚接头(在溶酶体中的代谢降解)(例如,曲妥珠单抗emtansine);肽接头(例如瓜氨酸-缬氨酸)(溶酶体蛋白酶如组织蛋白酶-B)(例如brentuximab vedotin):参见,例如,WO-A-2004/010957,WO-A-2006/060533和WO-A-2007/024536。还已经描述了借助尺寸排阻层析(SEC)的抗体-药物缀合物的纯化[参见,例如,Liu等人,Proc.Natl.Acad.Set(USA),93:8618-8623(1996),和Chari等人,Cancer Research,52:127-131(1992)]。
曲妥珠单抗(Herceptin)是干预HER2/neu受体的单克隆抗体。其主要用途是治疗某些乳腺癌。HER受体是嵌入细胞膜中并将分子信号从细胞外部(称为EGF的分子)传达到细胞内部,并开启或关闭基因的蛋白。HER蛋白刺激细胞增殖。在一些癌症中,尤其是某些类型的乳腺癌中,HER2过表达,并且引起癌细胞不可控地复制。
HER2基因在20-30%的早期乳腺癌中扩增,这使得它在细胞膜中过表达表皮生长因子(EGF)受体。在一些类型的癌症中,HER2可以在没有生长因子到达并结合受体的情况下发出信号,使得其在细胞中的作用是组成型的;然而,在这种情况下曲妥珠单抗不是有效的。
当其正常发挥作用时,HER2通路促进细胞生长和分裂;然而当其过表达时,细胞生长加速超过其正常极限。在一些类型的癌症中,利用该通路促进了快速的细胞生长和增殖以及由此的肿瘤形成。在癌细胞中,HER2蛋白可以比在正常细胞中多表达多至100倍(2百万相对20,000/细胞)。这种过表达导致强烈和持续的增殖信号传导和因此的肿瘤形成。HER2的过表达还引起检查点(checkpoint)的失活,导致增殖的甚至更大的增加。
发明概述
存在对不基于目前已经用作有效荷载的细胞毒性药物家族的新型活性药物缀合物的需求。本发明解决了该需求。其还提供用于制备本发明的药物缀合物的新型药物接头化合物,用于制备本发明的新型药物缀合物的方法,含有所述药物缀合物的药物组合物和它们作为抗肿瘤剂的用途,以及用于治疗癌症的包含本发明的药物缀合物的试剂盒。
在本发明的第一方面,提供一种药物缀合物,其包含与药物缀合物的剩余部分共价连接的药物部分,所述化合物具有式[D-(X)b-(AA)w-(L)-]n-Ab其中:
D是具有下式(I)的药物部分或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
A选自
R1,R2和R3各自独立地选自氢,ORa,OCORa,OCOORa,NRaRb,NRaCORb,NRaC(=NRa)NRaRb,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基和取代的或未取代的C2-C12炔基,其中所述任选的取代基是一个或多个取代基Rx;
R3’选自氢,CORa,COORa,CONRaRb,S(O)Ra,SO2Ra,P(O)(Ra)Rb,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基和取代的或未取代的C2-C12炔基,其中所述任选的取代基是一个或多个取代基Rx;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自由以下各项组成的组:氢,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基和取代的或未取代的C2-C12炔基,其中所述任选的取代基是一个或多个取代基Rx;
R11选自由以下各项组成的组:氢,CORa,COORa,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基和取代的或未取代的C2-C12炔基,或R11和R12可以同它们所连接的相应N原子和C原子一起形成具有一个或多个环并且在所述一个或多个环中任选地包含除NR11的氮原子之外的一个或多个另外的选自氧、氮和硫原子的杂原子的5至14元取代的或未取代的不饱和或饱和杂环基,其中所述任选的取代基是一个或多个取代基Rx;
R13和R14各自独立地选自由以下各项组成的组:氢,CORa,COORa,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基,取代的或未取代的C2-C12炔基和取代的或未取代的C4-C12烯炔基,其中所述任选的取代基是一个或多个取代基Rx;
Ra和Rb各自独立地选自由以下各项组成的组:氢,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基,取代的或未取代的C2-C12炔基,一个或多个环的具有6至18个碳原子的取代的或未取代的芳基,和具有一个或多个环的5至14元取代的或未取代的不饱和或饱和杂环基,其中所述任选的取代基是一个或多个取代基Rx;
取代基Rx选自由以下各项组成的组:可以任选地被至少一个基团Ry取代的C1-C12烷基,可以任选地被至少一个基团Ry取代的C2-C12烯基,可以任选地被至少一个基团Ry取代的C2-C12炔基,卤素原子,氧代基,硫基,氰基,硝基,ORy,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,S(O)Ry,SO2Ry,P(O)(Ry)ORz,NRyRz,NRyCORz,NRyC(=O)NRyRz,NRyC(=NRy)NRyRz,一个或多个环的具有6至18个碳原子的可以任选地被选自由Ry,ORy,OCORy,OCOORy,NRyRz,NRyCORz和NRyC(=NRy)NRyRz组成的组的一个或多个可以相同或不同的取代基取代的芳基,包含被如上文限定的任选地取代的芳基取代的具有1至12个碳原子的烷基的芳烷基,包含被如上文限定的任选地取代的芳基取代的具有1至12个碳原子的烷氧基的芳烷氧基,和具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的5至14元饱和或不饱和杂环基,所述杂环基任选地被一个或多个取代基Ry取代,并且在任何给定基团上存在多于一个任选的取代基的情况下,所述任选的取代基Ry可以相同或不同;
Ry和Rz各自独立地选自由以下各项组成的组:氢,C1-C12烷基,被至少一个卤素原子取代的C1-C12烷基,包含被一个或多个环的具有6至18个碳原子的芳基取代的C1-C12烷基的芳烷基和包含被具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的5至14元不饱和或饱和杂环基取代的C1-C12烷基的杂环烷基;
每个虚线表示任选的另外的键;
每个波形线表示基团A与药物部分的剩余部分的共价连接点;
X是延伸基团;
每个AA独立地是氨基酸单元;
L是接头基团;
w是范围为0至12的整数;
b是0或1的整数;
Ab是包含至少一个抗原结合位点的部分;并且
n是基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分的比例并且在1至20的范围内
如我们将在下文更详细解释和举例说明的,本发明的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物代表了解决以上概述的需要除了基于目前已经用作有效荷载的显示优异抗肿瘤活性的三种主要细胞毒性药物家族的那些之外的另外的药物缀合物的问题的突破。
在本发明的第一方面的优选实施方案中,提供根据权利要求1的药物缀合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中D是选自式(Ia)和(Ib)的药物部分:
其中(Ia)和(Ib)的波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
A选自
其中部分A的波形线表示与药物部分的剩余部分的共价连接点;
R1,R2,R3,R3’,R4,R5,R6,R7,R8,R9,R10,R11,R12,R13,Ra,Rb,Rx,Ry和Rz各自如本发明的第一方面中所限定;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢,取代的或未取代的C1-C12烷基,取代的或未取代的C2-C12烯基和取代的或未取代的C2-C12炔基,其中所述任选的取代基是一个或多个取代基Rx;
R18选自由以下各项组成的组:氢,可以任选地被至少一个基团Rx取代的C1-C12烷基,一个或多个芳环的具有6至18个碳原子的芳基,所述芳基任选地被一个或多个取代基Rx取代,和具有一个或多个环的5至14元取代的或未取代的不饱和或饱和杂环基,其中所述任选的取代基是一个或多个取代基Rx;
R27选自氢,取代的或未取代的C1-C12烷基和卤素;
并且每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在。
在本发明的第二方面,提供式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是选自由以下各项组成的式的组的接头:
波形线各自表示与如果存在的(AA)w,或与X的共价连接点;
G选自卤素,-O-甲磺酰基和-O-甲苯磺酰基;
J选自卤素,羟基,-N-琥珀酰亚胺基氧基,-O-(4-硝基苯基),-O-五氟苯基,-O-四氟苯基和-O-C(O)-OR20;
R19选自-C1-C12亚烷基-,-C3-C8碳环,-O-(C1-C12亚烷基),一个或多个环的可以任选地被一个或多个取代基Rx取代的-C6-C18亚芳基,-C1-C12亚烷基-C6-C18亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C18亚芳基-C1-C12亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C3-C8碳环)-,-(C3-C8碳环)-C1-C12亚烷基-,-C5-C14杂环-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C5-C14杂环)-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(C5-C14杂环)-C1-C12亚烷基-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(OCH2CH2)r-和-CH2-(OCH2CH2)r-,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
R20是C1-C12烷基或一个或多个芳环的具有6至18个碳原子的芳基,所述芳基任选地被一个或多个取代基Rx取代;
r是范围为1-10的整数;并且
D,X,AA和w各自如本文发明的第一方面中所限定。
在本发明的第三方面,提供根据本发明的第一方面的药物缀合物,其用作药物。
在本发明的第四方面,提供根据本发明的第一方面的药物缀合物,其用于治疗癌症,并且更优选选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌的癌症。最优选的癌症选自结直肠癌,乳腺癌,白血病,淋巴瘤,和卵巢癌。
在本发明的第五方面,提供药物组合物,其包含根据本发明的第一方面的药物缀合物和药用载体。
在本发明的第六方面,提供预防或治疗癌症的方法,其包括向需要其的患者施用有效量的根据本发明的第一方面的药物缀合物。优选地,所述癌症选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌。最优选的癌症选自结直肠癌,乳腺癌,白血病,淋巴瘤和卵巢癌。
在本发明的第七方面,提供根据本发明的第一方面的药物缀合物在制备药物中的用途,所述药物用于治疗癌症,并且更优选选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌的癌症。最优选的癌症选自结直肠癌,乳腺癌,白血病,淋巴瘤和卵巢癌。
在本发明的第八方面,提供试剂盒,其包含治疗有效量的根据本发明的第一方面的药物缀合物和药用载体。所述试剂盒用于治疗癌症,并且更优选选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌的癌症。最优选的癌症选自结直肠癌,乳腺癌,白血病,淋巴瘤,和卵巢癌。
本发明的第九方面提供制备根据本发明的第一方面的药物缀合物的方法,其包括将包含至少一个抗原结合位点的部分Ab和式(I),(Ia)或(Ib)的药物D缀合,Ab和D如本发明的第一方面中所限定。
优选实施方案详述
在本发明的化合物中,R1,R2,R3,R3’,R4,R5,R6,R7,R8,R9,R10,R11,R12,R13,R14,R15,R16,R17,R17’,R18,R18’,R20,R24,R24’,R25,R26,R27,Ra,Rb,Rx,Ry和Rz的定义中的烷基可以是具有1至12个碳原子的直链或支链烷基,并且它们优选为具有1至6个碳原子的烷基,更优选为甲基,乙基或异丙基,并且最优选甲基。在M和Q的定义中,它们可以是具有1至6个碳原子的直链或支链烷基。
在本发明的化合物中,R1,R2,R3,R3’,R4,R5,R6,R7,R8,R9,R10,R11,R12,R13,R14,R15,R16,R17,R17’,R18’,R24,R24’,R25,R26,Ra,Rb和Rx的定义中的烯基是支链的或无支链的,并且可以具有一个或多个双键和2至12个碳原子。优选地,它们具有2至6个碳原子,并且更优选地,它们是具有2,3或4个碳原子的支链的或无支链的烯基。
在本发明的化合物中,R1,R2,R3,R3’,R4,R5,R6,R7,R8,R9,R10,R11,R12,R13,R14,R15,R16,R17,R17’,R18’,R24,R24’,R25,R26,Ra,Rb和Rx的定义中的炔基是支链的或无支链的,并且可以具有一个或多个三键和2至12个碳原子。优选地,它们具有2至6个碳原子,并且更优选地,它们是具有2,3或4个碳原子的支链的或无支链的炔基。
在本发明的化合物中,R13和R14的定义中的烯炔基(alkenynyl)是支链的或无支链的,并且可以具有一个或多个双键和一个或多个三键。优选地,它们具有4至12个碳原子,并且更优选它们是具有6至10个碳原子的支链的或无支链的炔基。
在本发明的化合物中,R27,Rx,Ry和Rz的定义中的卤素取代基包括F,Cl,Br和I,优选Cl。
在本发明的化合物中,Rx,Ra,Rb,R18,和可以由R11和R12与它们所连接的氮原子和碳原子一起形成的杂环基的定义中的5至14元饱和或不饱和杂环基是具有一个或多个环,在所述一个或多个环中包含至少一个氧、氮或硫原子的杂环基。杂环基是可以是杂芳族基或杂脂环族基的基团,其后者可以是部分不饱和的,芳族和脂环族杂环基含有1至3个分离的或稠合的环。优选地,杂芳族基和杂脂环族基含有5至10个环原子。在本发明的化合物中的合适的杂芳族基含有一个,两个或三个选自N,O或S原子的杂原子并且包括,例如,喹啉基,包括8-喹啉基,异喹啉基,香豆素基,包括8-香豆素基,吡啶基,吡嗪基,吡唑基,嘧啶基,呋喃基,吡咯基,噻吩基,噻唑基,异噻唑基,三唑基,四唑基,异唑基,唑基,咪唑基,吲哚基,异吲哚基,吲唑基,吲嗪基,酞嗪基,蝶啶基,嘌呤基,二唑基,噻二唑基,呋咱基,哒嗪基,三嗪基,噌啉基,苯并咪唑基,苯并呋喃基,苯并呋咱基,苯并噻吩基,苯并噻唑基,苯并唑基,喹唑啉基,喹喔啉基,萘啶基和呋喃吡啶基。在本发明的化合物中的合适的杂脂环族基含有一个,两个或三个选自N,O或S原子的杂原子并且包括,例如,吡咯烷基,四氢呋喃基,二氢呋喃基,四氢噻吩基,四氢硫代吡喃基,哌啶基,吗啉基,硫代吗啉基,噻烷基,哌嗪基,氮杂环丁基(azetidinyl),氧杂环丁基(oxetanyl),硫杂环丁基(thietanyl),高哌啶基(homopiperidyl),氧杂环庚基(oxepanyl),硫杂环庚基(thiepanyl),氧杂氮杂基,二氮杂基,硫杂氮杂基,1,2,3,6-四氢吡啶基,2-吡咯啉基,3-吡咯啉基,吲哚啉基,2H-吡喃基,4H-吡喃基,二烷基,1,3-二氧杂环戊基(1,3-dioxolanyl),吡唑啉基,二硫烷基,二硫杂环戊基(dithiolanyl),二氢吡喃基,二氢噻吩基,二氢呋喃基,吡唑烷基,咪唑啉基,咪唑烷基,3-氮杂双环[3.1.0]己基,3-氮杂双环[4.1.0]庚基,3H-吲哚基,和喹嗪基。
在本发明的化合物中,R18,R20,Ra,Rb,和Rx的定义中的芳基,是含有分离的和/或稠合的芳基并且具有6至18个环原子的单或多环化合物并且是任选地取代的。典型的芳基含有1至3个分离的或稠合的环。优选芳基含有6至12个碳环原子。特别优选的芳基包括取代的或未取代的苯基,取代的或未取代的萘基,取代的或未取代的联苯基,取代的或未取代的菲基和取代的或未取代的蒽基,并且最优选取代的或未取代的苯基,其中取代基如上文所示,取决于芳基是取代基R20,R28,Ra和Rb中的一种或其是取代基Rx。
在本发明的化合物中,Rx,Ry和Rz的定义中的芳烷基包含被一个或多个如上文所限定和举例说明的芳基取代的如上文所限定和举例说明的烷基。优选的实例包括任选地取代的苄基,任选地取代的苯基乙基和任选地取代的萘甲基。
在本发明的化合物中,Rx的定义中的芳烷氧基包含被一个或多个如上文所限定和举例说明的芳基取代的具有1至12个碳原子的烷氧基。优选地,所述烷氧基部分具有1至6个碳原子并且所述芳基含有6至约12个碳环原子,并且最优选所述芳烷氧基是任选地取代的苄氧基,任选地取代的苯基乙氧基和任选地取代的萘基甲氧基。
在本发明的化合物中,Ry和Rz的定义中的杂环烷基包含被一个或多个如上文所限定和举例说明的杂环基取代的如上文所限定和举例说明的烷基。优选地,所述杂环烷基包含被1或2个环原子的具有5至10个环原子的杂环基取代的并且可以是芳族的,部分饱和的或完全饱和的具有1至6个碳原子的烷基。更优选地,所述杂环烷基包含被选自由以下各项组成的组的杂环基取代的甲基或乙基:吡咯烷基,咪唑烷基,哌啶基,哌嗪基,吗啉基,四氢呋喃基,烷基,硫烷基,8-喹啉基,异喹啉基,吡啶基,吡嗪基,吡唑基,嘧啶基,呋喃基,吡咯基,噻吩基,噻唑基,异噻唑基,三唑基,四唑基,异唑基,唑基和苯并咪唑。
在本发明的化合物中,R19的定义中的亚烷基是具有1至12个碳原子的直链或支链的亚烷基并且M和X的定义中的亚烷基是具有1至6个碳原子的直链或支链的亚烷基。优选地,R19的定义中的亚烷基是具有1至8个碳原子的直链或支链的亚烷基,更优选具有1至6个碳原子的直链或支链的亚烷基。对于M,优选的是具有1至3个碳原子的直链或支链的亚烷基。在X的定义中,X的定义中的亚烷基优选为具有2至4个碳原子的直链或支链的亚烷基。
在本发明的化合物中,R19和M的定义中的碳环基团是具有3至8个碳原子的环烷基,所述环烷基在环烷基环的任意位置具有两个共价键,所述共价键将所述环烷基连接于药物缀合物的其余部分。优选地,R19和M的定义中的碳环是具有3至7个碳原子的环烷基,并且更优选具有5至7个碳原子的碳环基团。
在本发明的化合物中,R19的定义中的亚芳基是一个或多个环的具有6至18个碳原子的芳基,所述芳基在芳环体系上的任意位置具有两个共价键,所述共价键将所述亚芳基连接于药物缀合物的其余部分。优选地,R19的定义中的亚芳基是一个或多个环的具有6至12个碳原子的芳基,所述芳基在芳环体系上的任意位置具有两个共价键,并且最优选它们是亚苯基。
在本发明的化合物中,R19的定义中的杂环基是含有1至3个具有5至14个环原子的分离的或稠合的环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的杂环基,其中在所述杂环基的环体系上的任意位置有两个共价键。所述杂环基是可以是杂芳基或杂脂环族基(后者可以是部分不饱和的)的基团。优选地,R19的定义中的杂环基是含有1至3个具有5至12个环原子的分离的或稠合的环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的杂环基,其中在所述杂环基的环体系上的任意位置有两个共价键。
在取代基上有多于一个任选的取代基Rx的情况下,每个取代基Rx可以相同或不同。
根据本发明的第一方面的优选的药物缀合物包括:
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L是选自由以下各项组成的组的接头基团:
其中
波形线表示与Ab(右侧的波形线)以及如果存在的(AA)w,或如果存在的(X)b,或药物部分(左侧的波形线)的共价连接点;
R19选自-C1-C12亚烷基-,-C3-C8碳环,-O-(C1-C12亚烷基),一个或多个环的可以任选地被一个或多个取代基Rx取代的-C6-C18亚芳基,-C1-C12亚烷基-C6-C18亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C18亚芳基-C1-C12亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C3-C8碳环)-,-(C3-C8碳环)-C1-C12亚烷基-,-C5-C14杂环-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C5-C14杂环)-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(C5-C14杂环)-C1-C12亚烷基-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(OCH2CH2)r-,和-CH2-(OCH2CH2)r-,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
M选自由以下各项组成的组:-C1-C6亚烷基-,-C1-C6亚烷基-(C3-C8碳环)-,-(CH2CH2O)s-,-C1-C6亚烷基-(C3-C8碳环)-CON(H或C1-6烷基)-C1-C6亚烷基-,可以任选地被一个或多个取代基Rx取代的亚苯基,其中亚苯基部分可以任选地被一个或多个取代基Rx取代的亚苯基-C1-C6亚烷基-和-C1-C6亚烷基-CON(H或C1-6烷基)C1-C6亚烷基-;
Q选自由以下各项组成的组:-N(H或C1-6烷基)亚苯基-和-N(H或C1-6烷基)-(CH2)s;
r是范围为1至10的整数;并且
s是范围为1至10的整数。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L选自由以下各项组成的组:
其中:
波形线表示与Ab(右侧的波形线)以及如果存在的(AA)w,或如果存在的(X)b,或药物部分(左侧的波形线)的共价连接点;
R19选自-C1-C12亚烷基-,-O-(C1-C12亚烷基),一个或多个环的可以任选地被一个或多个取代基Rx取代的-C6-C12亚芳基,-C1-C12亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C12亚芳基-C1-C12亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C5-C12杂环-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C5-C12杂环)-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(C5-C12杂环)-C1-C12亚烷基-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(OCH2CH2)r-,和-CH2-(OCH2CH2)r-,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;并且
M选自由以下各项组成的组:-C1-C6亚烷基-,-C1-C6亚烷基-(C3-C8碳环)-和可以任选地被一个或多个取代基Rx取代的亚苯基;并且
r是范围为1-6的整数。
·选自式(IV)和(V)的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物:
其中:
X是如本发明的第一方面中所限定的延伸基团;
每个AA独立地是如本发明的第一方面中所限定的氨基酸单元;
w是范围为0至12的整数;
b是0或1的整数;
D是药物部分;
Ab是包含至少一个抗原结合位点的部分;
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分的比例,并且在1至20的范围内;
R19选自-C1-C8亚烷基-,-O-(C1-C8亚烷基),-C1-C8亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C12亚芳基-C1-C8亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;并且
M选自由以下各项组成的组:-C1-C3亚烷基-和-C1-C3亚烷基-(C5-C7碳环)-。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其选自式(IV)和(V):
其中:
X是延伸基团;
每个AA独立地是氨基酸单元;
w是范围为0至12的整数;
b是0或1的整数;
D是药物部分;
Ab是包含至少一个抗原结合位点的部分;
n是其中L如(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分的比例,并且在1至20的范围内;
R19选自-C1-C6亚烷基-,亚苯基-C1-C6亚烷基-,其中的亚苯基可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,其中无论是单独的或与碳链中的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,具有6至12个碳原子的芳基,卤素原子,硝基和氰基,并且优选R19是-C1-C6亚烷基;并且
M是-C1-C3亚烷基-(C5-C7碳环)-。
·优选的是,在式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物的定义中,L如对以上所述基团的优选的定义中限定的并且(AA)w具有式(II):
其中波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;并且
R21,每次出现时,选自由以下各项组成的组:氢,甲基,异丙基,异丁基,仲丁基,苄基,对羟基苄基,-CH2OH,-CH(OH)CH3,-CH2CH2SCH3,-CH2CONH2,-CH2COOH,-CH2CH2CONH2,-CH2CH2COOH,-(CH2)3NHC(=NH)NH2,-(CH2)3NH2,-(CH2)3NHCOCH3,-(CH2)3NHCHO,-(CH2)4NHC(=NH)NH2,-(CH2)4NH2,-(CH2)4NHCOCH3,-(CH2)4NHCHO,-(CH2)3NHCONH2,-(CH2)4NHCONH2,-CH2CH2CH(OH)CH2NH2,2-吡啶基甲基-,3-吡啶基甲基-,4-吡啶基甲基-,苯基,环己基,
并且w是范围为0至12的整数。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L是如对于以上所述基团的优选的定义中限定的并且(AA)w具有式(II),其中:
R21,每次出现时,选自由以下各项组成的组:氢,甲基,异丙基,仲丁基,苄基,吲哚基甲基,-(CH2)3NHCONH2,-(CH2)4NH2,-(CH2)3NHC(=NH)NH2和-(CH2)4NHC(=NH)NH2;并且
w是范围为0至6的整数。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L是如对于以上所述基团的优选的定义中限定的,其中w是0或2,并且当w是2时,则(AA)w具有式(III),其中:
波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
R22选自甲基,苄基,异丙基,仲丁基和吲哚基甲基;并且
R23选自甲基,-(CH2)4NH2,-(CH2)3NHCONH2和-(CH2)3NHC(=NH)NH2。
·另外,优选的是,在式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物的定义中,L和AA是如对于以上所述基团的优选的定义中限定的并且X是选自由以下各项组成的组的延伸基团:
-CONH-(C1-C6亚烷基)NH-;
-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-;
-CONH-(C1-C6亚烷基)NH-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-;
-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-;
-COCH2NH-COCH2-NH-;
-COCH2NH-;
-CONH-(C1-C6亚烷基)S-;
-CONH-(C1-C6亚烷基)NHCO(C1-C6亚烷基)S-;
-(C1-C6亚烷基)NHCO(C1-C6亚烷基)S-;
-(C1-C6亚烷基)S-;
-(C1-C6亚烷基)NH-;和
-(C1-C6亚烷基)NH-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L和AA是如对于以上所述基团的优选的定义中限定的,并且X是选自由以下各项组成的组的延伸基团:
-CONH-(C2-C4亚烷基)NH-;
-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基;
-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;
-COCH2NH-COCH2-NH-;
-CONH-(C2-C4亚烷基)S-;
-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-;
-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-;
-(C2-C4亚烷基)S-;
-(C2-C4亚烷基)NH-;和
-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-。
·根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物,其中L和AA是如对于以上所述基团的优选的定义中限定的并且X是选自由以下各项组成的组的延伸基团:
-CONH(CH2)3NHCOOCH2-亚苯基-NH-;
-CONH(CH2)3NH-;
-CONH(CH2)3-S-;
-CONH(CH2)3NHCO(CH2)2S-;
-(CH2)3NHCO(CH2)2S-;
-(CH2)3S-;
-(CH2)3NH-;和
-(CH2)3NHCOOCH2-亚苯基-NH-。
·优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R2和R3各自独立地选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R2和R3各自是氢。
·另一个优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R1选自氢,ORa和OCORa,其中Ra选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R1是氢或甲氧基。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R3’选自氢,CORa,和取代的或未取代的C1-C6烷基,其中Ra是取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R3’是氢。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢,取代的和未取代的甲基,取代的和未取代的异丙基和取代的和未取代的叔丁基,其中所述任选的取代基是一个或多个取代基Rx。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R5,R7,R8,R9和R10各自是氢。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R4和R6各自是甲基。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R12是异丙基,叔丁基或苄基。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,AA和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R11和R13各自独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R11和R13各自是氢。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢,取代的或未取代的C1-C6烷基,取代的或未取代的C2-C6烯基和取代的或未取代的C2-C6炔基,其中所述任选的取代基是一个或多个取代基Rx,更优选R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz,和NRyCORz,其中Ry和Rz各自选自氢原子和具有1至6个碳原子的烷基,还更优选R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地是氢或C1-C6烷基,并且最优选R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自是氢或甲基。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R18选自氢,可以任选地被一个或多个取代基Rx取代的C1-C6烷基,一个或多个芳环的具有6至12个碳原子的芳基,所述芳基任选地被一个或多个取代基Rx取代,和具有一个或多个环的5至10元不饱和或饱和杂环基,所述杂环基任选地被一个或多个取代基Rx取代,其中所述取代基Rx选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,卤素原子,具有1至6个碳原子的烷基氨基和具有1至6个碳原子的二烷基氨基,更优选R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基和可以任选地被至少一个基团Rx取代的苯基,并且最优选R18是氢或苯基,特别是氢。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中R27选自氢原子,卤素原子或取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx,并且更优选R27选自氢原子和氯原子。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中由一条或多条虚线连接的每对碳原子通过双键键合。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
R1选自氢,ORa和OCORa,其中Ra选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R2和R3各自独立地选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R3’选自氢,CORa,和取代的或未取代的C1-C6烷基,其中Ra是取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R11和R13独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26独立地选自由以下各项组成的组:
氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz和NRyCORz,其中Ry和Rz各自选自氢原子和具有1至6个碳原子的烷基;
R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基,一个或多个芳环的具有6至12个碳原子的芳基,所述芳基任选地被一个或多个取代基Rx取代,和具有一个或多个环的5至10元不饱和或饱和杂环基,所述杂环基任选地被一个或多个取代基Rx取代,其中取代基Rx选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,卤素原子,具有1至6个碳原子的烷基氨基和具有1至6个碳原子的二烷基氨基;
R27选自氢,卤素和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;并且
每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
R1是氢或甲氧基;
R2和R3各自是氢;
R3’是氢;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢,取代的或未取代的甲基,取代的或未取代的异丙基和取代的或未取代的叔丁基,其中所述任选的取代基是一个或多个取代基Rx;
R11和R13各自是氢;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:
氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz和NRyCORz,其中Ry和Rz各自选自氢原子,和具有1至6个碳原子的烷基。
R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基和可以任选地被至少一个基团Rx取代的苯基;
R27是氢原子或氯原子;并且
每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是式(Ia)或(Ib)的化合物,或其药用盐、
酯、溶剂化物、互变异构体或立体异构体,其中:
R1是氢或甲氧基;
R2和R3各自是氢;
R3’是氢;
R5,R7,R8,R9和R10各自是氢;
R4和R6各自是甲基;
R11和R13各自是氢;
R12是异丙基,叔丁基或苄基;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢和C1-C6烷基,优选氢和甲基;
R18选自氢和苯基,优选氢;
R27是氢原子或氯原子;并且
由一条或多条虚线连接的每对碳原子通过双键键合。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是选自以下各项的式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是选自以下各项的式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w和X如上文所限定并且其中D是选自以下各项的式(I),(Ia)或(Ib)的化合物,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且其中包含至少一个抗原结合位点的部分Ab是抗原结合肽。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是抗体,单域抗体或其抗原结合片段。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是单克隆,多克隆抗体或双特异性抗体,并且其中抗体或其抗原结合片段源自任何物种,优选人,小鼠或兔。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是选自由以下各项组成的组的抗体或其抗原结合片段:人抗体,人抗体的抗原结合片段,人源化抗体,人源化抗体的抗原结合片段,嵌合抗体,嵌合抗体的抗原结合片段,糖基化抗体和糖基化抗原结合片段。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是抗体或其抗原结合片段,其中所述抗体或其抗原结合片段是选自由以下各项组成的组的抗原结合片段:Fab片段,Fab’片段,F(ab’)2片段和Fv片段。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是抗体或其抗原结合片段,其中所述抗体或其抗原结合片段是免疫特异结合癌细胞抗原,病毒抗原,产生与自身免疫病相关的自身免疫抗体的细胞的抗原,微生物抗原的单克隆抗体,并且优选免疫特异结合癌细胞抗原的单克隆抗体。
·进一步优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物是这样的,其中L,(AA)w,X和D如上文所限定,并且包含至少一个抗原结合位点的部分Ab是选自由以下各项组成的组的抗体:阿昔单抗(Abciximab),阿仑单抗(Alemtuzumab),巴利昔单抗(Basiliximab),贝伐单抗(Bevacizumab),西妥昔单抗(Cetuximab),达克珠单抗(Daclizumab),Glembatumumab,吉姆单抗(Gemtuzumab),替伊莫单抗(Ibritumomab),伊珠单抗(Inotuzumab),拉贝珠单抗(Labetuzumab),Lorvotuzumab,米拉珠单抗(Milatuzumab),尼妥珠单抗(Nimotuzumab),奥马珠单抗(Omalizumab),帕利珠单抗(Palivizumab),帕尼单抗(Panitumumab),Pinatuzumab,利妥昔单抗(Rituximab),Vorsetuzumab,曲妥珠单抗(Trastuzumab),抗CD4抗体,抗CD5抗体和抗CD13抗体,或其免疫活性部分,其中优选所述抗体选自阿昔单抗,阿仑单抗,巴利昔单抗,贝伐单抗,西妥昔单抗,达克珠单抗,Glembatumumab,吉姆单抗,替伊莫单抗,伊珠单抗,拉贝珠单抗,Lorvotuzumab,米拉珠单抗,尼妥珠单抗,奥马珠单抗,帕利珠单抗,帕尼单抗,Pinatuzumab,利妥昔单抗,Vorsetuzumab,曲妥珠单抗和抗CD4抗体,或其免疫活性部分,并且还更优选阿昔单抗,阿仑单抗,巴利昔单抗,贝伐单抗,西妥昔单抗,达克珠单抗,吉姆单抗,替伊莫单抗,尼妥珠单抗,奥马珠单抗,帕利珠单抗,帕尼单抗,利妥昔单抗和曲妥珠单抗,或其免疫活性部分。在这些中,特别优选的是曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分;或所述抗体选自曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,尤其是曲妥珠单抗或其免疫活性部分;或所述抗体选自抗CD5抗体和抗CD13抗体或其免疫活性部分,尤其是抗CD13抗体或其免疫活性部分。
·特别优选的根据本发明的第一方面的式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物包含以下各项:(a)根据本发明的第一方面的药物缀合物,其中:L选自由以下各项组成的组:
并且
其中:
波形线表示与Ab(右侧的波形线)以及如果存在的(AA)w,或如果存在的(X)b,或药物部分(左侧的波形线)的共价连接点;
R19选自-C1-C12亚烷基-,-O-(C1-C12亚烷基),一个或多个环的可以任选地被一个或多个取代基Rx取代的-C6-C12亚芳基,-C1-C12亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C12亚芳基-C1-C12亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C5-C12杂环-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C5-C12杂环)-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(C5-C12杂环)-C1-C12亚烷基-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(OCH2CH2)r-和-CH2-(OCH2CH2)r-,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
M选自由以下各项组成的组:-C1-C6亚烷基-,-C1-C6亚烷基-(C3-C8碳环)-和可以任选地被一个或多个取代基Rx取代的亚苯基;
r是范围为1-6的整数;
(AA)w具有式(II):
其中波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
R21,每次出现时,选自由以下各项组成的组:氢,甲基,异丙基,异丁基,仲丁基,苄基,对羟基苄基,-CH2OH,-CH(OH)CH3,-CH2CH2SCH3,-CH2CONH2,-CH2COOH,-CH2CH2CONH2,-CH2CH2COOH,-(CH2)3NHC(=NH)NH2,-(CH2)3NH2,-(CH2)3NHCOCH3,-(CH2)3NHCHO,-(CH2)4NHC(=NH)NH2,-(CH2)4NH2,-(CH2)4NHCOCH3,-(CH2)4NHCHO,-(CH2)3NHCONH2,-(CH2)4NHCONH2,-CH2CH2CH(OH)CH2NH2,2-吡啶基甲基-,3-吡啶基甲基-,4-吡啶基甲基-,苯基,环己基,
w是范围为0至12的整数;
其中X是选自以下各项的延伸基团:-CONH-(C1-C6亚烷基)NH-,-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-,-CONH-(C1-C6亚烷基)NH-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-,-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-,-COCH2NH-COCH2-NH-,-COCH2-NH-,-CONH-(C1-C6亚烷基)S-,-CONH-(C1-C6亚烷基)NHCO(C1-C6亚烷基)S-,-(C1-C6亚烷基)NHCO(C1-C6亚烷基)S-,-(C1-C6亚烷基)S-,-(C1-C6亚烷基)NH-和-(C1-C6亚烷基)NH-COO-CH2-(可以任选地被一个或多个取代基Rx取代的亚苯基)-NH-;
D是式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
R1选自氢,ORa和OCORa,其中Ra选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R2和R3各自独立地选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R3’选自氢,CORa,和取代的或未取代的C1-C6烷基,其中Ra是取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R11和R13独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:
氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz和NRyCORz,其中Ry和Rz各自选自氢原子和具有1至6个碳原子的烷基;
R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基,一个或多个芳环的具有6至12个碳原子的芳基,所述芳基任选地被一个或多个取代基Rx取代,和具有一个或多个环的5至10元不饱和或饱和杂环基,所述杂环基任选地被一个或多个取代基Rx取代,其中所述取代基Rx选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,卤素原子,具有1至6个碳原子的烷基氨基和具有1至6个碳原子的二烷基氨基;
R27选自氢,卤素和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在;
包含至少一个抗原结合位点的部分Ab是抗体或其抗原结合片段并且其选自由以下各项组成的组:人抗体,人抗体的抗原结合片段,人源化抗体,人源化抗体的抗原结合片段,嵌合抗体,嵌合抗体的抗原结合片段,糖基化抗体和糖基化抗原结合片段;并且
n是基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在1至12的范围内。
(b)选自式(IV)和(V)的根据本发明的第一方面的药物缀合物:
其中:
R19选自-C1-C8亚烷基-,-O-(C1-C8亚烷基),-C1-C8亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,和-C6-C12亚芳基-C1-C8亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
M选自由以下各项组成的组:-C1-C3亚烷基-和-C1-C3亚烷基-(C5-C7碳环)-;
(AA)w具有式(II)
其中:
波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
R21,每次出现时,选自由以下各项组成的组:氢,甲基,异丙基,仲丁基,苄基,吲哚基甲基,-(CH2)3NHCONH2,-(CH2)4NH2,-(CH2)3NHC(=NH)NH2和-(CH2)4NHC(=NH)NH2;
w是0至6的整数;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-,-COCH2NH-COCH2-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;
D是式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
R1是氢或甲氧基;
R2和R3各自是氢;
R3’是氢;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢,取代的和未取代的甲基,取代的和未取代的异丙基和取代的和未取代的叔丁基,其中所述任选的取代基是一个或多个取代基Rx;
R11和R13各自是氢;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:
氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz,NRyCORz,其中Ry和Rz各自选自氢原子和具有1至6个碳原子的烷基。
R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基,和任选地被一个或多个取代基RX取代的苯基;
R27是氢原子或氯原子;
每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在;
包含至少一个抗原结合位点的部分Ab是抗体或其抗原结合片段,其中所述抗体或抗原结合片段是免疫特异结合癌细胞抗原,病毒抗原,产生与自身免疫病相关的自身免疫抗体的细胞的抗原,微生物抗原的单克隆抗体,并且优选免疫特异结合癌细胞抗原的单克隆抗体;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至8的范围内。
(c)根据第一方面的药物缀合物,其选自式(IV)和(V):
其中:
R19选自-C1-C6亚烷基-,-亚苯基-C1-C6亚烷基-,其中的亚苯基可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,其中无论是单独的或与碳链中的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,具有6至12个碳原子的芳基,卤素原子,硝基和氰基,并且优选R19是C1-C6亚烷基;
M是-C1-C3亚烷基-(C5-C7碳环)-;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
R22选自甲基,苄基,异丙基,仲丁基和吲哚基甲基;
R23选自甲基,-(CH2)4NH2,-(CH2)3NHCONH2和-(CH2)3NHC(=NH)NH2;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基或氰基)-NH-,-COCH2NH-COCH2-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;
D是式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
R1是氢或甲氧基;
R2和R3各自是氢;
R3’是氢;
R5,R7,R8,R9和R10各自是氢;
R4和R6各自是甲基;
R11和R13各自是氢;
R12是异丙基,叔丁基或苄基;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢和C1-C6烷基,优选氢和甲基;
R18选自氢和苯基,优选氢;
R27是氢原子或氯原子;
通过一条或多条虚线连接的各个碳对通过双键键合;
包含至少一个抗原结合位点的部分Ab是选自由以下各项组成的组的单克隆抗体:阿昔单抗,阿仑单抗,巴利昔单抗,贝伐单抗,西妥昔单抗,达克珠单抗,Glembatumumab,吉姆单抗,替伊莫单抗,伊珠单抗,拉贝珠单抗,Lorvotuzumab,米拉珠单抗,尼妥珠单抗,奥马珠单抗,帕利珠单抗,帕尼单抗,Pinatuzumab,利妥昔单抗,Vorsetuzumab,曲妥珠单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分,优选其是选自由以下各项组成的组的单克隆抗体:阿昔单抗,阿仑单抗,巴利昔单抗,贝伐单抗,西妥昔单抗,达克珠单抗,Glembatumumab,吉姆单抗,替伊莫单抗,伊珠单抗,拉贝珠单抗,Lorvotuzumab,米拉珠单抗,尼妥珠单抗,奥马珠单抗,帕利珠单抗,帕尼单抗,Pinatuzumab,利妥昔单抗,Vorsetuzumab,曲妥珠单抗和抗CD4抗体,或其免疫活性部分,并且还更优选阿昔单抗,阿仑单抗,巴利昔单抗,贝伐单抗,西妥昔单抗,达克珠单抗,吉姆单抗,替伊莫单抗,尼妥珠单抗,奥马珠单抗,帕利珠单抗,帕尼单抗,利妥昔单抗和曲妥珠单抗,或其免疫活性部分。在这些中,特别优选的是选自以下各项的抗体:曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体,或其免疫活性部分,更优选所述抗体选自曲妥珠单抗,抗CD13抗体,和抗CD4抗体,抗CD5抗体或其免疫活性部分;或选自曲妥珠单抗,利妥昔单抗和抗CD4抗体的抗体,或其免疫活性部分,优选曲妥珠单抗或其免疫活性部分;或选自抗CD5抗体和抗CD13抗体的抗体或其免疫活性部分,特别是抗CD13抗体或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内。
(d)选自式(IV)和(V)的根据本发明的第一方面的药物缀合物:
其中:
R19是-C3-C6亚烷基-;
M是-C1-C3亚烷基-(C5-C7碳环)-;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,其中波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-,-COCH2NH-COCH2-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;
D是选自以下组的式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分,并且更优选其选自曲妥珠单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分,或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,优选曲妥珠单抗或其免疫活性部分;或其选自抗CD5抗体和抗CD13抗体或其免疫活性部分,特别是抗CD13抗体或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内。
(e)选自式(IV)和(V)的根据本发明的第一方面的药物缀合物:
其中:
R19是-C3-C6亚烷基-;
M是-C1-C3亚烷基-(C5-C7碳环)-;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,并且波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-,-COCH2NH-COCH2-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;
D是选自以下各项的式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分;更优选其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,并且最优选其是曲妥珠单抗或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分的比例,并且在3至5的范围内。
(f)根据本发明的第一方面的药物缀合物,其具有式(IV):
其中:
R19是C5亚烷基-;
b是1;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,并且波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;并且
X是选自-CONH(CH2)3NHCOOCH2-亚苯基-NH-,和-CONH(CH2)3NH-的延伸基团;
或具有式(V)
其中M是-甲基-亚环己基-;
b是1;
w是0;并且
X是选自-CONH(CH2)3S-和-CONH(CH2)3NHCO(CH2)2S-的延伸基团;
D是选自以下各项的式(Ia)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab是曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分,更优选其选自曲妥珠单抗,抗CD13抗体,抗CD4抗体和抗CD5抗体,或其免疫活性部分;或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体,或其免疫活性部分,并且最优选其是曲妥珠单抗或其免疫活性部分;或其选自抗CD5抗体和抗CD13抗体或其免疫活性部分,并且最优选其是抗CD13抗体或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内,并且优选4。
(g)根据本发明的第一方面的药物缀合物,其具有式(IV):
其中R19是-C5亚烷基-;
b是1;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,并且波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;并且
X是选自-(CH2)3NHCOOCH2-亚苯基-NH-,和--(CH2)3NH-的延伸基团;
或具有式(V)
其中M是-甲基-亚环己基-;
b是1;
w是0;并且
X是选自-(CH2)3S-和-(CH2)3NHCO(CH2)2S-的延伸基团;
D是选自以下各项的式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分;并且更优选其选自曲妥珠单抗,抗CD13抗体,抗CD4抗体,和抗CD5抗体或其免疫活性部分;或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体,或其免疫活性部分;并且最优选其是曲妥珠单抗或其免疫活性部分;或其选自抗CD5抗体和抗CD13抗体或其免疫活性部分,特别是抗CD13抗体或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内,并且优选4。
h)根据本发明的第一方面的药物缀合物,其具有式(IV):
其中R19是-C5亚烷基-;
b是1;
w是0或2,并且在w是2的情况下,(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,并且波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;并且
X是选自-(CH2)3NHCOOCH2-亚苯基-NH-和-(CH2)3NH-的延伸基团;
或具有式V)
其中M是-甲基-亚环己基-;
b是1;
w是0;并且
X是选自-(CH2)3S-和-(CH2)3NHCO(CH2)2S-的延伸基团:
D是下式的药物部分
,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体,或其免疫活性部分;并且最优选其是曲妥珠单抗或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内,并且优选4。
i)根据本发明的第一方面的药物缀合物,其具有式(IV):
其中R19是-C5亚烷基-;
b是1;
w是0或2,并且在w是2的情况下,(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,并且波形线表示与如果存在的(X)b,或药物部分(左侧的波形线)以及与接头(右侧的波形线)的共价连接点;并且
X是选自-CONH(CH2)3NHCOOCH2-亚苯基-NH-和-CONH(CH2)3NH-的延伸基团;
或具有式(V)
其中M是-甲基-亚环己基-;
b是1;
w是0;并且
X是选自-CONH(CH2)3S-和-CONH(CH2)3NHCO(CH2)2S-的延伸基团:
D是下式的药物部分
或其药用盐、酯、溶剂化物、互变异构体或立体异构体,
其中波形线表示与如果存在的(X)b,或如果存在的(AA)w,或接头基团L的共价连接点;
包含至少一个抗原结合位点的部分Ab选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体,或其免疫活性部分;并且更优选其选自曲妥珠单抗或其免疫活性部分;并且
n是其中L如式(IV)或(V)中所限定的基团[D-(X)b-(AA)w-(L)-]与包含至少一个抗原结合位点的部分Ab的比例,并且在3至5的范围内,并且优选4。
j)根据本发明的第一方面的抗体药物缀合物,其选自由以下各项组成的组:
其中各自选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分,优选曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,并且最优选曲妥珠单抗或其免疫活性部分;或备选地抗CD5抗体和抗CD13抗体或其免疫活性部分,并且最优选抗CD13抗体或其免疫活性部分。更优选抗体药物缀合物选自由以下各项组成的组:
其中选自曲妥珠单抗和抗CD13抗体或其免疫活性部分,优选曲妥珠单抗或其免疫活性部分;或优选抗CD13抗体或其免疫活性部分,
其中是曲妥珠单抗或其免疫活性部分,
其中是曲妥珠单抗或其免疫活性部分,
其中选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分,优选曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,并且最优选曲妥珠单抗或其免疫活性部分;或备选地抗CD5抗体和抗CD13抗体,或其免疫活性部分,并且最优选抗CD13抗体或其免疫活性部分,
其中是曲妥珠单抗或其免疫活性部分,
其中选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体,和抗CD13抗体或其免疫活性部分,优选曲妥珠单抗,利妥昔单抗和抗CD4抗体或其免疫活性部分,并且最优选曲妥珠单抗或其免疫活性部分;或备选地抗CD5抗体和抗CD13抗体或其免疫活性部分,并且最优选抗CD13抗体或其免疫活性部分,
其中是曲妥珠单抗或其免疫活性部分,和
其中是曲妥珠单抗或其免疫活性部分。
特别优选地,根据本发明的抗体药物缀合物应该是分离或纯化的形式。
优选的根据本发明的第二方面的式D-X-(AA)w-(L1)b的化合物包括:
·根据本发明的第二方面的式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是下式的接头:
其中:
波形线表示与如果存在的(AA)w,或与X的共价连接点;
R19选自-C1-C12亚烷基-,-O-(C1-C12亚烷基),一个或多个环的可以任选地被一个或多个取代基Rx取代的-C6-C12亚芳基,-C1-C12亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C12亚芳基-C1-C12亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C5-C12杂环-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-C1-C12亚烷基-(C5-C12杂环)-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(C5-C12杂环)-C1-C12亚烷基-,其中所述杂环基可以是具有一个或多个环并且在所述一个或多个环中包含至少一个氧、氮或硫原子的饱和或不饱和基团,所述基团任选地被一个或多个取代基Rx取代,-(OCH2CH2)r-和-CH2-(OCH2CH2)r-,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
r是范围为1-6的整数;并且
D,X,AA和w各自如本发明的第一方面中所限定。
·根据本发明的第二方面的式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是下式的接头:
其中:
波形线表示与如果存在的(AA)w,或与X的共价连接点;
R19选自-C1-C8亚烷基-,-O-(C1-C8亚烷基),-C1-C8亚烷基-C6-C12亚芳基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,-C6-C12亚芳基-C1-C8亚烷基-,其中的亚芳基是一个或多个环的,其可以任选地被一个或多个取代基Rx取代,其中无论是单独的或与碳链的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个取代基Rx取代;
(AA)w具有式(II):
其中波形线表示与X(左侧的波形线)以及与L1或与氢原子(右侧的波形线)的共价连接点;
其中R21,每次出现时,选自由以下各项组成的组:氢,甲基,异丙基,仲丁基,苄基,吲哚基甲基,-(CH2)3NHCONH2,-(CH2)4NH2,-(CH2)3NHC(=NH)NH2和-(CH2)4NHC-(=NH)NH2,并且w是范围为0至6的整数;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-,-COCH2NH-COCH2-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)-NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;并且
D是式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体,其中:
其中(Ia)和(Ib)的波形线表示与X的共价连接点;
A选自
其中部分A的波形线表示与药物部分的其余部分的共价连接点;
R1选自氢,ORa和OCORa,其中Ra选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R2和R3各自独立地选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R3’选自氢,CORa,和取代的或未取代的C1-C6烷基,其中Ra是取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R4,R5,R6,R7,R8,R9,R10和R12各自独立地选自氢和取代的和未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R11和R13各自独立地选自氢和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:
氢和取代的或未取代的C1-C6烷基,其中任选的取代基选自由以下各项组成的组:具有1至6个碳原子的烷氧基,羟基,氧代基,卤素原子,OCORy,OCOORy,CORy,COORy,OCONRyRz,CONRyRz,NRyRz,NRyCORz,其中Ry和Rz各自选自氢原子和具有1至6个碳原子的烷基。
R18选自氢,可以任选地被至少一个基团Rx取代的C1-C6烷基,和任选地被一个或多个取代基RX取代的苯基;
R27选自氢,卤素和取代的或未取代的C1-C6烷基,其中所述任选的取代基是一个或多个取代基Rx;
并且每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在。
·根据本发明的第二方面的式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是下式的基团:
其中:
波形线表示与如果存在的(AA)w,或与X的共价连接点;
R19选自-C1-C6亚烷基-,亚苯基-C1-C6亚烷基-,其中的亚苯基可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,其中无论是单独的或与碳链中的另一部分连接的,上述亚烷基取代基各自可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,具有6至12个碳原子的芳基,卤素原子,硝基和氰基,并且优选R19是C1-C6亚烷基;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中波形线表示与X(左侧的波形线)以及与L1或与氢原子(右侧的波形线)的共价连接点;
R22选自甲基,苄基,异丙基,仲丁基和吲哚基甲基;
R23选自甲基,-(CH2)4NH2,-(CH2)3NHCONH2和-(CH2)3NHC(=NH)NH2;
X是选自以下各项的延伸基团:-CONH-(C2-C4亚烷基)NH-,-CONH(C2-C4亚烷基)NHCOO-CH2-(可以任选地被一个或多个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO-(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;并且
D是式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中(Ia)和(Ib)的波形线表示与X的共价连接点;
A选自
其中部分A的波形线表示与药物部分的其余部分的共价连接点;
R1是氢或甲氧基;
R2和R3各自是氢;
R3’是氢;
R5,R7,R8,R9和R10各自是氢;
R4和R6各自是甲基;
R12是异丙基,叔丁基或苄基;
R11和R13各自是氢;
R15,R16,R17,R17’,R18’,R24,R24’,R25和R26各自独立地选自由以下各项组成的组:氢和C1-C6烷基,优选氢和甲基;
R18选自氢和苯基,并且优选氢;
R27是氢或卤素;
并且每个虚线表示任选的另外的键,但当在R25所连接的C原子与R26和R27所连接的C原子之间存在三键时,则R25以及R26或R27之一不存在。
·根据本发明的第二方面的式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是下式的接头:
其中:
波形线表示与如果存在的(AA)w,或与X的共价连接点;
R19是-C3-C6亚烷基-;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
R22是异丙基,R23是-(CH2)3NHCONH2,其中波形线表示与X(左侧的波形线)以及与L1或与氢原子(右侧的波形线)的共价连接点;
X是选自由以下各项组成的组的延伸基团:-CONH-(C2-C4亚烷基)NH-,-COO-CH2-亚苯基-NH-,其中所述亚苯基可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基,-CONH-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-,-CONH-(C2-C4亚烷基)S-,-CONH-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)NHCO(C1-C3亚烷基)S-,-(C2-C4亚烷基)S-,-(C2-C4亚烷基)NH-和-(C2-C4亚烷基)NH-COO-CH2-(可以任选地被一至四个选自由以下各项组成的组的取代基Rx取代的亚苯基:具有1至6个碳原子的烷基,具有1至6个碳原子的烷氧基,卤素原子,硝基和氰基)-NH-;并且
D是选自以下组的式(Ia)或式(Ib)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体:
其中波形线表示与X的共价连接点。
·根据本发明的第二方面的式D-X-(AA)w-L1或式D-X-(AA)w-H的化合物,其中:
L1是下式的基团:
其中:
波形线表示与如果存在的(AA)w,或与X的共价连接点;
R19是-C5亚烷基-;
w是0或2,并且在w是2的情况下,则(AA)w具有式(III):
其中R22是异丙基,R23是-(CH2)3NHCONH2,其中波形线表示与X(左侧的波形线)以及与L1或与氢原子(右侧的波形线)的共价连接点;
X是选自-CONH(CH2)3NHCOOCH2-亚苯基-NH-,和-CONH(CH2)3NH-,-CONH(CH2)3-S-和-CONH(CH2)3NHCO(CH2)2S-的延伸基团;并且
D是式(Ia)的药物部分,或其药用盐、酯、溶剂化物、互变异构体或立体异构体选自:
其中波形线表示与X的共价连接点。
·式D-X-(AA)w-L1的化合物,其选自:
·式D-X-(AA)w-H的化合物,其选自:
本发明的药物缀合物中的术语“药用盐,酯,溶剂化物,互变异构体或立体异构体”是指任何药用盐,酯,溶剂化物,水合物或立体异构形式或当施用于患者时能够直接或间接地提供本文所述的化合物的任何其他化合物。然而,将理解,非药用盐也落在本发明的范围内,因为那些可以用于制备药用盐。盐,前药和衍生物的制备可以通过本领域中已知的方法进行。
例如,本文提供的化合物的药用盐通过常规化学方法从含有碱性或酸性部分的母体化合物合成。通常,这样的盐,例如,通过将这些化合物的游离酸或碱形式与化学计量量的适当的碱或酸在水中或在有机溶剂中或在二者的混合物中反应来制备。通常,非水介质如醚,乙酸乙酯,乙醇,异丙醇或乙腈是优选的。酸加成盐的实例包括无机酸加成盐如,例如,盐酸盐,氢溴酸盐,氢碘酸盐,硫酸盐,硝酸盐,磷酸盐,和有机酸加成盐如,例如,乙酸盐,三氟乙酸盐,马来酸盐,富马酸盐,柠檬酸盐,草酸盐,琥珀酸盐,酒石酸盐,苹果酸盐,扁桃酸盐,甲磺酸盐和对苯磺酸盐。碱加成盐的实例包括无机盐如,例如,钠盐,钾盐,钙盐,铵盐,和有机碱盐如,例如,乙二胺,乙醇胺,N,N-二亚烷基乙醇胺,三乙醇胺和碱性氨基酸盐。
本发明的药物缀合物可以是作为游离化合物或作为溶剂化物(例如水合物)的晶体形式并且意在两种形式都在本发明的范围内。溶剂化的方法通常是本领域已知的。
作为本发明的药物缀合物的前药的任何化合物均在本发明的范围和精神内。术语“前药”以其最宽泛的意义使用并且包括在体内被转变为本发明的化合物的那些衍生物。本领域技术人员将会想到这样的衍生物,并且其包括,例如,其中游离羟基被转变为酯衍生物的化合物。很多合适的前药是本领域技术人员公知的,并且可以在例如,Burger“Medicinal Chemistry and Drug Discovery(药物化学和药物发现)第6版(Donald J.Abraham编,2001,Wiley)和“Design and Applications of Prodrugs(前药的设计和应用)”(H.Bundgaard编,1985,Harwood Academic Publishers)中找到,其内容通过引用结合在本文中。
关于本发明的化合物,药用酯不特别限制,并且可以由本领域普通技术人员选择。在所述酯的情况下,优选这样的酯可以由生物过程如体内水解裂解。构成所述酯的基团(当其酯表示为-COOR时,以R示出的基团)可以是,例如,C1-C4烷氧基C1-C4烷基如甲氧基乙基,1-乙氧基乙基,1-甲基-1-甲氧基乙基,1-(异丙氧基)乙基,2-甲氧基乙基,2-乙氧基乙基,1,1-二甲基-1-甲氧基甲基,乙氧基甲基,丙氧基甲基,异丙氧基甲基,丁氧基甲基或叔丁氧基甲基;C1-C4烷氧基化的C1-C4烷氧基C1-C4烷基如2-甲氧基乙氧基甲基;C6-C10芳氧基C1-C4烷基如苯氧基甲基;卤化的C1-C4烷氧基C1-C4烷基如2,2,2-三氯乙氧基甲基或双(2-氯乙氧基)甲基;C1-C4烷氧基羰基C1-C4烷基如甲氧基羰基甲基;氰基C1-C4烷基如氰基甲基或2-氰基乙基;C1-C4烷基硫代甲基如甲基硫代甲基或乙基硫代甲基;C6-C10芳基硫代甲基如苯基硫代甲基或萘基硫代甲基;可以任选地的被一个或多个卤素原子取代的C1-C4烷基磺酰基C1-C4低级烷基,如2-甲磺酰基乙基或2-三氟甲磺酰基乙基;C6-C10芳基磺酰基C1-C4烷基如2-苯磺酰基乙基或2-甲苯磺酰基乙基;C1-C7脂族酰氧基C1-C4烷基如甲酰氧基甲基,乙酰氧基甲基,丙酰氧基甲基,丁酰氧基甲基,新戊酰氧基甲基,戊酰氧基甲基,异戊酰氧基甲基,己酰氧基甲基,1-甲酰氧基乙基,1-乙酰氧基乙基,1-丙酰氧基乙基,1-丁酰氧基乙基,1-新戊酰氧基乙基,1-戊酰氧基乙基,1-异戊酰氧基乙基,1-己酰氧基乙基,2-甲酰氧基乙基,2-乙酰氧基乙基,2-丙酰氧基乙基,2-丁酰氧基乙基,2-新戊酰氧基乙基,2-戊酰氧基乙基,2-异戊酰氧基乙基,2-己酰氧基乙基,1-甲酰氧基丙基,1-乙酰氧基丙基,1-丙酰氧基丙基,1-丁酰氧基丙基,1-新戊酰氧基丙基,1-戊酰氧基丙基,1-异戊酰氧基丙基,1-己酰氧基丙基,1-乙酰氧基丁基,1-丙酰氧基丁基,1-丁酰氧基丁基,1-新戊酰氧基丁基,1-乙酰氧基戊基,1-丙酰氧基戊基,1-丁酰氧基戊基,1-新戊酰氧基戊基或1-新戊酰氧基己基;C5-C6环烷基羰基氧基C1-C4烷基如环戊基羰基氧基甲基,环己基羰基氧基甲基,1-环戊基羰基氧基乙基,1-环己基羰基氧基乙基,1-环戊基羰基氧基丙基,1-环己基羰基氧基丙基,1-环戊基羰基氧基丁基或1-环己基羰基氧基丁基;C6-C10芳基羰基氧基C1-C4烷基如苯酰氧基甲基;C1-C6烷氧基羰基氧基C1-C4烷基如甲氧基羰基氧基甲基,1-(甲氧基羰基氧基)乙基,1-(甲氧基羰基氧基)丙基,1-(甲氧基羰基氧基)丁基,1-(甲氧基羰基氧基)戊基,1-(甲氧基羰基氧基)己基,乙氧基羰基氧基甲基,1-(乙氧基羰基氧基)乙基,1-(乙氧基羰基氧基)丙基,1-(乙氧基羰基氧基)丁基,1-(乙氧基羰基氧基)戊基,1-(乙氧基羰基氧基)己基,丙氧基羰基氧基甲基,1-(丙氧基羰基氧基)乙基,1-(丙氧基羰基氧基)丙基,1-(丙氧基羰基氧基)丁基,异丙氧基羰基氧基甲基,1-(异丙氧基羰基氧基)乙基,1-(异丙氧基羰基氧基)丁基,丁氧基羰基氧基甲基,1-(丁氧基羰基氧基)乙基,1-(丁氧基羰基氧基)丙基,1-(丁氧基羰基氧基)丁基,异丁氧基羰基氧基甲基,1-(异丁氧基羰基氧基)乙基,1-(异丁氧基羰基氧基)丙基,1-(异丁氧基羰基氧基)丁基,叔丁氧基羰基氧基甲基,1-(叔丁氧基羰基氧基)乙基,戊氧基羰基氧基甲基,1-(戊氧基羰基氧基)乙基,1-(戊氧基羰基氧基)丙基,己氧基羰基氧基甲基,1-(己氧基羰基氧基)乙基或1-(己氧基羰基氧基)丙基;C5-C6环烷氧基羰基氧基C1-C4烷基如环戊氧基羰基氧基甲基,1-(环戊氧基羰基氧基)乙基,1-(环戊氧基羰基氧基)丙基,1-(环戊氧基羰基氧基)丁基,环己氧基羰基氧基甲基,1-(环己氧基羰基氧基)乙基,1-(环己氧基羰基氧基)丙基或1-(环己氧基羰基氧基)丁基;[5-(C1-C4烷基)-2-氧代-1,3-二氧戊环-4基-]甲基如(5-甲基-2-氧代-1,3-二氧戊环-4基-)甲基,(5-乙基-2-氧代-1,3-二氧戊环-4基-)甲基,(5-丙基-2-氧代-1,3-二氧戊环-4基-)甲基,(5-异丙基-2-氧代-1,3-二氧戊环-4基-)甲基或(5-丁基-2-氧代-1,3-二氧戊环-4基-)甲基;[5-(可以任选地被一个或多个C1-C4烷基,C1-C4烷氧基或卤素原子取代的苯基)-2-氧代-1,3-二氧戊环-4基-]甲基如(5-苯基-2-氧代-1,3-二氧戊环-4基-)甲基,[5-(4-甲基苯基)-2-氧代-1,3-二氧戊环-4基-]甲基,[5-(4-甲氧基苯基)-2-氧代-1,3-二氧戊环-4基-]甲基,[5-(4-氟苯基)-2-氧代-1,3-二氧戊环-4基-]甲基或[5-(4-氯苯基)-2-氧代-1,3-二氧戊环-4基-]甲基;或可以任选地被一个或多个C1-C4烷基或C1-C4烷氧基取代的酞基,如酞基,二甲基酞基或二甲氧基酞基,并且优选为新戊酰氧基甲基,酞基或(5-甲基-2-氧代-1,3-二氧戊环-4基-)甲基,并且更优选(5-甲基-2-氧代-1,3-二氧戊环-4基-)甲基。
本文涉及的任何化合物意在表示这样的特定化合物以及某些变体或形式。尤其是,本文涉及的化合物可以具有不对称中心并且因此以不同对映体形式存在。本文涉及的化合物的所有光学异构体和立体异构体,和其混合物,被认为在本发明的范围内。因此本文涉及的任意给定化合物意在表示外消旋体,一个或多个对映体形式,一个或多个非映体形式,一个或多个阻转异构形式中的任一种,和其混合物。特别是,式[D-(X)b-(AA)w-(L)]n-Ab的药物缀合物以及D-X-(AA)w-L1或D-X-(AA)w-H的化合物可以包括取决于其不对称性的对映体或非对映体。关于双键的立体异构也是可能的,因此在一些情况下,分子可以以(E)-异构体或(Z)-异构体存在。如果分子含有数个双键,各个双键将具有其自己的立体异构,其可以是相同的或与分子的其他双键的立体异构不同的。单个异构体和异构体的混合物落在本发明的范围内。
此外,本文涉及的化合物可以以几何异构体(即顺式和反式异构体),以互变异构体,或以阻转异构体存在。具体地,术语互变异构体是指化合物的平衡存在并且容易从一种异构形式转变为另一种的两种以上结构异构体之一。常见的互变异构对是胺-亚胺,酰胺-酰亚胺,酮-烯醇,内酰胺-内酰亚胺等。此外,本文涉及的任意化合物意在表示当这样的形式存在于介质中时的水合物,溶剂化物和多晶形物,和其混合物。此外,本文涉及的化合物可以以同位素标记的形式存在。本文涉及的化合物的所有几何异构体,互变异构体,阻转异构体,水合物,溶剂化物,多晶形物,和同位素标记的形式,和其混合物被认为在本发明的范围内。
在本发明的化合物中,Ab是包含至少一个抗原结合位点的部分。在备选的实施方案中,Ab可以是能够结合靶细胞,优选动物细胞并且更优选人细胞的任何合适的药剂。这样的药剂的实例包括淋巴因子,激素,生长因子和营养运输分子(例如运铁蛋白)。
其中Ab是包含至少一个抗原结合位点的部分,所述部分优选是抗原结合肽或多肽。在优选的实施方案中,所述部分是抗体或其抗原结合片段。
本发明的药物缀合物中的术语‘抗体’是指任何免疫球蛋白,优选全长免疫球蛋白。优选地,该术语涵盖单克隆抗体,多克隆抗体,多特异性抗体,如双特异性抗体,和其抗体片段,只要它们呈现所需生物活性即可。抗体可以源自任何物种,但优选是啮齿类动物,例如大鼠或小鼠,人或兔来源的。备选地,所述抗体,优选单克隆抗体,可以是人源化的,嵌合的或其抗体片段。术语‘嵌合抗体’还可以包括"灵长化的"抗体,其包含源自非人灵长类动物(例如,旧世界猴(Old World Monkey),猿等)的可变结构域抗原结合序列和人恒定区序列。免疫球蛋白还可以属于免疫球蛋白分子的任何类型(例如IgG,IgE,IgM,IgD,和IgA),种类(例如,IgGl,IgG2,IgG3,IgG4,IgAl和IgA2)或亚类。
术语‘单克隆抗体’是指由B系细胞,通常是杂交瘤的单个克隆产生的,基本上同源的抗体分子群体(即包含该群体的个体抗体除了可能少量存在的可能的天然存在的突变之外是相同的)。重要的是,各个单克隆具有相同的抗原特异性——即其针对抗原上的单一决定簇。
单克隆抗体的产生可以通过本领域已知的方法进行。然而,作为实例,单克隆抗体可以由杂交瘤方法(Kohler等人(1975)Nature 256:495),人B细胞杂交瘤技术(Kozbor等人,1983,Immunology Today 4:72),或EBV-杂交瘤技术(Cole等人,1985,Monoclonal Antibodies and Cancer Therapy(单克隆抗体和癌症治疗),Alan R.Liss,Inc.,pp.77-96)制备。备选地,单克隆抗体可以使用重组DNA方法制备(参见,US 4816567)或使用在Clackson等人(1991)Nature,352:624-628;Marks等人(1991)J.MoI.Biol.,222:581-597中描述的技术从噬菌体抗体文库分离。
多克隆抗体是针对不同决定簇(表位)的抗体。该异质抗体群体可以使用本领域公知的各种工序源自免疫动物的血清。
术语‘双特异性抗体’是指由两种不同单克隆抗体构成的人工抗体。它们可以被设计为结合单个抗原上的两个相邻表位,从而增加亲合力和特异性二者,或结合两种不同抗原,用于许多应用,但特别用于募集细胞毒性T-和天然杀伤(NK)细胞或再靶向毒素,放射性核素或细胞毒性药物,用于癌症治疗(Holliger&Hudson,Nature Biotechnology,2005,9,23)。双特异性抗体可以在一个臂上具有第一结合特异性的杂交免疫球蛋白重链,并且在另一个臂上具有杂交免疫球蛋白重链-轻链对(提供第二结合特异性)。该不对称结构有利于将所需双特异性化合物与不希望的免疫球蛋白链组合中分离,因为仅双特异性分子的一半中的免疫球蛋白轻链的存在提供容易的分离途径(WO 94/04690;Suresh等人,Methods in Enzymology,1986,121:210;Rodrigues等人,1993,J.of Immunology 151:6954-6961;Carter等人,1992,Bio/Technology 10:163-167;Carter等人,1995,J.of Hematotherapy 4:463-470;Merchant等人,1998,Nature Biotechnology 16:677-681。
制备杂交的或双特异性抗体的方法是本领域中已知的。在一种方法中,双特异性抗体可以通过化学交联或遗传融合两种不同Fab或scFv组件将两种杂交瘤融合为单个‘细胞杂交瘤(quadroma)’而产生(Holliger&Hudson,Nature Biotechnology,2005,9,23)。
术语‘嵌合’抗体是指其中不同部分源自不同动物物种的抗体。例如,嵌合抗体可以使可变区源自小鼠并且使恒定区源自人。相比之下,‘人源化抗体’主要来源于人,尽管其含有非人部分。具体地,人源化抗体是其中来自受体的高变区的残基被具有所需特异性、亲和力和性能(capacity)的非人物种(供体抗体)如小鼠,大鼠,兔或非人灵长类动物的高变区的残基替代的人免疫球蛋白(受体抗体)。在一些情况下,人免疫球蛋白的框架区(FR)残基被相应非人残基替代。此外,人源化抗体可以包含未在受体抗体或供体抗体中发现的残基。进行这些修饰以进一步改善抗体性能。总的来说,人源化抗体将包含基本上全部至少一个,并且通常两个可变结构域,其中全部或基本上全部高变环对应于非人免疫球蛋白的那些高变环并且全部或基本上全部FR是人免疫球蛋白序列的那些FR。人源化抗体还将任选地包含至少一部分免疫球蛋白恒定区(Fc),通常人免疫球蛋白的恒定区(Fc)。
重组抗体如嵌合和人源化单克隆抗体可以通过本领域已知的重组DNA技术生产。完全人抗体可以使用不能表达内源免疫球蛋白重链和轻链基因,但可以表达人重链和轻链基因的转基因小鼠产生。转基因小鼠利用选择的抗原以通常方式免疫。针对该抗原的单克隆抗体可以使用常规杂交瘤技术获得。转基因小鼠包含的人免疫球蛋白转基因在B细胞分化期间重排,并随后经历类型转换和体细胞突变。因此,使用这样的技术,可能产生治疗上有用的IgG,IgA,IgM和IgE抗体。对于这种用于产生人抗体的技术的概览,参见Lonberg和Huszar(1995,Int.Rev.Immunol.13:65-93)。对于这种用于产生人抗体和人单克隆抗体的技术和产生这样的抗体的方案的详细讨论,参见,例如,美国专利号5625126;5633425;5569825;5661016;5545806;其各自通过引用以其整体结合于本文。其他人抗体可以从例如,Abgenix,Inc.(Freemont,CA)和Genpharm(San Jose,CA)商购获得。
本发明的药物缀合物中的术语‘抗原结合片段’是指其中抗体的该抗原结合片段保留相应全长抗体的抗原结合功能的全长抗体部分。抗原结合片段可以包含抗体的可变区的一部分,所述部分包含至少一个,两个,优选三个选自CDR1,CDR2和CDR3的CDR。抗原结合片段还可以包含免疫球蛋白轻链和重链的一部分。抗体片段的实例包括Fab,Fab',F(ab')2,scFv,di-scFv,和BiTE(双特异性T细胞衔接物(engager)),Fv片段(包括纳米抗体(nanobody),双抗体(diabody),双抗体-Fc融合物,三抗体(triabody)和,四抗体(tetrabody));微抗体(minibody);线性抗体;由Fab表达文库产生的片段,抗独特型(抗Id)抗体,CDR(互补决定区),和免疫特异地结合靶抗原如癌细胞抗原,病毒抗原或微生物抗原的上述任一种的表位结合片段,单链或单域抗体分子(包括仅重链抗体,例如,骆驼VHH结构域和鲨鱼V-NAR);和由抗体片段形成的多特异性抗体。为了比较,被称为‘抗体’的全长抗体是包含VL和VH结构域,以及完全轻链和重链恒定结构域的抗体。
抗体还可以具有一种或多种效应子功能,其是指归因于抗体的Fc区(根据改变受体结合的本领中的方法改造的天然序列Fc区或氨基酸序列变体Fc区)的生物活性。抗体效应子功能的实例包括CIq结合;补体依赖性细胞毒性;Fc受体结合;抗体依赖性细胞介导的细胞毒性(ADCC);吞噬(phagocytosis);细胞表面受体(例如,B细胞受体;BCR)的下调,等。
抗体还可以是免疫特异结合靶抗原如癌细胞抗原,病毒抗原,或微生物抗原的抗体或结合肿瘤细胞的其他抗体的功能活性片段,衍生物或类似物。就此而言,功能活性意为:片段,衍生物或类似物能够诱导抗独特型抗体,该抗体识别与该片段,衍生物或类似物源自其中的抗体所识别的相同的抗原。具体地,在示例性实施方案中,免疫球蛋白分子的独特型的抗原性可以通过删除框架区和特异性识别抗原的CDR序列的C末端的CDR序列而增强。为了确定哪个CDR序列结合抗原,含有CDR序列的合成的肽可以用于通过本领域已知的任意结合测定方法(例如,BIA core测定)的利用抗原的结合测定,参见,例如,Kabat等人,1991,Sequences of Proteins of Immunological Interest(免疫学感兴趣的蛋白质的序列),第五版,National Institute of Health,Bethesda,Md;Kabat E等人,1980,J.of Immunology 125(3):961-969)。
术语‘抗体’还可以包括抗体的,或其功能活性片段的融合蛋白,例如其中抗体通过共价键(例如,肽键),在不是抗体的另一蛋白(或其部分,如所述蛋白的至少10,20或50个氨基酸部分)的氨基酸序列的N末端或C末端融合。抗体或其片段可以与另一蛋白在恒定结构域的N末端共价连接。
此外,本发明的抗体或抗原结合片段可以包括二者中任一个如通过共价连接任意类型的分子(只要这样的共价连接允许抗体保留其抗原结合免疫特异性即可)被修饰的抗体或其抗原结合片段的类似物和衍生物。修饰的实例包括糖基化,乙酰化,聚乙二醇化(pegylation),磷酸化,酰胺化,通过已知保护/封闭基团(blocking group)衍生化,蛋白水解裂解,与细胞抗体单元或其他蛋白的连接等。多种化学修饰中的任一种可以通过已知技术进行,包括但不限于化学裂解,乙酰化,甲酰化,在衣霉素(tunicamycin)的存在下的代谢合成等。此外,该类似物或衍生物可以含有一个或多个非天然氨基酸。
本发明的抗体或抗原结合片段还可以在抗体的Fc结构域中具有修饰(例如,替代,删除或添加)。具体地,所述修饰可以在Fc-铰链区中并且导致增加的对FcRn受体的结合(WO 97/34631)。
在一个实施方案中,本发明的药物缀合物中的抗体可以是任何抗体或其抗原结合片段,优选用于治疗疾病,优选癌症的单克隆抗体。所述癌症可以是乳腺癌(breast cancer),结直肠癌(colorectal cancer),子宫内膜癌(endometrial cancer),肾癌(kidney cancer),黑素瘤(melanoma),白血病(leukaemia),肺癌(lung cancer),多发性骨髓瘤(multiple myeloma),淋巴瘤(lymphoma)(例如霍奇金病(Hodgkin's disease)和非霍奇金淋巴瘤(non-Hodgkin's Lymphoma)),实体瘤(solid tumor)如肉瘤(sarcoma)和癌(carcinoma),黑素瘤(melanoma),间皮瘤(mesothelioma),骨肉瘤(osteosarcoma),卵巢癌(ovarian cancer)和肾癌(renal cancer)。在优选的实施方案中,所述癌症是肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌。在更优选的实施方案中,所述癌症是结直肠癌,乳腺癌,白血病,淋巴瘤,和卵巢癌。
可以用于治疗癌症的抗体包括,但不限于,针对以下抗原的抗体:CA125(卵巢),CA15-3(癌),CA19-9(癌),L6(癌),Lewis Y(癌),Lewis X(癌),胎蛋白(癌),CA242(结直肠),胎盘碱性磷酸酶(癌),前列腺特异性抗原(前列腺),前列腺酸性磷酸酶(前列腺),表皮生长因子(癌)例如EGF受体2蛋白(乳腺癌),MAGE-I(癌),MAGE-2(癌),MAGE-3(癌),MAGE-4(癌),抗运铁蛋白受体(癌),p97(黑素瘤),MUCl-KLH(乳腺癌),CEA(结直肠),gplOO(黑素瘤),MARTl(黑素瘤),PSA(前列腺),IL-2受体(T细胞白血病和淋巴瘤),CD20(非霍奇金淋巴瘤),CD52(白血病),CD33(白血病),CD22(淋巴瘤),人绒毛膜促性腺激素(癌),CD38(多发性骨髓瘤),CD40(淋巴瘤),粘蛋白(癌),P21(癌),MPG(黑素瘤),和Neu癌基因产物(癌)。一些具体的有用抗体包括,但不限于,BR96mAb(Trail,P.A.,等人Science(1993)261,212-215),BR64(Trail,PA,等人Cancer Research(1997)57,100-105,针对CD40抗原的mAb,如S2C6mAb(Francisco,J.A.,等人Cancer Res.(2000)60:3225-3231),针对CD70抗原的mAb,如1F6mAb,和针对CD30抗原的mAb,如AClO(Bowen,M.A.,等人(1993)J.Immunol.,151:5896-5906;Wahl等人,2002Cancer Res.62(13):3736-42)。可以使用很多其他的结合肿瘤相关抗原的内在化抗体,并且已经被综述(Franke,A.E.,等人Cancer Biother Radiopharm.(2000)15:459-76;Murray,J.L.,(2000)Semin Oncol,27:64-70;Breitling,F.,和Dubel,S.,Recombinant Antibodies(重组抗体),John Wiley,和Sons,New York,1998)。
其他肿瘤相关抗原包括,但不限于,BMPR1B,E16,STEAP1,STEAP2,0772P.MPF,Napi3b,Sema5b,PSCA hlg,ETBR,MSG783,TrpM4,CRIPTO,CD21,CD79b,FcRH2,HER2,NCA,MDP,IL20Rα,短蛋白聚糖(Brevican),EphB2R,ASLG659,PSCA,GEDA,BAFF-R,CD79A,CXCR5,HLA-DOB,P2X5,CD72,LY64,FCRH1,IRTA2和TENB2。
在备选的实施方案中,本发明的药物缀合物中的抗体可以是抗体或其抗原结合片段,优选免疫特异结合病毒抗原,微生物抗原或产生与自身免疫病相关的自身免疫抗体的细胞的抗原的单克隆抗体。
病毒抗原可以包括,但不限于,能够引发免疫应答的任何病毒肽,多肽或蛋白如HIV gpl20,HIV nef,RSV F糖蛋白,流感病毒神经氨酸酶,流感病毒血凝素,HTLV tax,单纯疱疹病毒糖蛋白(例如,Gb,Gc,Gd,和Ge)和乙型肝炎表面抗原)。
微生物抗原可以包括,但不限于,能够引发免疫应答的任何微生物肽,多肽,蛋白,糖,多糖,或脂质分子(例如,细菌,真菌,致病原生动物,或酵母多肽,包括例如,LPS和荚膜多糖)。
在另一个实施方案中,所述抗体可以是已知用于治疗或预防病毒或微生物感染——即感染性疾病的任意抗体。这样的抗体的实例包括,但不限于,PRO542(Progenies),其是可用于治疗HIV感染的CD4融合抗体;OsTAVIR(Protein Design Labs,Inc.,CA),其是可用于治疗乙型肝炎病毒的人抗体;PROTOVIR.(蛋白Design Labs,Inc.,CA),其是用于治疗巨细胞病毒(CMV)的人源化IgG1抗体;和抗LPS抗体。
可用于治疗感染性疾病的其他抗体包括,但不限于,针对来自以下各项的抗原的抗体:致病细菌菌株(化脓性链球菌(Streptococcus pyogenes),肺炎链球菌(Streptococcus pneumoniae),淋病奈瑟菌(Neisseria gonorrheae),脑膜炎双球菌(Neisseria meningitidis),白喉杆菌(Corynebacterium diphtheriae),肉毒杆菌(Clostridium botulinum),产气荚膜梭菌(Clostridium perfringens),破伤风杆菌(Clostridium tetani),流感嗜血杆菌(Hemophilusinfluenzae),肺炎克雷伯氏杆菌(Klebsiella pneumoniae),臭鼻克雷伯氏杆菌(Klebsiella ozaenas),鼻硬结克雷伯氏菌(Klebsiella rhinoscleromotis),金黃色葡萄球菌(Staphylococcus aureus),霍乱弧菌(Vibrio colerae),大肠杆菌(Escherichia coli),铜绿假单胞菌(Pseudomonas aeruginosa),胎儿弯曲杆菌(弧菌)(Campylobacter(Vibrio)fetus),嗜水气单胞菌(Aeromonas hydrophila),蜡状芽孢杆菌(Bacillus cereus),迟钝爱德华菌(Edwardsiella tarda),小肠结肠炎耶尔森杆菌(Yersinia enterocolitica),鼠疫耶尔森菌(Yersinia pestis),假结核耶尔森菌(Yersinia pseudotuberculosis),痢疾志贺菌(Shigella dysenteriae),弗氏志贺菌(Shigella flexneri),索氏志贺菌(Shigella sonnei),鼠伤寒沙门氏菌(Salmonella typhimurium),苍白密螺旋体(Treponema pallidum),雅司密螺旋体(Treponema pertenue),斑点病密螺旋体(Treponema carateneum),奋森氏疏螺旋体(Borrelia vincentii),伯氏疏螺旋体(Borrelia burgdorferi),出血性黄疸型钩端螺旋体(Leptospira icterohernorrhagiae),结核分枝杆菌(Mycobacterium tuberculosis),卡氏肺囊虫(Pneumocystis carinii),土拉热弗朗西丝氏菌(Francisella tularensis),流产布鲁氏菌(Brucella abortus),猪布鲁氏菌(Brucella suis),马尔他布鲁氏菌(Brucella melitensis),支原体属(Mycoplasma spp.),普氏立克次体(Rickettsia prowazeki),恙虫热立克次体(Rickettsia tsutsugumushi),衣原体属(Chlamydia spp.));致病真菌菌株(粗球孢子菌(Coccidioides immitis),烟曲霉菌(Aspergillus fumigatus),白色念珠菌(Candida albicans),皮炎芽生菌(Blastomyces dermatitidis),新型隐球菌(Cryptococcus neoformans),夹膜组织胞浆菌(Histoplasma capsulatum));原生动物(溶组织内阿米巴(Entomoeba histolytica),刚地弓形虫(Toxoplasma gondii),口腔毛滴虫(Trichomonas tenas),肠毛滴虫(Trichomonas hominis),阴道滴虫(Trichomonas vaginalis),冈比亚锥虫(Tryoanosoma gambiense),罗得西亚锥虫(Trypanosoma rhodesiense),克氏锥虫(Trypanosoma cruzi),杜氏利什曼原虫(Leishmania donovani),热带利什曼原虫(Leishmania tropica),巴西利什曼原虫(Leishmania braziliensis),卡氏肺孢子虫肺炎(Pneumocystis pneumonia),间日疟原虫(Plasmodium vivax),恶性疟原虫(Plasmodium falciparum),三日疟原虫(Plasmodium malaria));或蠕虫(Helminiths)(蠕形住肠蛲虫(Enterobius vermicularis),毛首鞭虫(Trichuris trichiura),人蛔虫(Ascaris lumbricoides),旋毛虫(Trichinella spiralis),粪类圆线虫(Strongyloides stercoralis),日本血吸虫(Schistosoma japonicum),曼氏血吸虫(Schistosoma mansoni),埃及血吸虫(Schistosoma haematobium),和钩虫(hookworms))。
可用于治疗病毒疾病的其他抗体包括,但不限于,针对致病病毒的抗原的抗体,作为实例,所述致病病毒包括但不限于:痘病毒科(Poxviridae),疱疹病毒科(Herpesviridae),单纯疱疹病毒1(Herpes Simplex virus 1),单纯疱疹病毒2(Herpes Simplex virus 2),腺病毒科(Adenoviridae),乳多空病毒科(Papovaviridae),肠道病毒科(Enteroviridae),小核糖核酸病毒科(Picornaviridae),微小病毒科(Parvoviridae),呼肠孤病毒科(Reoviridae),逆转录病毒科(Retroviridae),流感病毒(influenza virus),副流感病毒(parainfluenza virus),腮腺炎(mumps),麻疹(measles),呼吸道合胞体病毒(respiratory syncytial virus),风疹(rubella),虫媒病毒科(Arboviridae),弹状病毒科(Rhabdoviridae),沙粒病毒科(Arenaviridae),甲型肝炎病毒(Hepatitis A virus),乙型肝炎病毒(Hepatitis B virus),丙型肝炎病毒(Hepatitis C virus),戊型肝炎病毒(Hepatitis E virus),非甲型/非乙型肝炎病毒(Non-A/Non-B Hepatitis virus),鼻病毒科(Rhinoviridae),冠状病毒科(Coronaviridae),轮状病毒科(Rotoviridae),和人免疫缺陷病毒(Human Immunodeficiency Virus)。
在备选实施方案中,本发明的药物缀合物的抗体还可以是已知用于治疗或预防自身免疫病的任何抗体,所述自身免疫病,如但不限于,Th2淋巴细胞相关病症(例如特应性皮炎(atopic dermatitis),特应性气喘(atopic asthma),鼻结膜炎(rhinoconjunctivitis),过敏性鼻炎(allergic rhinitis),奥曼综合征(Omenn's syndrome),系统性硬化病(systemic sclerosis),和移植物抗宿主病(graft versus host disease));Th1淋巴细胞相关病症(例如类风湿性关节炎(rheumatoid arthritis),多发性硬化(multiple sclerosis),银屑病(psoriasis),斯耶格伦氏综合征(Sjorgren's syndrome),桥本甲状腺炎(Hashimoto's thyroiditis),格雷夫氏病(Grave's disease),原发性胆汁性肝硬化(primary biliary cirrhosis),韦格纳肉芽肿(Wegener's granulomatosis),和结核(tuberculosis));活化的B淋巴细胞相关病症(例如系统性红斑狼疮(systemic lupus erythematosus),古德帕斯丘综合征(Goodpasture's syndrome),类风湿性关节炎,和I型糖尿病);和慢性活动性肝炎(Active Chronic Hepatitis),爱迪生氏病(Addison's Disease),过敏性肺泡炎(Allergic Alveolitis),过敏反应(Allergic Reaction),过敏性鼻炎,阿尔波特氏综合征(Alport's Syndrome),过敏反应(Anaphlaxis),强直性脊柱炎(Ankylosing Spondylitis),抗磷脂抗体综合征(Anti-phosholipid Syndrome),关节炎(Arthritis),蛔虫病(Ascariasis),曲霉病(Aspergillosis),特应性过敏(Atopic Allergy),特应性皮炎(Atropic Dermatitis),萎缩性鼻炎(Atropic Rhinitis),白塞病(Behcet's Disease),养鸟人肺(Bird-Fancier's Lung),支气管哮喘(Bronchial Asthma),卡普兰综合征(Caplan's Syndrome),心肌病(Cardiomyopathy),乳糜泻(Celiac Disease),恰加斯氏病(Chagas'Disease),慢性肾小球肾炎(Chronic Glomerulonephritis),耳蜗前庭综合征(Cogan's Syndrome),冷凝集素病(Cold Agglutinin Disease),先天性风疹感染(Congenital Rubella Infection),CREST综合征,克劳恩病(Crohn's Disease),冷球蛋白血症(Cryoglobulinemia),库欣综合征(Cushing's Syndrome),皮肌炎(Dermatomyositis),盘状狼疮(Discoid Lupus),心肌梗死后综合征(Dresser's Syndrome),依顿-兰伯特综合征(Eaton-Lambert Syndrome),艾柯病毒(Echovirus)感染,脑脊髓炎(Encephalomyelitis),内分泌性眼病(Endocrine opthalmopathy),EB病毒(Epstein-Barr Virus)感染,Equine Heaves,狼疮(Erythematosis),埃文斯综合征(Evan's Syndrome),费尔蒂综合征(Felty's Syndrome),纤维肌痛(Fibromyalgia),富克斯睫状体炎(Fuch's Cyclitis),胃萎缩(Gastric Atrophy),胃肠道过敏(Gastrointestinal Allergy),巨细胞动脉炎(Giant Cell Arteritis),肾小球肾炎(Glomerulonephritis),古德帕斯丘综合征,移植物抗宿主疾病(Graft v.Host Disease),格雷夫斯病,吉兰-巴雷综合征(Guillain-Barre Disease),桥本甲状腺炎,溶血性贫血(Hemolytic Anemia),过敏性紫癜(Henoch-Schonlein Purpura),特发性肾上腺萎缩(Idiopathic Adrenal Atrophy),特发性肺纤维化(Idiopathic Pulmonary Fibritis),IgA肾病,炎性肠疾病(Inflammatory Bowel Diseases),胰岛素依赖型糖尿病(Insulin-dependent Diabetes Mellitus),幼年型关节炎(Juvenile Arthritis),幼年型糖尿病(Juvenile Diabetes Mellitus)(I型),兰伯特-依顿综合征(Lambert-Eaton Syndrome),蹄叶炎(Laminitis),扁平苔藓(Lichen Planus),狼疮样肝炎(Lupoid Hepatitis),淋巴细胞减少狼疮(Lupus Lymphopenia),美尼尔病(Meniere's Disease),混合性结缔组织病(Mixed Connective Tissue Disease),多发性硬化,重症肌无力(Myasthenia Gravis),恶性贫血(Pernicious Anemia),多腺性综合征(Polyglandular Syndromes),老年性痴呆(Presenile Dementia),原发性无丙种球蛋白血症(Primary Agammaglobulinemia),原发性胆汁性肝硬化,银屑病,银屑病关节炎(Psoriatic Arthritis),雷诺现象(Raynauds Phenomenon),反复流产(Recurrent Abortion),赖特综合征(Reiter's Syndrome),风湿热(Rheumatic Fever),类风湿关节炎,Sampter's综合征,血吸虫病(Schistosomiasis),施密特氏综合征(Schmidt's Syndrome),硬皮病(Scleroderma),Shulman's综合征,斯耶格伦氏综合征,全身肌强直综合征(Stiff-Man Syndrome),Sympathetic Ophthahnia,系统性红斑性狼疮,无脉病(Takayasu's Arteritis),巨细胞动脉炎(Temporal Arteritis),甲状腺炎(Thyroiditis),血小板减少症(Thrombocytopenia),甲状腺毒症(Thyrotoxicosis),中毒性表皮坏死松解症(Toxic Epidermal Necrolysis),B型胰岛素抗性,I型糖尿病,溃疡性结肠炎(Ulcerative Colitis),葡萄膜炎(Uveitis),白癫风(Vitiligo),特发性巨球蛋白血(Waldenstrom's Macroglobulemia)和韦格纳肉芽肿。
对用于产生自身免疫抗体的细胞的抗原免疫特异的抗体可以通过本领域技术人员已知的任何方法获得,所述方法如,例如,化学合成或重组表达技术。自身免疫抗体的实例包括,但不限于,抗核抗体;抗ds DNA;抗ss DNA,抗心磷脂(Cardiolipin)抗体IgM,IgG;抗磷脂抗体IgM,IgG;抗SM抗体;抗线粒体抗体;甲状腺抗体;微粒体抗体;甲状腺球蛋白(Thyroglobulin)抗体;抗SCL-70;抗-Jo;抗U1RNP;抗-La/SSB;抗SSA;抗SSB;抗Perital细胞抗体;抗组蛋白;抗RNP;C-ANCA;P-ANCA;抗着丝粒;抗纤维蛋白,和抗GBM抗体。
在另一个实施方案中,本发明的药物缀合物的抗体可以是结合在活化的淋巴细胞上表达的受体或受体复合物二者的抗体,如与自身免疫病相关的抗体。受体或受体复合物可以包含免疫球蛋白基因超家族成员,TNF受体超家族成员,整联蛋白,白细胞介素,细胞因子受体,趋化因子受体,主要组织相容性蛋白,凝集素,或补体控制蛋白。合适的免疫球蛋白超家族成员的非限制性实例是CD2,CD3,CD4,CD5,CD8,CD13,CD19,CD22,CD28,CD79,CD90,CD152/CTLA-4,PD-I,和ICOS。合适的TNF受体超家族成员的非限制性实例是CD27,CD40,CD95/Fas,CD134/OX40,CD137/4-1BB,TNF-Rl,TNFR-2,RANK,TACI,BCMA,骨保护素(osteoprotegerin),Apo2/TRAEL-Rl,TRAIL-R2,TRAIL-R3,TRABL-R4,和APO-3。合适的整联蛋白的非限制性实例是CDl Ia,CDlIb,CDlIc,CD18,CD29,CD41,CD49a,CD49b,CD49c,CD49d,CD49e,CD49f,CD103,和CD104。合适的凝集素的非限制性实例是C型,S型,和I型凝集素。
结合目标分子靶点或抗原,例如,ErbB2抗原的抗体是能够以足够的亲和力结合该抗原以致所述抗体可用于靶向表达所述抗原的细胞的抗体。在抗体是结合ErbB2的抗体的情况下,与其他ErbB受体相对,其通常优先地结合ErbB2,并且可以是不显著与其他蛋白如EGFR,ErbB 3或ErbB4交叉反应的抗体。在这样的实施方案中,如通过荧光活化细胞分选(FACS)分析或放射免疫沉淀(RIA)确定的,抗体对这些非ErbB2蛋白(例如,结合内源受体的细胞表面)的结合程度将小于10%。有时,抗ErbB2抗体将不显著与大鼠neu蛋白交叉反应,例如,如在Schecter等人,Nature 312:513(1984)和Drebin等人,Nature 312:545-548(1984)中描述的。
在另一个实施方案中,本发明的药物缀合物的抗体或靶点可以选自下表中的抗体或靶点。这样的抗体是对靶抗原免疫特异的并且可以商购获得或通过本领域已知的任意方法如,例如,重组表达技术生产。
表1:治疗性单克隆抗体
除以上之外,本发明的药物抗体缀合物的抗体可以是Vitaxin,其是用于治疗肉瘤的人源化抗体;Smart IDlO,其是用于治疗非霍奇金淋巴瘤的人源化抗HLA-DR抗体;Oncolym,其是用于治疗非霍奇金淋巴瘤的放射标记的鼠抗HLA-DrlO抗体;和Allomune,其是用于治疗霍奇金病或非霍奇金淋巴瘤的人源化抗CD2mAb。
本发明的药物缀合物的抗体也可以是已知用于治疗任何疾病,优选癌症的任何抗体片段。此外,这样的抗体片段是对靶抗原免疫特异的并且可以商购获得或通过本领域已知的任何方法如,例如,重组表达技术生产。可获得的这样的抗体的实例包括来自下表的任一形式。
表2:治疗性单克隆抗体片段
(Holliger&Hudson,Nature Biotechnology,2005,9,23)。
在优选的实施方案中,本发明的药物缀合物中的抗体可以结合由ErbB基因编码的受体。所述抗体可以特异结合选自EGFR,HER2,HER3和HER4的ErbB受体。优选地,药物缀合物中的抗体可以特异结合HER2受体的细胞外结构域并且抑制过表达HER2受体的肿瘤细胞的生长。药物缀合物的抗体可以是单克隆抗体,例如鼠单克隆抗体,嵌合抗体,或人源化抗体。优选地,人源化抗体可以是huMAb4D5-1,huMAb4D5-2,huMAb4D5-3,huMAb4D5-4,huMAb4D5-5,huMAb4D5-6,huMAb4D5-7或huMAb4D5-8(曲妥珠单抗),特别优选曲妥珠单抗。所述抗体还可以是抗体片段,例如Fab片段。
其他优选的抗体包括:
(i)抗CD4抗体。药物缀合物的抗体可以是单克隆抗体,例如鼠单克隆抗体,嵌合抗体,或人源化抗体;
(ii)抗CD5抗体。药物缀合物的抗体可以是单克隆抗体,例如鼠单克隆抗体,嵌合抗体,或人源化抗体;
(iii)抗CD13抗体。药物缀合物的抗体可以是单克隆抗体,例如鼠单克隆抗体,嵌合抗体,或人源化抗体;和
(iv)抗CD20抗体。药物缀合物的抗体可以是单克隆抗体,例如鼠单克隆抗体,嵌合抗体,或人源化抗体。优选地,所述人源化抗体是利妥昔单抗或其抗体片段,例如Fab片段。
用于制备药物抗体缀合物的方法
本发明的药物抗体缀合物可以根据本领域公知的技术制备。使用不同方法将包含至少一个抗原结合位点抗体的部分如抗体与多种不同药物缀合的过程之前已经在例如,WO-A-2004/010957,WO-A-2006/060533和WO-A-2007/024536中描述并举例说明,其内容通过引用结合于本文。这些包括使用以其可以随后连接于所述部分如抗体的方式使药物、毒素或放射性核素衍生的接头基团。通常通过以下三种途径之一连接于所述部分如抗体:在部分还原抗体中的二硫化物基团后通过半胱氨酸中的游离硫醇基;通过抗体中的赖氨酸中的游离氨基;和通过抗体中的丝氨酸和/或苏氨酸中的游离羟基。连接方法根据所述部分如抗体上的连接位点而改变。也已经描述了通过尺寸排阻层析(SEC)纯化抗体-药物缀合物[参见,例如,Liu等人,Proc.Natl.Acad.Set(USA),93:8618-8623(1996),和Chari等人,Cancer Research,52:127-131(1992)]。
如之前指出的,本发明的药物缀合物的药物有效荷载是二氢吡喃-2-酮和四氢吡喃-2-酮衍生物,其已经被公开或落在国际公布号WO-A-2007/144423和WO-A-2009/080761的范围内,其内容通过引用于其中结合于本文。这些化合物根据这些国际申请中描述和举例说明的方法来合成。
如之前指出的,在本发明的第九方面,提供用于制备根据本发明的第一方面的药物缀合物的方法,所述方法包括将包含至少一个抗原结合位点的部分Ab和式(I),(Ia)或(Ib)的药物D缀合,Ab和D如本发明的第一方面中所限定。
用于制备本发明的药物缀合物的方法的一个实例包括如下制备本发明的式(G)或(H)的药物抗体缀合物:
所述方法包括以下步骤:
(i)将式(Ia)-H的药物(D),其中(Ia)的定义中的取代基如上文限定:
与其中X1和X2是离去基团的式X2-C(O)-X1的化合物反应,得到式(B)的化合物:
并且-(C=O)X1部分的连接点是连接于与R18’相同的碳的游离羟基。
(ii)将步骤(i)中产生的式(B)的化合物与式H2N-(CH2)1-6NH2的二胺反应,得到式(C)的化合物:
(iii)或将步骤(ii)中产生的式(C)的化合物与式(D’)的化合物反应:
得到式(F)的化合物:
或
将步骤(ii)中产生的化合物(C)与6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯反应,得到(E):
(iv)部分还原要缀合的抗体中的一个或多个二硫键,得到还原的具有游离硫醇基的抗体Ab-SH:
并且
(v)将部分还原的具有游离硫醇基的抗体Ab-SH与步骤(iv)中产生的式(E)或(F)的化合物反应,分别得到所需的式(G)或(H)的药物抗体缀合物:
优选地,与步骤(i)中的药物(A)反应的式X2-C(O)-X1的化合物是1,1’羰基二咪唑。
优选地,步骤(ii)中的二胺具有式NH2-(CH2)2-4-NH2,并且更优选其是亚丙基-1,3-二胺。
在该方法的一个优选的实施方案中,将中间体(C)与式(D’)的化合物反应,其中R23是-(CH2)3-NH-CO-NH2并且R22是异丙基。
在该方法的另一优选的实施方案中,所述抗体选自曲妥珠单抗,利妥昔单抗,抗CD4抗体,抗CD5抗体和抗CD13抗体或其免疫活性部分,或其选自曲妥珠单抗,利妥昔单抗和抗CD4抗体,或其免疫活性部分,并且最优选其是曲妥珠单抗或其免疫活性部分;或其选自抗CD5抗体和抗CD13抗体,或其免疫活性部分,并且最优选其是抗CD13抗体或其免疫活性部分。此外,该单克隆抗体的部分还原使用三[2-羧基乙基]膦盐酸盐(TCEP)进行。
用于制备本发明的药物抗体缀合物的方法的另一个实例,包括如下制备式(O)或(P)的药物抗体缀合物
所述方法包括以下步骤:
(i)以下两项之一:
将式(Ia)-H的药物(D),其中(Ia)-H的定义中的取代基如上文限定:
与式X2-C(O)-X1其中X1和X2是离去基团的化合物反应,得到式(B)的化合物:
并且X1(CO)部分的连接点是连接于与R18’相同的碳原子的游离羟基,或者
(b)将如上文限定的式(Ia)-H的所述药物(A)与氯甲酸4-硝基-苯酯反应,得到式(J)的化合物:
并且(4-硝基苯基)-O-CO-基团的连接点与对于上述(a)中的X1(CO)部分的连接点相同;
(ii)以下两项之一:
(c)将步骤(i)中产生的式(B)的化合物与式H2N-(CH2)1-6NH2的二胺反应,得到式(C)的化合物:
并且然后将得到的式(C)的化合物与式Me-S-S-(CH2)1-3-CO2H的化合物反应,得到式(K)的化合物
或者
(d)将步骤(i)中产生的化合物(J)与式H2N-(CH2)1-3SH的氨基烷基硫代化合物反应,得到式(L)的化合物:
(iii)将在步骤(ii)中产生的(K)或(L)与二硫苏糖醇在二硫化物还原条件下反应,分别得到式(M)和(N)的化合物:
(iv)将要缀合的抗体与琥珀酰亚胺基-4-(N-马来酰亚胺基甲基)环己烷-1-甲酸酯反应,以将所述抗体在一个或多个赖氨酸基团处用琥珀酰亚胺基-4-(N-马来酰亚胺基甲基)环己烷-1-羰基衍生化:
(v)将在步骤(iv)中产生的衍生化抗体与步骤(iii)中产生的(M)或(N)反应,得到所需的式(O)或(P)的药物抗体缀合物:
至于更早的方法,式X2-C(O)-X1的化合物优选为1,1’-羰基二咪唑。类似地,式(B)的二胺化合物优选为NH2-(CH2)2-4-NH2,并且更优选亚丙基-1,3-二胺。
在本发明的一个优选的一个实施方案中,与式(C)的化合物反应以得到式(K)的化合物的化合物是3-(甲基二硫烷基)丙酸。
在另一个优选的实施方案中,与式(J)的化合物反应得到式(L)的化合物的氨基烷基硫代化合物是3-氨基丙烷-1-硫醇。
在与药物接头部分的连接是通过在部分还原包含至少一个抗原结合位点的部分如单克隆抗体中的二硫化物基团后的半胱氨酸中的游离硫醇基的情况下,部分还原通常通过首先稀释至合适的浓度并将溶液缓冲,之后通过添加合适的还原剂如三[2-羧基乙基]膦盐酸盐(TCEP)或二硫苏糖醇(DTT)的方式部分还原二硫键来进行。通过选择要还原的部分如单克隆抗体和还原剂的适当比例,反应条件和还原时间,可以获得所需游离硫醇基与部分的比例,例如四个游离硫醇基/单克隆抗体。
然后将如上文所述制备的部分还原的部分如具有游离硫醇基的部分还原的单克隆抗体与式D-X-(AA)w-L1(其中这样的化合物中的基团L是马来酰亚胺基,其自由与硫醇基反应)的本发明的药物接头化合物反应。通过本领域已知的任何合适的方法,例如通过尺寸排阻层析(SEC)纯化得到的药物抗体缀合物[参见,例如,Liu等人,Proc.Natl.Acad.Set(USA),93:8618-8623(1996),和Chari等人,Cancer Research,52:127-131(1992)]。
在本发明的一个优选的实施方案中,部分还原的单克隆抗体是曲妥珠单抗或抗CD13抗体或其免疫活性部分,优选曲妥珠单抗或其免疫活性部分;或优选抗CD13抗体或其免疫活性部分。
在本发明的备选实施方案中,包含至少一个抗原结合位点的部分如单克隆抗体中的赖氨酸可以首先与琥珀酰亚胺基-4-(N-马来酰亚胺基甲基)环己烷-1-甲酸酯反应。抗体上的游离胺基可以与N-羟基琥珀酰亚胺酯反应,得到马来酰亚胺活化的抗体:
马来酰亚胺-活化的抗体可以随后与具有反应性硫醇基部分的式D-X-(AA)w-H的化合物反应。
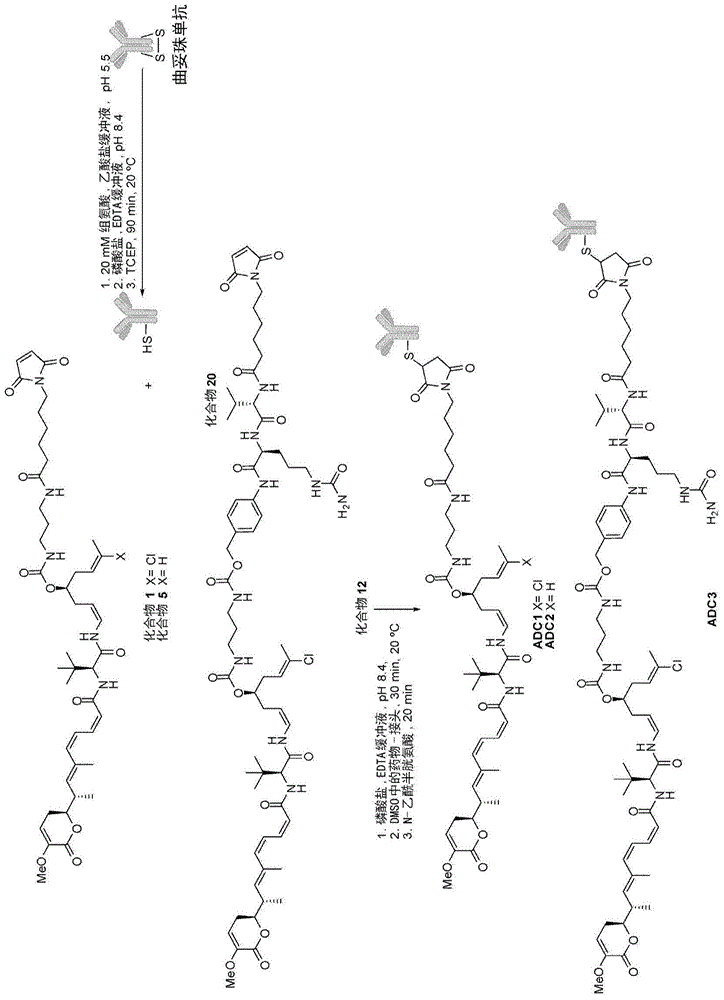
通过部分还原抗体中的二硫化物基团后经半胱氨酸中的游离硫醇基和经用马来酰亚胺基活化后的抗体中的赖氨酸中的游离氨基缀合来制备本发明的式[D-(X)b-(AA)w-(L)-]n-Ab的药物抗体缀合物的方法的两个具体实例显示在图1和2中。
包含本发明的药物抗体缀合物的组合物及其用途
在本发明的第五个方面,提供药物组合物,其包含根据本发明的药物缀合物和药用载体。具有本发明的通式[D-(X)b-(AA)w-(L)-]n-Ab的药物缀合物的施用形式的实例包括但不限于口服、局部、肠胃外、舌下、直肠、阴道、眼和鼻内。肠胃外施用包括皮下注射,静脉内,肌肉内,胸骨内注射或输注技术。优选地,所述组合物肠胃外施用。可以配制本发明的药物组合物式,从而允许本发明的药物缀合物在将组合物施用于动物,优选人时是可生物利用的。组合物可以采用一个或多个剂量单位的形式,其中例如,片剂可以是单个剂量单位,并且气雾剂形式的本发明的药物抗体缀合物的容器可以含有多个剂量单位。
药用载体或媒介物(vehicle)可以是颗粒的,从而组合物为,例如,片剂或粉末形式。一种或多种载体可以是液体,而组合物为,例如,口服糖浆或可注射液体。此外,一种或多种载体可以是气体的,从而提供可用于例如,吸入施用的气雾剂组合物。术语"载体"是指本发明的药物抗体缀合物利用其施用的稀释剂,辅药或赋形剂。这样的药物载体可以是液体,如水和油,包括石油、动物油、植物油或合成来源的油,如花生油、大豆油、矿物油、芝麻油等。载体可以是盐水、阿拉伯胶、明胶、淀粉糊、滑石、角蛋白、硅胶、尿素等。此外,可以使用辅助剂、稳定剂、增稠剂、润滑剂和着色剂。在一个实施方案中,当施用于动物时,本发明的药物抗体缀合物或组合物和药用载体是无菌的。当本发明的药物抗体缀合物静脉内施用时,水是优选的载体。还可以使用盐水溶液、水性葡萄糖和甘油溶液作为液体载体,特别是用于可注射溶液。合适的药物载体还包括赋形剂如淀粉、葡萄糖、乳糖、蔗糖、明胶、麦芽、大米、面粉、白垩、硅胶、硬脂酸钠、单硬脂酸甘油酯、滑石、氯化钠、脱脂奶粉、甘油、丙烯、乙二醇、水、乙醇等。如果需要,本组合物还可以含有少量的湿润剂或乳化剂,或pH缓冲剂。
当意在口服施用时,组合物优选为固体或液体形式,其中半固体、半液体、悬浮液和凝胶形式包括在本文所认为的固体或液体的形式内。
作为口服施用的固体组合物,可以将组合物配制为粉末、颗粒、压制的片剂、丸剂、胶囊、口香糖、薄脆饼或类似形式。这样的固体组合物通常含有一个或多个惰性稀释剂。此外,可以存在以下各项中的一种或多种:粘合剂如羧甲基纤维素,乙基纤维素,微晶纤维素,或明胶;赋形剂如淀粉,乳糖或糊精,崩解剂如海藻酸,海藻酸钠,玉米淀粉等;润滑剂如硬脂酸镁;助流剂如胶体二氧化硅;增甜剂如蔗糖或糖精;调味剂如薄荷,水杨酸甲酯或柑橘调味剂;和着色剂。
当组合物是胶囊(例如明胶胶囊)的形式时,除上述类型的材料外,其可以含有液体载体如聚乙二醇,环糊精或脂肪油。
组合物可以是液体的形式,例如酏剂(elixir),糖浆,溶液,乳剂或悬浮液。液体可以用于口服施用或用于通过注射递送。当意在口服施用时,组合物可以包含增甜剂,防腐剂,染料/着色剂和增味剂中的一种或多种。在用于通过注射施用的组合物中,还可以包含表面活性剂、防腐剂、湿润剂、分散剂、悬浮剂、缓冲液、稳定剂和等渗剂中的一种或多种。
优选的施用途径是肠胃外施用,包括但不限于,皮内,肌肉内,腹膜内,静脉内,皮下,鼻内,硬膜外,鼻内,脑内,心室内,鞘内,阴道内或经皮。优选的施用方式取决于执业医生的判断,并且部分取决于医学状况的位点(如癌症位点)。在更优选的实施方案中,本发明的药物抗体缀合物静脉内施用。
本发明的液体组合物,不论他们是溶液,悬浮液或其他类似形式,还可以包括以下各项中的一种或多种:用于注射的无菌稀释剂如水,盐水溶液,优选生理盐水,林格氏溶液,等渗氯化钠,非发挥性油如合成的单或二甘油酯,聚乙二醇,甘油,或其他溶剂;抗菌剂如苄醇或对羟苯甲酸甲酯;和用于调节渗涨度的试剂如氯化钠或葡萄糖。肠胃外组合物可以装入由玻璃、塑料或其他材料制成的安瓿瓶、一次性注射器或多剂量瓶中。生理盐水是优选的辅药。
在特定疾病或病状的治疗中有效的本发明的药物缀合物的量将取决于疾病或病状的性质,并且可以通过标准临床技术确定。此外,可以任选地使用体外或体内测定,帮助鉴定最佳的剂量范围。要用于组合物的准确剂量还将取决于施用途径,和疾病或病症的严重度,并且应该根据执业医生的判断和各个患者的情况确定。
组合物包含有效量的本发明的药物缀合物,从而将获得合适的剂量。化合物的正确剂量将根据以下各项而变化:具体的制剂,应用模式,和其待治疗的特异性位点,宿主和疾病,例如癌症,以及(如果是这样的话)何种类型的肿瘤。还应当考虑其他因素如年龄,体重,性别,膳食,施用时间,排泄率,宿主状况,药物组合,反应敏感性和疾病的严重度。施用可以在最大耐受剂量内连续或周期性进行。
通常,该量为组合物的重量的至少约0.01%的本发明的药物缀合物。当意在口服施用时,该量可以在组合物的重量的约0.1%至约80%的范围内改变。优选的口服组合物可以包含以组合物的重量计约4%至约50%的本发明的药物缀合物。
制备优选的本发明的组合物,使得肠胃外剂量单位含有以重量计约0.01%至约2%的本发明的药物缀合物。
对于静脉内施用,所述组合物可以包含约通常约0.1mg/kg至约250mg/kg的动物体重,优选地,约0.1mg/kg至约20mg/kg之间的动物体重,并且更优选约1mg/kg至约10mg/kg的动物体重。
本发明的药物缀合物或组合物可以通过任何方便的途径施用,例如通过输注或推注注射(bolus injection),通过经上皮或粘膜皮肤内膜吸收。
在具体实施方案中,可以希望将一种或多种本发明的药物缀合物或组合物局部施用于需要治疗的区域。在一个实施方案中,可以在癌,肿瘤或肿瘤新生或肿瘤新生前组织的位点(或前位点)通过直接注射进行施用。在另一个实施方案中,可以通过在自身免疫病的表现的位点(或前位点)通过直接注射施用。
还可以例如通过使用吸入器或喷雾器,和利用气雾化剂配制,或通过氟代烃或合成的肺表面活性剂灌注使用肺部施用。在某些实施方案中,可以利用传统的粘合剂和载体如甘油三酯将本发明的药物抗体缀合物或组合物配制为栓剂。
本组合物可以采用溶液、悬浮液、乳剂、片剂、丸剂、小球、胶囊、含液体胶囊、粉末、缓释制剂、栓剂、乳剂、气雾剂、喷雾、悬浮液、或适于使用的任何其他形式。合适的药物载体的其他实例在E.W.Martin的"Remington's Drugs Sciences(雷明顿药物科学)"中描述。
可以使用药物领域公知的方法制备药物组合物。例如,意在通过注射施用的组合物可以通过将本发明的药物缀合物与水组合从而形成溶液来制备。可以加入表面活性剂以促进均质溶液或悬浮液的形成。
我们已经发现,本发明的药物缀合物和组合物在癌症的治疗中特别有效。
因此,如之前所述,本发明的第六方面提供治疗受癌症影响的需要其的患者,尤其是人的方法,所述方法包括向受影响的个体施用治疗有效量的本发明的药物缀合物或组合物。本发明的第四方面提供根据本发明的第一方面的药物缀合物,其用于治疗癌症,并且更优选选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤和淋巴瘤的癌症。
本发明的药物缀合物和组合物可用于抑制肿瘤细胞或癌细胞的增殖(multiplication),或用于治疗动物中的癌症。本发明的药物缀合物和组合物可以因此在用于治疗动物癌症的多种环境中使用。包含含有至少一个抗原结合位点的药物-接头-部分的本发明的缀合物可以用于将药物或药物单位递送至肿瘤细胞或癌细胞中。在不受理论限制的情况下,在一个实施方案中,本发明的药物缀合物的包含至少一个抗原结合位点的部分结合癌细胞或肿瘤细胞相关抗原或与癌细胞或肿瘤细胞相关抗原相连,并且本发明的药物缀合物可以经由受体介导的内吞被摄入肿瘤细胞或癌细胞内部。抗原可以附着于肿瘤细胞或癌细胞或可以是与肿瘤细胞或癌细胞相连的细胞外基质蛋白。一旦在细胞内部,接头单元内的一个或多个特定序列被一种或多种肿瘤细胞或癌细胞相关蛋白酶或水解酶水解切割,导致药物或药物-接头化合物的释放。随后释放的药物或药物接头化合物在细胞中自由迁移并诱导细胞毒活性。在备选的实施方案中,药物或药物单元在肿瘤细胞或癌细胞外部从本发明的药物缀合物上切割,并且药物或药物接头化合物随后穿透细胞。
在一个实施方案中,包含至少一个抗原结合位点的部分结合肿瘤细胞或癌细胞。在另一个实施方案中,包含至少一个抗原结合位点的部分结合肿瘤细胞或癌细胞表面上的肿瘤细胞或癌细胞抗原。在又一个实施方案中,包含至少一个抗原结合位点的部分结合作为与肿瘤细胞或癌细胞相连的细胞外基质蛋白的肿瘤细胞或癌细胞抗原。
包含至少一个抗原结合位点的部分对特定肿瘤细胞或癌细胞的特异性对于确定最有效治疗的那些肿瘤或癌症而言可能是重要的。例如,具有曲妥珠单抗单元的本发明的药物缀合物可以用于治疗抗原阳性癌,包括白血病,肺癌,结肠癌,淋巴瘤(例如霍奇金病,非霍奇金淋巴瘤),实体瘤如,肉瘤和癌,多发性骨髓瘤,肾癌和黑素瘤。癌症可以优选为肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤或卵巢癌。例如,具有利妥昔单抗单元的本发明的药物缀合物可以用于治疗表达CD-20的肿瘤如血液系统癌症,包括白血病和淋巴瘤。例如,具有抗CD4抗体单元的本发明的药物缀合物可以用于治疗表达CD-4的肿瘤如血液系统癌症,包括淋巴瘤。例如,具有抗CD5抗体单元的本发明的药物缀合物可以用于治疗表达CD-5的肿瘤如血液系统癌症,包括白血病和淋巴瘤。例如,具有抗CD13抗体单元的本发明的药物缀合物可以用于治疗表达CD-13的肿瘤如血液系统癌症,包括白血病和淋巴瘤。
可以用本发明的药物缀合物治疗的其他特定癌症类型包括,但不限于:血源性癌症,包括所有形式的白血病;淋巴瘤,如霍奇金病,非霍奇金淋巴瘤和多发性骨髓瘤。
尤其是,本发明的药物缀合物和组合物在治疗癌症如肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌方面显示优异的活性。
本发明的药物缀合物和组合物提供特异肿瘤或癌症靶向的缀合,因此降低这些缀合物的一般毒性。接头单元使血液中的药物抗体缀合物稳定,其仍然能够被细胞内肿瘤特异的蛋白酶和水解酶切割,释放药物。
本发明的药物缀合物和组合物可以施用于还经历作为对于癌症的治疗的手术的动物。在本发明的一个实施方案中,另外的治疗方法是放射治疗。
在本发明的具体实施方案中,本发明的药物缀合物或组合物与化疗剂或与放射治疗同时施用。在另一个具体实施方案中,化疗剂或放射治疗在施用本发明的药物缀合物或组合物之前或之后施用,优选在施用本发明的药物抗体缀合物或组合物之前或之后至少一小时,五小时,12小时,一天,一周,一个月,更优选数月(例如多至三个月)施用。
化疗剂可以在连续的时期内施用,可以施用本领域已知的任一种化疗剂或化疗剂的组合。
关于放射,可以使用任意放射治疗方案,这取决于要治疗的癌症的类型。例如,但不限于,可以施用x射线辐射;尤其是,对于深层肿瘤,可以使用高能兆伏级(大于1MeV能量的辐射),并且对于皮肤癌,可以使用电子束和常电压(orthovoltage)x-射线辐射。还可以施用发出-射线的放射性同位素,如镭、钴和其他元素的放射性同位素。
在本发明的第八方面,提供试剂盒,其包含治疗有效量的根据本发明的第一方面的药物缀合物和药用载体。
在一个实施方案中,根据此方面的试剂盒用于治疗癌症,并且更优选选自肺癌,结直肠癌,乳腺癌,胰腺癌,肾癌,白血病,多发性骨髓瘤,淋巴瘤和卵巢癌的癌症。
附图简述
通过举例的方式,在附图中图解说明本发明,其中:
图1是根据本发明的一种方法的示意图,其中通过游离硫醇基与抗体缀合;
图2是根据本发明的另一方法的示意图,其中通过抗体游离赖氨酸基团与抗体缀合;
图3是ADC1针对不同癌细胞系的代表性的剂量响应曲线;
图4表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC1处理不同细胞系后细胞存活的百分数的直方图;
图5是ADC2针对不同癌细胞系的代表性的剂量响应曲线;
图6表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC2处理不同细胞系后细胞存活的百分数的直方图;
图7是ADC3针对不同癌细胞系的代表性的剂量响应曲线;
图8表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC3处理不同细胞系后细胞存活的百分数的直方图;
图9是ADC4针对不同癌细胞系的代表性的剂量响应曲线;
图10表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC4处理不同细胞系后细胞存活的百分数的直方图;
图11是ADC5针对不同癌细胞系的代表性的剂量响应曲线;
图12表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC5处理不同细胞系后细胞存活的百分数的直方图;
图13是ADC6针对不同癌细胞系的代表性的剂量响应曲线;
图14表明显示在单独利用mAb(10μg/mL)或10或1μg/mL的ADC6处理不同细胞系后细胞存活的百分数的直方图;
图15是ADC7针对不同癌细胞系的代表性的剂量响应曲线;
图16表明显示在单独利用mAb(50μg/mL)或50或1μg/mL的ADC7处理不同细胞系后细胞存活的百分数的直方图;
图17是ADC8针对不同癌细胞系的代表性的剂量响应曲线;
图18表明显示在单独利用mAb(50μg/mL)或50或10μg/mL的ADC8处理不同细胞系后细胞存活的百分数的直方图;
图19是ADC9针对不同癌细胞系的代表性的剂量响应曲线;
图20表明显示在单独利用mAb(50μg/mL)或50或0.1μg/mL的ADC9处理不同细胞系后细胞存活的百分数的直方图;
图21是ADC10针对不同癌细胞系的代表性的剂量响应曲线;
图22表明显示在单独利用mAb(50μg/mL)或50或1μg/mL的ADC10处理不同细胞系后细胞存活的百分数的直方图;
图23是ADC11针对不同癌细胞系的代表性的剂量响应曲线;
图24表明显示在单独利用mAb(50μg/mL)或50或1μg/mL的ADC11处理不同细胞系后细胞存活的百分数的直方图;
图25是ADC12针对不同癌细胞系的代表性的剂量响应曲线;
图26表明显示在单独利用mAb(50μg/mL)或1或0.1μg/mL的ADC12处理不同细胞系后细胞存活的百分数的直方图;
图27是ADC13针对不同癌细胞系的代表性的剂量响应曲线;
图28表明显示在单独利用mAb(50μg/mL)或1或0.1μg/mL的ADC13处理不同细胞系后细胞存活的百分数的直方图;
图29是ADC14针对不同癌细胞系的代表性的剂量响应曲线;
图30表明显示在单独利用mAb(50μg/mL)或1μg/mL的ADC14处理不同细胞系后细胞存活的百分数的直方图;
图31是ADC16针对不同癌细胞系的代表性的剂量响应曲线;
图32表明显示在单独利用mAb(50μg/mL)或1μg/mL和0.1μg/mL的ADC16处理不同细胞系后细胞存活的百分数的直方图;
图33是ADC17针对不同癌细胞系的代表性的剂量响应曲线;
图34表明显示在单独利用mAb(50μg/mL)或1μg/mL和0.1μg/mL的ADC17处理不同细胞系后细胞存活的百分数的直方图;
图35是ADC14针对两种Raji细胞克隆的代表性的剂量响应曲线;
图36表明显示在单独利用mAb(50μg/mL)或10μg/mL的ADC14处理不同细胞系后细胞存活的百分数的直方图;
图37是ADC15针对两种Raji细胞克隆的代表性的剂量响应曲线;并且
图38表明显示在单独利用mAb(50μg/mL)或10μg/mL的ADC15处理不同细胞系后细胞存活的百分数的直方图。
实施例
通过以下非限制性实施例的方式进一步描述本发明。在实施例中,使用以下缩写:
CDI,1,1’-羰基二咪唑
DIPEA,二异丙基乙胺
Hex,己烷
EtOAc,乙酸乙酯
DCM,二氯甲烷
NMP,N-甲基-2-吡咯烷酮
DMF,二甲基甲酰胺
EDC,N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺盐酸盐
EDTA,乙二胺四乙酸
MeOH,甲醇
DTT,二硫苏糖醇
Py,吡啶
THF,四氢呋喃
TCEP,三[2-羧基乙基]膦盐酸盐
MC,6-马来酰亚胺基己酰基
Fmoc,9-芴基甲氧基羰基
Cit,瓜氨酸
Val,缬氨酸
DMSO,二甲亚砜
Trt,三苯基甲基
HOBt,1-羟基苯并三唑
DIPCDI,N,N’-二异丙基碳二亚胺
TFA,三氟乙酸
PABOH,4-氨基苄醇
bis-PNP,双(4-硝基苯基)碳酸酯
NAC,N-乙酰半胱氨酸
SEC,尺寸排阻层析
HPLC,高效液相色谱
ADC,抗体药物缀合物
ATCC,美国典型培养物保藏中心(American Type Culture Collection)
DMEM,达尔伯克改良伊格尔培养基(Dulbecco’s Modified Eagle’s Medium)
RPMI,罗斯维尔帕克纪念研究所(Rosmell Park Memorial Institute)培养基
ITS,胰岛素-运铁蛋白-亚硒酸钠培养基补充物
FCS,胎牛血清
SRB,磺酰罗丹明B
PBS,磷酸盐缓冲盐水
DR,剂量响应
UV,紫外光
SMCC,琥珀酰亚胺基-4-(N-马来酰亚胺基甲基)环己烷-1-甲酸酯
LAR,接头与抗体比例
制备实施例
化合物9的制备:MC-Val-Cit-PABC-PNP
反应方案
(a)化合物10的制备:MC-Val-Cit-OH
化合物10
将Cl-TrtCl-树脂(20g,1.49mmol/g)(Iris Biotech,Ref.:BR-1065,2-氯三苯甲基氯树脂(200-400目,1%DVB,1.0-1.6mmol/g),CAS 42074-68-0)置于滤板中。将100mL的DCM加入至树脂中并将混合物搅拌1h。通过在真空下过滤除去溶剂。加入Fmoc-Cit-OH(11.83g,29.78mmol)和DIPEA(17.15mL,98.45mmol)在DCM(80mL)中的溶液并且将混合物搅拌10min。之后加入DIPEA(34.82mmol,199.98mmol)并且将混合物搅拌1h。在搅拌15分钟后通过加入MeOH(30mL)终止反应。对因此产生的Fmoc-Cit-O-TrtCl-树脂进行以下洗涤/处理:DCM(5x 50mL x 0.5min),DMF(5x 50mL x 0.5min),哌啶:DMF(1:4,1x 1min,2x 10min),DMF(5x 50mL x 0.5min),DCM(5x 50mL x 0.5min)。最终哌啶洗涤得到NH2-Cit-O-TrtCl-树脂。计算加载量:1.15mmol/g。
将上文产生的NH2-Cit-O-TrtCl-树脂用DMF(5x 50mL x 0.5min)洗涤,并且将Fmoc-Val-OH(31.22g,91.98mmol),HOBt(11.23g,91.98mmol)在DMF(100mL)中的溶液加入NH2-Cit-O-TrtCl-树脂中,搅拌并加入DIPCDI(14.24mL,91.98mmol)并且将混合物搅拌1.5h。通过用DMF(5x50mL x 0.5min)洗涤将反应终止。将因此产生的Fmoc-Val-Cit-O-TrtCl-树脂用哌啶:DMF(1:4,1x 1min,2x 10min)处理并且用DMF(5x 50mL x 0.5min)洗涤。最终哌啶洗涤得到NH2-Val-Cit-O-TrtCl-树脂。
将6-马来酰亚胺基己酸(MC-OH)(9.7g,45.92mmol),HOBt(6.21g,45.92mmol)在DMF(100mL)中的溶液加入上文产生的NH2-Val-Cit-O-TrtCl-树脂中,搅拌并加入DIPCDI(7.12mL,45.92mmol),并且将混合物搅拌1.5h。通过用DMF(5x 50mL x 0.5min)和DCM(5x 50mL x 0.5min)洗涤终止反应。
通过用TFA:DCM(1:99,5x 100mL)处理将肽从树脂切割。将树脂用DCM(7x 50mL x 0.5min)洗涤。在减压下将合并的滤液蒸发至干燥,并且将获得的固体用Et2O研碎并过滤,获得化合物10(7.60g,71%),为白色固体。
1H NMR(500MHz,DMSO-d6):δ12.47(s,1H),8.13(d,J=7.3Hz,1H),7.74(d,J=9.0Hz,1H),6.99(s,2H),5.93(s,1H),5.35(s,2H),4.20(dd,J=9.0,6.8Hz,1H),4.15-4.07(m,1H),3.36(t,J=7.0Hz,2H),3.00-2.88(m,2H),2.21-2.12(m,1H),2.11-2.03(m,1H),1.98-1.86(m,1H),1.74-1.62(m,1H),1.61-1.50(m,1H),1.50-1.31(m,6H),1.21-1.11(m,2H),0.84(d,J=6.8Hz,3H),0.80(d,J=6.8Hz,3H)。
ESI-MS m/z:对于C21H33N5O7计算:467.2。实验值:468.3(M+H)+。
(b)化合物11的制备:MC-Val-Cit-PABOH
化合物11
向化合物10(1.6g,3.42mmol)和4-氨基苄醇(PABOH)(0.84g,6.84mmol)在DCM(60mL)中的溶液中加入HOBt(0.92g,6.84mmol)在DMF(5mL)中的溶液。加入DIPCDI(1.05mL,6.84mmol),将反应混合物在23℃搅拌2h,加入Et2O(150mL),并在真空下将获得的固体在滤板中过滤,获得化合物11(1.31g,67%)。
1H NMR(500MHz,DMSO-d6):δ9.88(s,1H),8.03(d,J=7.6Hz,1H),7.77(dd,J=12.2,8.5Hz,1H),7.53(d,J=8.2Hz,2H),7.21(d,J=8.2Hz,2H),6.99(s,3H),6.01-5.92(m,1H),5.39(s,2H),5.07(s,1H),4.41(s,2H),4.39-4.31(m,1H),4.23-4.12(m,1H),3.36(t,J=7.0Hz,2H),3.06-2.97(m,1H),2.96-2.90(m,1H),2.22-2.03(m,2H),2.01-1.88(m,1H),1.76-1.62(m,1H),1.63-1.28(m,6H),1.25-1.11(m,2H),0.84(d,J=6.9Hz,3H),0.81(d,J=6.8Hz,3H)。
ESI-MS m/z:对于C28H40N6O7计算:572.3。实验值:573.3(M+H)+。
(c)化合物9的制备:MC-Val-Cit-PAB-PNP
化合物9
向化合物11(500mg,0.87mmol)和双(4-硝基苯基)碳酸酯(bis-PNP)(2.64g,8.72mmol)在DCM:DMF(8:2,25mL)中的溶液加入DIPEA(0.45mL,2.61mmol)。将反应混合物在23℃搅拌20h并且倒在硅胶柱上(DCM:CH3OH,从50:1至10:1),以获得纯的目标化合物9(364mg,57%)。
Rf=0.40(CH2Cl2:CH3OH,9:1)。
1H NMR(400MHz,CDCl3/CD3OD):δ9.45(s,1H),8.23(d,J=8.3Hz,2H),7.59(d,J=8.5Hz,2H),7.35(d,J=8.3Hz,2H),7.34(d,J=8.5Hz,2H),6.65(s,2H),5.20(s,2H),4.56(dt,J=10.5,5.4Hz,1H),4.15(d,J=7.2Hz,1H),3.46(dd,J=8.0,6.4Hz,2H),3.16-2.89(m,2H),2.21(dd,J=8.3,6.6Hz,2H),2.06-1.97(m,1H),1.90-1.83(m,1H),1.73-1.46(m,7H),1.34-1.20(m,2H),0.91(d,J=6.7Hz,3H),0.90(d,J=6.7Hz,3H)。
13C NMR(125MHz,CDCl3/CD3OD)δ174.4,172.4,171.1,170.6,160.5,155.5,152.5,145.3,138.7,134.1,129.9,129.5,125.2,121.8,120.0,70.6,59.0,53.2,37.5,35.8,30.6,29.6,29.3,28.1,26.2,26.2,25.1,19.1,18.1。
ESI-MS m/z:对于C35H43N7O11计算:737.3。实验值:738.3(M+H)+。
实施例1
化合物1的制备
(a)化合物3的制备
向化合物2(化合物30a,如WO 2007144423中所述制备,其内容通过引用结合于此)(1.014g,1.8mmol)在DCM(45mL)中的溶液中加入1,1’-羰基二咪唑(876mg,5.4mmol)。在23℃搅拌过夜后,将反应混合物在真空下浓缩。将获得的残留物在用于急骤色谱(flash chromatography)的系统(SiO2,Hex:EtOAc混合物,99:1至85:15)中纯化,获得纯的化合物3(1.176g,86%)。
1H NMR(500MHz,CDCl3):δ8.39(d,J=9.0Hz,1H),8.12(bs,1H),7.40(bs,1H),7.30(t,J=11.5Hz,1H),7.08(bs,1H),6.91(t,J=12.0Hz,1H),6.86(t,J=10.0Hz,1H),6.22(d,J=9.0Hz,1H),6.18(d,J=11.0Hz,1H),5.67(d,J=12.0Hz,1H),5.63-5.61(m,2H),5.28(d,J=11.5Hz,1H),4.94-4.91(m,1H),4.81-4.76(m,1H),4.42(d,J=9.0Hz,1H),4.23-4.19(m,1H),3.66(s,3H),2.87-2.82(m,1H),2.58-2.46(m,3H),2.42-2.35(m,3H),2.08(s,3H),1.83(s,3H),1.16(d,J=6.6Hz,3H),1.06(s,9H)。
13C NMR(125MHz,CDCl3):δ168.5,166.4,161.5,148.7,145.2,140.4,137.6,137.0,134.4,133.9,133.0,130.9,124.2,123.9,121.0,120.5,117.03,110.0,108.1,104.1,81.7,77.8,60.4,55.4,37.2,34.5,26.6,26.3,21.0,17.1,16.6。
ESI-MS m/z:对于C34H45ClN4O7计算:656.30。实验值:657.3(M+H)+。
(b)化合物4的制备
向如上文步骤(a)中所述制备的化合物3(1.160g,1.78mmol)在DCM(45mL)中的溶液中加入丙烷1,3-二胺(0.19mL,2.22mmol)。将反应混合物在23℃搅拌过夜,并随后在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiNH2,DCM:CH3OH,100:0至97:3)中纯化,获得纯的化合物4(800mg,68%)。
1H NMR(300MHz,CDCl3):δ8.90(d,J=11.7Hz,1H),7.34-7.26(m,1H),6.99-6.74(m,2H),6.50(d,J=9.0Hz,1H),6.15(d,J=12.9Hz,1H),5.83(t,J=11.5Hz,1H),5.70(d,J=11.5Hz,1H),5.68-5.57(m,2H),5.27(d,J=9.4Hz,1H),4.80(q,J=8.3Hz,1H),4.52-4.44(m,2H),4.24-4.17(m,1H),3.66(s,3H),3.39-3.17(m,2H),2.93-2.82(m,1H),2.78(t,J=6.5Hz,2H),2.50-2.34(m,2H),2.34-2.24(m,2H),2.19-1.99(m,2H),2.06(s,3H),1.84(s,3H),1.72-1.50(m,2H),1.16(d,J=6.6Hz,3H),1.04(s,9H)。
13C NMR(125MHz,CDCl3):δ168.4,166.1,161.5,156.7,145.2,139.9,137.1,134.0,133.9,131.8,124.3,124.2,122.5,120.9,108.1,105.5,81.8,74.3,60.6,55.4,39.81,39.3037.2,34.7,33.1,31.5,29.6,26.7,26.2,21.0,17.1,16.6。
ESI-MS m/z:对于C34H51ClN4O7计算:662.30。实验值:663.3(M+H)+。
(c)化合物1的制备
向上述步骤(b)中所述制备的化合物4(52mg,0.078mmol)和6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯(27.1mg,0.088mmol)在DCM(2mL)中的溶液中加入DIPEA(15μL,0.086mmol)。将反应混合物在23℃搅拌过夜并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的目标化合物1(29.7mg,44%)。
1H NMR(500MHz,CDCl3):δ8.86(d,J=10.8Hz,1H),7.30(t,J=11.6Hz,1H),6.90(td,J=11.5,1.2Hz,1H),6.78(t,J=9.7Hz,1H),6.68(bs,2H),6.63(d,J=9.4Hz,1H),6.51(t,J=6.4Hz,1H),6.16(d,J=11.8Hz,1H),5.76(t,J=6.4Hz,1H),5.72(d,J=11.6Hz,1H),5.65(dd,J=6.4,2.9Hz,1H),5.62-5.57(m,1H),5.29(d,J=11.1Hz,1H),4.81(q,J=8.3Hz,1H),4.52-4.48(m,2H),4.24(ddd,J=11.4,7.3,4.3Hz,1H),3.66(s,3H),3.50(t,J=7.3Hz,2H),3.33-3.10(m,4H),2.93-2.81(m,1H),2.45-2.31(m,5H),2.17(t,J=7.6Hz,2H),2.10-1.98(m,1H),2.07(s,3H),1.84(s,3H),1.72-1.54(m,8H),1.16(d,J=6.6Hz,3H),1.05(s,9H)。
13C NMR(125MHz,CDCl3):δ173.8,170.8,168.3,166.5,161.6,157.1,145.1,140.4,137.5,134.2,134.1,134.0,131.9,124.2,124.0,122.5,120.6,108.3,106.0,81.8,74.5,60.6,55.4,37.7,37.6,37.2,36.3,36.1,34.7,33.4,31.0,29.8,28.3,26.7,26.3,26.2,25.6,21.0,17.2,16.6。
ESI-MS m/z:对于C44H62ClN5O10计算:855.42。实验值:856.5(M+H)+。
实施例2
化合物5的制备
(a)化合物7的制备
向化合物6(化合物30b,如WO 2007144423中所述制备,其内容通过引用结合于此)(750mg,1.42mmol)在DCM(35.5mL)中的溶液中加入1,1’-羰基二咪唑(691mg,4.26mmol)。在23℃搅拌过夜后,将反应混合物在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物7(717mg,81%)。
1H NMR(300MHz,CDCl3):δ8.24(d,J=11.0Hz,1H),8.09(s,1H),7.40(s,1H),7.36-7.23(m,1H),7.05(s,1H),6.95-6.83(m,2H),6.34(d,J=9.2Hz,1H),6.14(d,J=11.8Hz,1H),5.74-5.57(m,3H),5.43-5.34(m,1H),5.28(d,J=10.2Hz,1H),4.98-4.88(m,1H),4.78(q,J=7.8Hz,1H),4.47(d,J=9.2Hz,1H),4.25-4.16(m,1H),3.64(s,3H),2.92-2.76(m,1H),2.59-2.37(m,6H),1.83(s,3H),1.64(d,J=6.7Hz,3H),1.14(d,J=6.7Hz,3H),1.03(s,9H)。
(b)化合物8的制备
向如上述步骤(a)中所述制备的化合物7(1.68g,2.7mmol)在DCM(80mL)中的溶液中加入丙烷1,3-二胺(0.27mL,3.24mmol)。将反应混合物在23℃搅拌过夜并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,DCM:CH3OH,100:0至97:3)中纯化,获得化合物8(854mg,50%)。
1H NMR(300MHz,CDCl3):δ8.90(d,J=11.7Hz,1H),7.39-7.18(m,1H),6.92-6.84(m,2H),6.50(d,J=9.0Hz,1H),6.15(d,J=12.9Hz,1H),5.75-5.67(m,2H),5.66-5.54(m,2H),5.46-5.33(m,1H),5.26(d,J=10.3Hz,1H),4.83(q,J=8.3Hz,1H),4.50-4.48(m,2H),4.30-4.04(m,1H),3.66(s,3H),3.39-3.17(m,2H),2.93-2.82(m,1H),2.78(t,J=6.5Hz,2H),2.50-2.34(m,3H),2.34-2.24(m,2H),2.19-1.99(m,1H),1.83(s,3H),1.72-1.50(m,2H),1.62(d,J=6.7Hz,3H),1.16(d,J=6.6Hz,3H),1.04(s,9H)。
13C NMR(125MHz,CDCl3):δ168.3,166.2,161.6,157.1,145.1,139.9,137.1,134.0,133.9,126.9,124.9,124.2,123.9,120.9,108.2,106.3,81.8,75.0,60.6,55.4,39.6,37.2,34.7,32.8,31.5,31.1,29.6,26.7,26.2,17.1,16.6,12.9。
ESI-MS m/z:对于C34H52N4O7计算:628.4。实验值:629.5(M+H)+。
(c)化合物5的制备
在23℃向如上述步骤(b)中所述制备的化合物8(150mg,0.24mmol)在DCM(8mL)中的溶液中加入6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯(88.3mg,0.28mmol)。将反应混合物在23℃搅拌18h,并且在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的目标化合物5(75mg,38%)。
1H NMR(500MHz,CDCl3):δ8.87(d,J=10.7Hz,1H),7.32-7.22(m,1H),6.89(t,J=11.6Hz,1H),6.78(t,J=9.7Hz,1H),6.68(s,2H),6.61(d,J=9.4Hz,1H),6.54(t,J=6.0Hz,1H),6.15(d,J=11.6Hz,1H),5.77-5.51(m,2H),5.64(dd,J=6.4,3.0Hz,1H),5.60-5.55(m,1H),5.38(ddd,J=13.0,8.8,6.6Hz,1H),5.28(d,J=10.0Hz,1H),4.83(q,J=8.3Hz,1H),4.59-4.44(m,2H),4.23(ddd,J=11.5,7.2,4.4Hz,1H),3.65(s,3H),3.49(t,J=7.2Hz,2H),3.29-3.12(m,4H),2.87-2.81(m,1H),2.48-2.32(m,5H),2.18-2.09(m,3H),1.88-1.82(m,1H),1.83(s,3H),1.67-1.55(m,7H),1.62(d,J=6.8Hz,3H),1.15(d,J=6.7Hz,3H),1.04(s,9H)。
13C NMR(125MHz,CDCl3):δ173.6,170.8,168.2,166.4,161.6,157.4,145.2,140.2,137.4,134.2,134.1,134.0,127.0,124.9,123.9,120.7,108.3,106.5,81.8,75.3,60.6,55.4,37.7,37.6,37.2,36.3,36.0,34.7,31.8,31.6,31.1,29.9,28.3,26.7,26.4,26.2,25.2,22.6,17.2,16.6。
ESI-MS m/z:对于C44H63N5O10计算:821.5。实验值:822.4(M+H)+。
实施例3
化合物12的制备
(a)化合物12的制备
在23℃,将DIPEA(25μL,0.14mmol)加入至如上述制备实施例中所示制备的化合物9(94.5mg,0.13mmol)和如上述实施例1(b)中所述制备的化合物4(85mg,0.13mmol)在NMP(6.5mL)中的溶液中。9h后,将反应混合物用H2O稀释并用EtOAc萃取。将合并的有机层经无水Na2SO4干燥,过滤,并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,DCM:CH3OH,100:0至90:10)中纯化。最后,通过半制备型HPLC(Symmetry C18,7μm,19x 150mm,梯度H2O+CH3CN,流速15mL/min,UV检测)实现目标化合物12(35.7mg,22%)的纯化。
1H NMR(500MHz,CDCl3/CD3OD):δ7.49(d,J=8.1Hz,2H),7.22(d,J=8.1Hz,2H),7.19(t,J=11.8Hz,1H),6.96(dd,J=23.5,8.9Hz,1H),6.84(t,J=11.5Hz,1H),6.70-6.64(m,1H),6.64(s,2H),6.10(d,J=11.6Hz,1H),5.93(t,J=6.2Hz,1H),5.82(t,J=6.2Hz,1H),5.69(d,J=11.4Hz,1H),5.61(dd,J=6.3,3.1Hz,1H),5.54(t,J=7.8Hz,1H),5.22(d,J=9.7Hz,1H),4.96(s,2H),4.75(q,J=8.1Hz,1H),4.55-4.49(m,2H),4.43-4.36(m,1H),4.23-4.10(m,2H),3.59(s,3H),3.44(t,J=7.2Hz,2H),3.18-3.04(m,8H),2.82-2.72(m,1H),2.49-2.34(m,3H),2.27(t,J=7.1Hz,2H),2.18(t,J=7.2Hz,2H),2.16-2.06(m,1H),2.01-1.95(m,1H),2.00(s,3H),1.87-1.79(m,1H),1.78(s,3H),1.73-1.40(m,11H),1.35-1.20(m,2H),1.09(d,J=10.0Hz,3H),0.96(s,9H),0.87(dd,J=6.8,4.3Hz,6H)。
13C NMR(125MHz,CDCl3):δ174.1,172.2,171.0,170.3,168.8,166.8,162.1,160.2,157.0,156.9,144.9,140.2,137.7,137.5,134.0,132.4,131.7,128.7,124.0,123.4,122.4,120.4,119.8,111.5,108.6,107.0,81.9,73.8,68.6,66.2,60.3,58.8,55.4,53.0,37.6,37.5,37.1,35.9,34.6,33.2,30.6,29.9,29.2,28.0,26.5,26.2,26.0,25.0,22.6,20.8,19.1,18.2,17.04,16.4。
ESI-MS m/z:对于C63H89ClN10O15计算:1260.6。实验值:1261.6(M+H)+。
实施例4
化合物13的制备
(a)化合物14的制备
向如上述实施例1(b)中所述制备的化合物4(110mg,0.17mmol)和3-(甲基二硫烷基)丙酸(34mg,0.22mmol)在DCM(5mL)中的溶液中加入N-(3-二甲基氨基丙基)-N′-乙基碳二亚胺盐酸盐(EDC)(47.8mg,0.22mmol)和N,N′-二异丙基乙胺(3.8μL,0.22mmol)。将反应混合物在23℃搅拌6h,用H2O稀释并用DCM萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物14(123mg,93%)。
1H NMR(500MHz,CDCl3):δ8.88(d,J=10.8Hz,1H),7.29-7.24(m,1H),6.90(t,J=11.5Hz,1H),6.82(t,J=9.1Hz,1H),6.63(t,J=6.1Hz,1H),6.49(d,J=9.4Hz,1H),6.16(dd,J=11.5,1.5Hz,1H),5.70(d,J=11.5Hz,1H),5.68-5.51(m,3H),5.29(d,J=9.7Hz,1H),4.81(q,J=8.2Hz,1H),4.52(d,J=9.5Hz,1H),4.52-4.43(m,1H),4.24(ddd,J=11.5,7.3,4.3Hz,1H),3.66(s,3H),3.37-3.21(m,3H),3.21-3.12(m,1H),2.97(t,J=7.2Hz,2H),2.90-2.81(m,1H),2.60(t,J=7.2Hz,2H),2.49-2.35(m,3H),2.39(s,3H),2.33(t,J=7.0Hz,2H),2.14-2.07(m,1H),2.07(s,3H),1.84(s,3H),1.73-1.64(m,2H),1.16(d,J=6.7Hz,3H),1.05(s,9H)。
13C NMR(125MHz,CDCl3):δ171.6,168.2,166.4,161.6,157.2,145.2,140.3,137.4,134.2,134.0,131.9,124.4,124.1,122.4,120.7,108.3,105.6,81.8,74.8,60.6,60.4,55.5,37.8,37.2,36.2,35.6,34.7,33.1,31.0,29.8,26.7,26.2,23.0,21.0,17.2,16.6。
(b)化合物13的制备
将如上述步骤(a)中所述制备的化合物14(100mg,0.125mmol)在EtOAc(4.3mL)和CH3OH(4.3mL)的混合物中的溶液用二硫苏糖醇(154.8mg,1.0mmol)在0.05M磷酸钾缓冲液(4.3mL)中(在pH 7.5,含有2mM乙二胺四乙酸(EDTA))的溶液处理。将混合物在23℃搅拌4h。将反应用0.2M磷酸钾缓冲液(13mL)在pH 6.0含有2mM EDTA的溶液处理并随后用EtOAc萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的粗产物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的目标化合物13(35mg,37%)。
1H NMR(300MHz,CDCl3):δ8.91(d,J=10.8Hz,1H),7.27-7.24(m,1H),6.91(t,J=11.5Hz,1H),6.82(t,J=9.7Hz,1H),6.67(t,J=6.1Hz,1H),6.49(d,J=9.4Hz,1H),6.16(d,J=11.6Hz,1H),5.71(d,J=11.6Hz,1H),5.66-5.57(m,3H),5.29(d,J=9.9Hz,1H),4.84(q,J=8.3Hz,1H),4.51(d,J=9.5Hz,1H),4.50-4.45(m,1H),4.24-4.20(m,1H),3.66(s,3H),3.36-3.12(m,4H),2.90-2.71(m,3H),2.64-2.24(m,7H),2.14-2.04(m,1H),2.06(s,3H),1.83(s,3H),1.73-1.68(m,2H),1.15(d,J=6.7Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C37H55ClN4O8S计算:750.3。实验值:773.2(M+Na)+。
实施例5
化合物15的制备
(a)化合物16的制备
在0℃向化合物2(化合物30a,如WO 2007144423中所述制备,其内容通过引用结合于此)(300mg,0.53mmol)在DCM(5mL)中的溶液中加入吡啶(85μL,1.06mmol)和氯甲酸4-硝基苯酯(214.7mg,1.06mmol)。将反应混合物在23℃搅拌1.5h,用10%柠檬酸稀释并用DCM萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物16(307mg,80%)。
1H NMR(500MHz,CDCl3):δ8.29(d,J=9.2Hz,2H),8.08(d,J=10.9Hz,1H),7.44(d,J=9.1Hz,2H),7.27-7.22(m,1H),6.92-6.83(m,2H),6.20(d,J=9.2Hz,1H),6.17(dd,J=11.6,1.4Hz,1H),5.67-5.58(m,3H),5.29(d,J=10.0Hz,1H),4.84(q,J=8.2Hz,1H),4.77-4.72(m,1H),4.41(d,J=9.3Hz,1H),4.22(ddd,J=11.5,7.5,4.4Hz,1H),3.67(s,3H),2.89-2.82(m,1H),2.54-2.33(m,6H),2.10(d,J=1.2Hz,3H),1.85(d,J=1.3Hz,3H),1.17(d,J=6.7Hz,3H),1.02(s,9H)。
13C NMR(125MHz,CDCl3):δ168.4,166.1,161.5,155.3,152.5,145.5,145.2,140.4,137.6,134.3,134.0,133.2,125.3,124.4,124.1,121.8,121.2,120.4,108.1,104.6,81.8,79.1,60.4,55.5,37.3,34.7,32.7,30.1,26.6,26.3,21.1,17.2,16.6。
(b)化合物17的制备
在23℃向化合物16(156.3mg,0.21mmol)在DCM(2.5mL)中的溶液中加入3-氨基丙烷-1-硫醇盐酸盐(44.8mg,0.26mmol)在DCM(2.5mL),三乙胺(58μL,0.34mmol)和DMF(0.1mL)中的悬浮液。将反应混合物在23℃搅拌3h,用H2O稀释并用DCM萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物17(80mg,95%)。
1H NMR(300MHz,CDCl3):δ8.68(d,J=10.6Hz,1H),7.28(t,J=11.6Hz,1H),6.89(t,J=11.5Hz,1H),6.76(t,J=9.6Hz,1H),6.65(d,J=9.1Hz,1H),6.13(d,J=11.7Hz,1H),5.87-5.51(m,4H),5.28(d,J=5.0Hz,1H),4.77(q,J=8.2Hz,1H),4.61-4.39(m,2H),4.29-4.00(m,1H),3.65(s,3H),3.31-3.18(m,2H),2.98-2.77(m,1H),2.68(t,J=7.4Hz,2H),2.55-2.22(m,6H),2.04(s,3H),2.00-1.80(m,2H),1.83(s,3H),1.15(d,J=6.6Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C68H98Cl2N6O14S2计算:1356.6。实验值:1357.3(M+H)+。
(c)化合物15的制备
将化合物17(59.4mg,0.044mmol)在EtOAc(1.5mL)和CH3OH(1.5mL)的混合物中的溶液用二硫苏糖醇(0.35mL,0.35mmol)在pH 7.5含有2mM乙二胺四乙酸(EDTA)的0.05M磷酸钾缓冲液(1.5mL)的溶液处理。将混合物在23℃搅拌4h。将反应用pH 6.0含有2mM EDTA的0.2M磷酸钾缓冲液的溶液处理并用EtOAc萃取(x3)。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的目标化合物15(31mg,59%)。
1H NMR(500MHz,CDCl3):δ8.66(d,J=10.7Hz,1H),7.29(t,J=11.2Hz,1H),6.91(t,J=11.5Hz,1H),6.83(t,J=9.7Hz,1H),6.38(d,J=9.4Hz,1H),6.17(d,J=11.8Hz,1H),5.70(d,J=11.4Hz,1H),5.65-5.51(m,2H),5.34(t,J=6.3Hz,1H),5.29(d,J=10.0Hz,1H),4.82(q,J=8.3Hz,1H),4.56-4.48(m,1H),4.45(d,J=9.3Hz,1H),4.22(ddd,J=11.4,7.5,4.3Hz,1H),3.67(s,3H),3.31(q,J=6.4Hz,2H),2.88-2.83(m,1H),2.55(q,J=7.7Hz,2H),2.47-2.30(m,5H),2.12-2.07(m,1H),2.08(s,3H),1.88-1.76(m,5H),1.17(d,J=6.6Hz,3H),1.06(s,9H)。
13C NMR(125MHz,CDCl3):δ168.2,166.2,161.5,156.7,145.2,140.2,137.3,134.2,134.0,132.0,124.4,124.2,122.3,120.8,108.1,105.5,81.8,74.5,60.6,55.4,39.6,37.3,34.6,33.9,33.3,30.8,26.7,26.3,21.8,21.1,17.2,16.7。
ESI-MS m/z:对于C34H50ClN3O7S计算:679.3。实验值:702.4(M+Na)+。
实施例6
化合物18的制备
(a)化合物19的制备
向如上述实施例2(b)中所述制备的化合物8(280mg,0.45mmol)和3-(甲基二硫烷基)丙酸(88mg,0.58mmol)在DCM(7.5mL)中的溶液中加入N-(3-二甲基氨基丙基)-N′-乙基碳二亚胺盐酸盐(EDC)(126mg,0.58mmol)和N,N′-二异丙基乙胺(0.1mL,0.58mmol)。将反应混合物在23℃搅拌3h,用H2O稀释并用DCM萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物19(240mg,71%)。
1H NMR(500MHz,CDCl3):δ8.91(d,J=10.8Hz,1H),7.34-7.20(m,1H),6.89(t,J=11.4Hz,1H),6.83-6.72(m,2H),6.51(d,J=9.5Hz,1H),6.16(d,J=11.3Hz,1H),5.70(d,J=11.5Hz,1H),5.64(dd,J=6.1,3.3Hz,1H),5.61-5.55(m,2H),5.47-5.33(m,1H),5.28(d,J=9.3Hz,1H),4.84(q,J=8.3Hz,1H),4.51(d,J=9.6Hz,1H),4.52-4.47(m,1H),4.27-4.19(m,1H),3.66(s,3H),3.37-3.21(m,3H),3.21-3.12(m,1H),2.96(t,J=7.2Hz,2H),2.90-2.81(m,1H),2.60(t,J=7.2Hz,2H),2.43-2.35(m,5H),2.39(s,3H),2.16-2.04(m,1H),1.84(s,3H),1.70-1.61(m,5H),1.16(d,J=6.7Hz,3H),1.05(s,9H)。
13C NMR(125MHz,CDCl3)δ171.5,168.1,166.3,157.4,145.2,140.3,137.4,134.2,134.0,127.0,124.9,124.0,120.8,108.3,106.3,81.8,75.5,60.6,55.4,53.4,37.8,37.2,36.2,35.9,34.7,33.1,31.8,31.2,29.8,26.7,26.2,23.0,17.2,16.6,13.0。
ESI-MS m/z:对于C38H58N4O8S2计算:763.4。实验值:762.4(M+H)+。
(b)化合物18的制备
将如上述步骤(a)中所述制备的化合物19(240mg,0.31mmol)在EtOAc(15mL)和CH3OH(22mL)的混合物中的溶液用二硫苏糖醇(0.79mL的1.0M溶液,0.79mmol)在pH 7.5含有2mM乙二胺四乙酸(EDTA)的0.05M磷酸钾缓冲液(17.4mL)的溶液处理。将混合物在23℃搅拌4h。将反应用pH 6.0含有2mM EDTA的0.2M磷酸钾缓冲液(21mL)的溶液处理并用乙酸乙酯萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的粗产物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的目标化合物18(105mg,47%)。
1H NMR(300MHz,CDCl3):δ8.93(d,J=10.6Hz,1H),7.30-7.22(m,1H),6.90(t,J=11.6Hz,1H)6.86-6.72(m,2H),6.48(d,J=9.4Hz,1H),6.16(d,J=11.6Hz,1H),5.70(d,J=11.4Hz,1H),5.65-5.55(m,3H),5.48-5.34(m,1H),5.29(d,J=9.9Hz,1H),4.84(q,J=9.4,8.7Hz,1H),4.52(d,J=9.5Hz,1H),4.57-4.44(m,1H),4.32-4.17(m,1H),3.66(s,3H),3.39-3.20(m,3H),3.22-3.09(m,1H),2.90-2.75(m,3H),2.50(t,J=6.9,2H),2.45-2.28(m,5H),2.16-2.08(m,1H),1.83(s,3H),1.72-1.65(m 2H),1.64(d,J=6.6Hz,3H)1.16(d,J=6.7Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C37H56N4O8S计算:716.4。实验值:717.3(M+H)+。
实施例7
化合物25的制备
(a)化合物23的制备
向化合物22(333mg,0.63mmol)(化合物71,如WO 2009080761中所述制备,其内容通过引用结合于此)在CH2Cl2(12.5mL)中的溶液中加入1,1’-羰基二咪唑(308mg,1.90mmol)。在23℃搅拌过夜后,将反应混合物在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物23(344mg,88%)。
1H NMR(300MHz,CDCl3):δ8.14(s,1H),8.11(d,J=10.8Hz,1H),7.43(s,1H),7.28(t,J=11.6Hz,1H),7.08(dd,J=1.7,0.9Hz,1H),6.96-6.81(m,2H),6.26(d,J=9.3Hz,1H),6.17(d,J=11.6Hz,1H),5.66(dt,J=11.4,1.4Hz,1H),5.62(dd,J=5.9,3.5Hz,1H),5.28(d,J=10.0Hz,1H),5.04-4.89(m,1H),4.80(q,J=8.3Hz,1H),4.40(d,J=9.3Hz,1H),4.21(ddd,J=10.3,7.5,5.3Hz,1H),3.65(s,3H),2.91-2.78(m,1H),2.68-2.49(m,3H),2.45-2.31(m,2H),1.89(t,J=2.5Hz,3H),1.84(s,3H),1.15(d,J=6.7Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C34H44N4O7计算:620.32。实验值:621.3(M+H)+。
(b)化合物24的制备
向如上述步骤(a)中所述制备的化合物23(0.130g,0.21mmol)在CH2Cl2(3.5mL)中的溶液中加入丙烷1,3-二胺(0.022mL,0.26mmol)。将反应混合物在23℃搅拌6小时并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,DCM:CH3OH,100:0至97:3)中纯化,获得化合物24(120mg,91%)。
1H NMR(300MHz,CDCl3):δ8.71(d,J=10.7Hz,1H),7.28(t,J=11.6Hz,1H),6.91-6.77(m,2H),6.44(d,J=9.4Hz,1H),6.14(d,J=11.6Hz,1H),5.77-5.57(m,3H),5.27(d,J=10.0Hz,1H),4.83(q,J=8.3Hz,1H),4.57-4.53(m,1H),4.46(d,J=12.4Hz,1H),4.31-4.11(m,1H),3.65(s,3H),3.31-3.24(m,2H),2.93-2.67(m,3H),2.56-2.24(m,6H),2.16(s,3H),1.83(s,3H),1.63(dd,J=9.6,3.5Hz,2H),1.15(d,J=6.6Hz,3H),1.04(s,9H)。
13C NMR (125MHz,CDCl3):δ168.3,166.1,161.6,156.5,145.2,140.0,137.2,134.1,134.0,124.3,124.2,120.9,108.1,106.0,81.8,78,4,74.5,73.2,60.6,55.4,39.8,39.2,37.3,34.8,33.0,30.9,30.2,26.7,26.3,17.2,16.7,3.6。
ESI-MS m/z:对于C34H50N4O7计算:626.37。实验值:627.3(M+H)+。
(c)化合物25的制备
向如上述步骤(b)中所述制备的化合物24(40mg,0.064mmol)在CH2Cl2(2mL)中的溶液中加入6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯(21.6mg,0.07mmol)。将反应混合物在23℃搅拌过夜并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物25(33.5mg,64%)。
1H NMR(400MHz,CDCl3):δ8.67(d,J=10.7Hz,1H),7.29-7.23(m,1H),6.90(t,J=11.5Hz,1H),6.80(t,J=9.6Hz,1H),6.68(s,2H),6.46(d,J=9.4Hz,1H),6.42(bs,1H),6.16(d,J=11.6Hz,1H),5.73-5.67(m,2H),5.64(dd,J=6.2,3.1Hz,1H),5.30(d,J=9.6Hz,1H),4.86(q,J=8.4Hz,1H),4.63-4.54(m,1H),4.44(d,J=9.4Hz,1H),4.30-4.18(m,1H),3.66(s,3H),3.50(t,J=7.2Hz,2H),3.34-3.10(m,3H),2.85(dt,J=9.9,6.9Hz,1H),2.54-2.37(m,4H),2.36-2.28(m,1H),2.16(t,J=7.6Hz,2H),1.84(d,J=1.3Hz,3H),1.83-1.81(m,3H),1.70-1.51(m,8H),1.34-1.23(m,2H),1.16(d,J=6.6Hz,3H),1.05(s,9H)。
13C NMR(100MHz,CDCl3):δ173.4,170.8,168.1,166.4,161.6,157.0,145.2,140.3,137.5,134.3,134.1,133.9,124.2,120.7,108.2,106.2,81.8,78.4,74.3,73.5,60.7,55.4,37.7,37.6,37.3,36.4,35.9,34.6,32.8,30.3,29.9,28.3,26.7,26.4,26.2,25.4,25.2,24.4,17.2,16.6,3.6。
ESI-MS m/z:对于C44H61ClN5O10计算:819.44。实验值:820.4(M+H)+。
实施例8
化合物27的制备
(a)化合物26的制备
向如上述实施例7(b)中所述制备的化合物24(70mg,0.11mmol)在CH2Cl2(2mL)中的溶液加入3-(甲基二硫烷基)丙酸N-羟基琥珀酰亚胺酯(36.2mg,0.12mmol)。将反应混合物在23℃搅拌16h并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物26(46.3mg,61%),为白色固体。
1H NMR(400MHz,CDCl3):δ8.72(d,J=10.8Hz,1H),7.29-7.19(m,1H),6.90(t,J=11.3Hz,1H),6.80(t,J=9.7Hz,1H),6.69(t,J=6.1Hz,1H),6.47(d,J=9.4Hz,1H),6.16(d,J=11.0Hz,1H),5.69(d,J=11.5Hz,1H),5.64(dd,J=6.3,3.1Hz,1H),5.30(d,J=0.5Hz,1H),4.86(q,J=8.4Hz,1H),4.60-4.54(m,1H),4.46(d,J=9.4Hz,1H),4.23(ddd,J=11.5,7.4,4.8Hz,1H),3.66(s,3H),3.33-3.22(m,3H),3.19-3.14(m,1H),2.96(t,J=7.2Hz,2H),2.66-2.54(m,1H),2.59(t,J=7.2Hz,2H),2.48-2.42(m,5H),2.40(s,3H),2.38-2.28(m,1H)1.83(s,3H),1.82(s,3H),1.73-1.64(m,2H),1.16(d,J=6.7Hz,3H),1.05(s,9H)。
13C NMR(100MHz,CDCl3):δ171.5,168.1,166.4,165.2,157.0,145.1,140.4,137.5,134.3,134.0,124.20,124.0,120.7,108.2,106.2,81.8,78.4,74.2,73.6,60.7,55.4,37.8,37.3,35.9,34.6,33.1,30.3,29.8,26.7,26.2,24.4,23.0,17.2,16.6,3.6。
(b)化合物27的制备
将如上述步骤(a)中所述制备的化合物26(44.3mg,0.064mmol)在EtOAc(3.6mL)和CH3OH(3.6mL)的混合物中的溶液用二硫苏糖醇(0.19mL的1.0M溶液,0.19mmol)在pH 7.5含有2mM乙二胺四乙酸(EDTA)的0.05M磷酸钾缓冲液(3.6mL)中的溶液处理。将混合物在23℃搅拌4h。将反应用pH 6.0含有2mM EDTA的0.2M磷酸钾缓冲液(21mL)的溶液处理,并随后用乙酸乙酯萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的粗产物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的目标化合物27(33.6mg,74%),为白色固体。
1H NMR(500MHz,CDCl3):δ8.73(d,J=10.7Hz,1H),7.27(t,J=11.5Hz,1H),6.91(td,J=11.5,1.1Hz,1H),6.94-6.86(m,1H),6.85-6.77(m,1H),6.43(d,J=9.4Hz,1H),6.17(dd,J=12.0,1.6Hz,1H),5.71(d,J=11.4Hz,1H),5.65(dd,J=6.5,2.9Hz,1H),5.54(t,J=6.3Hz,1H),5.30(d,J=10.5Hz,1H),4.84(q,J=8.3Hz,1H),4.53(d,J=9.5Hz,1H),4.48-4.44(m,1H),4.24(ddd,J=11.5,7.3,4.2Hz,1H),3.67(s,3H),3.336-3.22(m,3H),3.21-3.10(m,1H),2.89-2.84(m,1H),2.80(dt,J=8.2,6.8Hz,2H),2.49(t,J=7.2Hz,2H),2.45-2.36(m,5H),2.31-2.28(m,1H),1.84(s,3H),1.83(s,3H),1.72-1.69(m,2H),1.16(d,J=6.7Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C37H54N4O8S计算:714.37。实验值:737.3(M+Na)+。
实施例9
利用曲妥珠单抗和化合物1的抗体-药物缀合物ADC1的制备
(a)部分还原曲妥珠单抗得到部分还原的曲妥珠单抗(化合物20)
利用20mM组氨酸/乙酸盐缓冲液(pH 5.5,30.5mL)将曲妥珠单抗(曲妥珠单抗,购自Roche,为白色冻干粉末,用于制备浓缩的输注用溶液)溶液(9.52mL,200mg,1.38μmol)稀释至5mg/mL的浓度,接着利用磷酸盐/EDTA缓冲液(pH 8.4,13mL)进行pH调节。抗体中的二硫键的部分还原通过加入5.17mM三[2-羧基乙基]膦盐酸盐(TCEP)溶液(689μL,3.562μmol,2.6eq.)进行。将还原反应在20℃搅拌90min。还原后立即进行Ellman测定,以得到4.1的游离硫醇基与抗体比例(FTAR),与按照计划的4.0的值非常接近。
(b)ADC1的制备
向如上述实施例7(a)中所述制备的部分还原的曲妥珠单抗化合物20(24.98mL,93.0mg,0.64μmol)的溶液中加入DMSO(1.25mL),接着加入如实施例1中所述制备的化合物1的新鲜制备的溶液(在DMSO中10mM,366μL,3.66μmol,5.7eq.)。在加入化合物1时,溶液变得非常混浊,因此另外加入DMSO(1mL)。将缀合反应在20℃搅拌30min并且浊度在缀合反应期间消失。通过加入N-乙酰半胱氨酸(NAC)(10mM,366μL,3.66μmol)接着将溶液搅拌20min来猝灭过量药物。将猝灭的缀合反应通过Vivaspin离心纯化并且将缓冲液用最终PBS制剂缓冲液交换。将最终目标产物ADC1浓缩至如通过UV确定的8.56mg/mL的终浓度并且获得7.4mL(63.3mg,0.43μmol,68.0%)ADC溶液。进行SEC HPLC运行以确定产物的纯度(61.4%)。
由于大量聚集体的存在,将ADC1在纯化仪系统上,使用HiLoad 16/600superdex 200柱的制备型凝胶过滤色谱进一步纯化。汇集后,通过UV确定终浓度(1.6mg/mL)并且通过SEC HPLC确定最终药物产物的纯度(90.9%),以获得8.65mL(13.7mg,0.09μmol,14.7%)的ADC溶液(ADC1)。
实施例10
利用曲妥珠单抗和化合物5的抗体-药物缀合物ADC2的制备
(a)部分还原曲妥珠单抗以得到部分还原的曲妥珠单抗(化合物20)
利用20mM组氨酸/乙酸盐缓冲液(pH,5.5,45.74mL)将曲妥珠单抗溶液(14.29mL,300mg,2.06μmol)稀释到5mg/mL的浓度,接着利用磷酸盐/EDTA缓冲液(pH 8.4,14.4mL)进行pH调节。通过加入5mM三[2-羧基乙基]膦盐酸盐(TCEP)溶液(1.07mL,5.36μmol,2.6eq.)进行抗体中二硫键的部分还原。将还原反应在20℃搅拌90min。在还原后立即进行Ellman测定,得到4.1的游离硫醇基与抗体比例(FTAR),与按照计划的4.0的值非常接近。
(b)ADC2的制备
向如上述实施例8(a)中所述制备的部分还原的曲妥珠单抗化合物20(23.6mL,93.8mg,0.645μmol)的溶液中加入DMSO(1.18mL),接着加入如实施例2中所述制备的化合物5的新鲜制备的溶液(在DMSO中10mM,368μL,3.68μmol,5.7eq.)。以10部分小心加入药物-接头。在第六部分后,溶液变得稍微混浊并且在缀合反应和猝灭期间浊度不消失。将缀合反应在20℃搅拌30min。通过加入N-乙酰半胱氨酸(NAC)(10mM,368μL,3.68μmol),通过将溶液搅拌46min猝灭过量的药物。将溶液经0.2μm注射器滤器过滤。通过Vivaspin离心将猝灭的缀合反应物浓缩的至16.5mg/mL并且经NAP-25柱纯化。进行SEC HPLC运行以确定产物的纯度(36.1%)。
由于大量聚集体的存在,在纯化仪系统上,使用HiLoad 16/600 superdex 200柱,通过制备型凝胶过滤色谱进一步纯化ADC2。汇集后,通过UV确定终浓度(3.7mg/mL)并且通过SEC HPLC确定最终目标ADC的纯度(78.3%)以获得5.3mL(19.4mg,19.4%)的ADC溶液(ADC2)。
实施例11
利用曲妥珠单抗和化合物12的抗体-药物缀合物ADC3的制备
(a)ADC3的制备
向如上述实施例10(a)中所述制备的部分还原的曲妥珠单抗化合物20(23.6mL,93.8mg,0.645μmol)的溶液中加入DMSO(1.18mL),接着加入如实施例3中所述制备的化合物12的新鲜制备的溶液(在DMSO中10mM,369μL,3.69μmol,5.7eq.)。以10部分小心加入药物-接头,然而第三部分后,溶液开始变得混浊。在加入最后两部分的过程中观察到高浊度。溶液不澄清,直到过滤步骤。将缀合反应在20℃搅拌31min。通过加入N-乙酰半胱氨酸(NAC)(10mM,369μL,3.69μmol)通过将反应混合物搅拌50min猝灭过量的药物。将猝灭的缀合反应溶液经0.2μm注射器滤器过滤并通过Vivaspin离心浓缩至14.2mg/mL。然后将其经NAP-25柱纯化。进行SEC HPLC运行,以确定产物的纯度(34.2%)。
由于大量聚集体的存在,通过制备型凝胶过滤色谱,在纯化仪系统上,使用HiLoad 16/600 superdex 200柱进一步纯化ADC3。汇集后,通过UV确定终浓度(2.3mg/mL)并且通过SEC HPLC确定最终药物产物的纯度(78.6%)以获得7.3mL(16.6mg,16.6%)的ADC溶液(ADC3)。
实施例12
分别利用曲妥珠单抗和化合物13,15和18的抗体-药物缀合物ADC 4,5和6的制备
(a)SMCC缀合于曲妥珠单抗(化合物21)
通过磷酸盐缓冲液(50mM磷酸盐,2mM EDTA,pH 6.5),使用NAP-25柱交换曲妥珠单抗溶液(262mg,1.8μmol)的缓冲液。向玻璃反应器中汇集的曲妥珠单抗溶液(16-17g/L)中加入DMSO(5%)。通过将SMCC(20.0mM,8.0eq.)加入至曲妥珠单抗溶液中起始接头缀合。将反应在18℃搅拌3小时。随后将反应混合物经NAP-25柱纯化,得到化合物21。进行反向Ellman测定,确定LAR为3.7。
(b)化合物13,15和18与曲妥珠单抗MCC的缀合:ADC4,ADC5和ADC6的制备。
对于缀合反应,在第一步中,利用磷酸盐缓冲液(50mM磷酸盐,2mM EDTA,pH 6.5)将抗体缀合物溶液化合物21稀释到10g/L的浓度。然后将DMSO(5%)加入至化合物21溶液中。通过缓慢将药物(10mM,6.3-6.6eq.)加入化合物21溶液中并在18℃搅拌四小时,进行与化合物13,15和18的缀合反应。缀合反应完成后,将反应混合物0.2μm过滤并且再次经NAP-25柱利用缓冲液交换纯化入1x PBS缓冲液中。进行SEC HPLC运行,确定产物的纯度并且通过UV测量终产物的浓度。
将利用化合物15的样品制备物的ADC分离,具有良好纯度(74.5%)并且获得56%(49mg)的产率,并且不需要进一步纯化。通过UV确定ADC5溶液(87mL)的终浓度(5.7mg/mL)。
然而在利用化合物13和18的两个样品制备物中,存在低分子种类。这些种类对于产物峰具有非常类似的保留时间并且在SEC柱不能很好地分离。为了去除仍然存在于溶液中的可能的药物剩余物,将溶液再次通过NAP-25柱。第一和第二次NAP-25纯化后的色谱图相同,因此种类不从仍然存在于溶液中的游离药物中产生并且必然有更大的来源。然后将样品通过尺寸排阻柱上的凝胶过滤色谱进一步纯化。
汇集后,通过UV确定终浓度(2.8和3.9mg/mL)并且通过SEC HPLC确定最终药物产物的纯度(62.3%和50.9%),分别获得6.7mL(19.3mg,22.0%)的ADC4溶液和6.8mL(26.7mg,30.5%)的ADC6溶液。
实施例13
利用曲妥珠单抗和化合物25和27的抗体-药物缀合物ADC 7和8的制备
(a)一般步骤
以分光光度法,通过监测其在280nm的吸光度,使用2.18E5M-1 cm-1的摩尔消光系数和150kDa的分子量检查抗体浓度。用于这些步骤的缓冲液是缓冲液A(50mM磷酸钠,pH 6.5,具有2mM EDTA)或缓冲液B(50mM磷酸钠pH 8.0)或磷酸盐水缓冲液(“PBS”)。假设药物-接头与马来酰亚胺连接体或与游离Cys的缀合反应是定量的,在通过Lys缀合的情况下从接头与抗体比例(“LAR”)推断药物与抗体比例(“DAR”),或在Cys-靶向的缀合的情况下,从游离Cys/mol抗体比例推断药物与抗体比例(“DAR”)。两种测定都基于5,5′-二硫双(2-硝基苯甲酸)(“DTNB”)与游离硫醇基形成有色的硫代硝基苯甲酸酯加合物的比色反应。对于LAR测定,通过混合等体积的200μM DTNB在缓冲液B中的溶液与200μM N-乙酰半胱氨酸在相同缓冲液中的溶液来预制加合物。然后将75μL的该混合物与75μL的测试样品混合,并且1h温育后,以分光光度法确定412nm的吸光度,并且将得到的值与从使用已知浓度的4-(N-马来酰亚胺基甲基)环己烷甲酸N-羟基琥珀酰亚胺酯(“SMCC”)的标准曲线获得那些相比较,以获得样品中马来酰亚胺的浓度。然后将该浓度针对抗体浓度以计算LAR。同样,通过将50μL的测试样品与150μL的在缓冲液B中的133μM DTNB混合,监测412nm的吸光度并且将得到的值与从使用已知浓度的Cys的标准曲线获得的那些比较,来确定游离的Cys:然后将推导的测试样品中游离Cys的浓度相对抗体浓度计算比例。
(b)抗体-药物缀合物的制备
当细胞毒性有效荷载缀合于Cys残基(如对于ADC7的制备利用化合物25)时,之前利用三(2-羧基乙基)膦盐酸盐(“TCEP”)还原抗体。简言之,将70μM(10.5mg/mL)的抗体在缓冲液B中的溶液适量的5mM的TCEP在水中的溶液混合,保持还原剂相对抗体2.5-倍过量。在20℃将混合物温育并搅拌60min,并且之后去除小等份的得到的还原的抗体以计算游离Cys与抗体的比例,同时将剩余的样品与适当体积的10mM化合物25在DMSO中的溶液混合,达到化合物相对抗体6-倍过量:考虑到还原的抗体通常存在少于6个游离Cys/蛋白分子,化合物与可接近游离Cys的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v)并且将混合物在20℃温育30min。之后,加入N-乙酰半胱氨酸以猝灭反应,使用适当体积的10mM在水中的溶液以匹配药物-接头的浓度。最后使用来自GEHealthcare的PD-10柱,通过在Sephadex G-25中凝胶过滤,将得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析,使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用利用Superdex 200 16/600制备型柱的相同色谱系统(运行与上述相同的方法)纯化单体。使用与母体抗体相同的摩尔消光系数,以分光光度法,通过监测其在280nm的吸光度,确定最终ADC浓度:如果ADC浓度低于2mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩,并且再次如上确定新浓度。
当细胞毒性有效荷载缀合于Lys残基(如对于ADC8的制备利用化合物27)时,事先将抗体用SMCC活化。简言之,将70μM(10.5mg/mL)抗体在缓冲液A中的溶液与适量的20mM SMCC在DMSO中的溶液混合以保持活化试剂相对抗体8-倍过量。如果需要加入DMSO以达到5%(v/v)的最终DMSO浓度。将混合物在18℃温育并搅拌3h,并且随后使用来自GE Healthcare的PD-10柱通过在Sephadex G-25上的凝胶过滤色谱,去除过量的SMCC。移除小的等份的得到的活化的抗体,计算LAR并且将得到的样品与适当体积的10mM的化合物27在DMSO中的溶液混合,达到化合物相对抗体8-倍过量:考虑到LAR值从不超过8,这保证化合物与可接近的反应位点的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v)。将混合物在18℃温育4h并且使用来自GE Healthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,将得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。以分光光度法,通过监测其在280nm的吸光度,使用与母体抗体相同的摩尔消光系数,确定最终ADC浓度:如果ADC浓度低于2mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
实施例14
抗CD4,抗CD5和抗CD13单克隆抗体的制备
按照通常在本领域中使用的公知步骤获得抗CD4,抗CD5和抗CD13单克隆抗体。简言之,将BALB/c小鼠用HPB-ALL细胞免疫(用于最后产生抗CD4抗体)或如Cebrian等人(1988,J.Exp.Med.168:1621-1637)所述用利用佛波醇12-肉豆蔻酸酯13-乙酸盐和可商购的抗CD3单克隆抗体的混合物活化的人T细胞免疫(用于最终产生抗CD5抗体)或利用分离自脐带的人内皮细胞免疫(用于最终产生抗CD13抗体)。为此,将1.5E7的相应细胞在第-45和-30天腹膜内注射入小鼠并且在第-3天静脉内注射。在第0天,根据标准技术将来自这些动物的脾移除并且将脾细胞与SP2小鼠骨髓瘤细胞以4:1的比例融合,产生相应杂交瘤并分散到96孔组织培养板(Costar Corp.,Cambridge,MA)上。2周后,收获杂交瘤培养上清液,并且通过流式细胞术测试其针对用于免疫步骤的细胞系的反应性。通过免疫荧光染色用作抗原的相应细胞测定阳性上清液。选择显示特定染色、免疫沉淀样式和细胞分布的杂交瘤,并通过有限稀释克隆和亚克隆。
一旦选择了克隆,将细胞在补充有10%(v/v)胎牛血清,2mM谷氨酰胺,100U/mL氨苄青霉素和100μg/mL链霉素的RPMI-1640培养基中在37℃培养3-4天,直到培养基变成淡黄色。此时,去除三分之二的培养基体积,在1,000xg离心10min以沉淀细胞并将上清液再次在3,000xg离心10min,用于进一步清除或经22μm孔径膜过滤。将澄清了的上清液利用55%饱和硫酸铵进行沉淀并且将得到的沉淀在100mM Tris-HCl pH 7.8(1mL/100mL的原澄清的上清液)中重悬并在4℃针对5L的具有150mM NaCl的100mM Tris-HCl pH 7.8透析16-24h,改变透析液至少三次。将透析的材料最后上样于蛋白A-Sepharose柱上,并且利用100mM乙酸钠pH 3.0或备选地用1M甘氨酸pH 3.0洗脱相应单克隆抗体。将那些含有抗体的级分用2M Tris-HCl pH 9.0中和并最后针对PBS透析并储存在-80℃备用。
实施例15
利用抗CD13和化合物1,12和13的抗体-药物缀合物ADC 9,10和11的制备
(a)一般步骤
在本文报道的所有方法中,使用2.25E5M-1 cm-1的摩尔消光系数和150kDa的分子量,以分光光度法,通过监测其在280nm的吸光度检查抗体浓度。用于这些步骤的缓冲液是缓冲液A(50mM磷酸钠,pH 6.5,具有2mM EDTA)或缓冲液B(50mM磷酸钠,pH 8.0)或磷酸盐水缓冲液(“PBS”)。假设药物-接头与马来酰亚胺连接体或与游离Cys的缀合反应是定量的,在通过Lys缀合的情况下从接头与抗体比例(“LAR”)推断药物与抗体比例(“DAR”),或在Cys-靶向的缀合的情况下,从游离Cys/mol抗体比例推断药物与抗体比例(“DAR”)。两种测定都基于5,5′-二硫双(2-硝基苯甲酸)(“DTNB”)与游离硫醇基形成有色的硫代硝基苯甲酸酯加合物的比色反应。对于LAR测定,通过混合等体积的200μM DTNB在缓冲液B中的溶液与200μM N-乙酰半胱氨酸在相同缓冲液中的溶液来预制加合物。随后将75μL的该混合物与75μL的测试样品混合,并且1h温育后,以分光光度法确定412nm的吸光度并且将得到的值与从使用已知浓度的4-(N-马来酰亚胺基甲基)环己烷甲酸N-羟基琥珀酰亚胺酯(“SMCC”)的标准曲线获得的那些相比,获得样品中马来酰亚胺的浓度。随后将浓度相对抗体浓度计算LAR。同样,通将50μL的测试样品与150μL的在缓冲液B中的133μM DTNB混合,监测412nm的吸光度并且将得到的值与从使用已知浓度的Cys的标准曲线获得的那些比较,来确定游离的Cys:然后将推导的测试样品中游离Cys的浓度相对抗体浓度计算比例。
(b)抗体-药物缀合物的制备
当细胞毒性有效荷载缀合于Cys残基(如对于ADC9的制备利用化合物1或对于ADC10的制备利用化合物12)时,之前将抗体用三(2-羧基乙基)膦盐酸盐(“TCEP”)还原。简言之,将70μM(10.5mg/mL)的抗体在缓冲液B中的溶液与适量的5mM TCEP在水中的溶液混合以保持还原剂相对抗体2.5-倍过量。将混合物在20℃温育并搅拌60min并且之后移除小等份的得到的还原的抗体以计算游离Cys与抗体的比例,同时将剩余的样品与适当体积的10mM药物接头(对于ADC9为化合物1或对于ADC10为化合物12)在DMSO中的溶液混合,达到化合物相对抗体6-倍过量:考虑到还原的抗体通常存在少于6个游离Cys/蛋白分子,化合物与可接近游离Cys的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v),并且将混合物在20℃温育30min。之后加入N-乙酰半胱氨酸以猝灭反应,使用适当体积的水中的10mM溶液以匹配药物-接头的浓度。最后使用来自GE Healthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,和得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析,使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。使用与母体抗体相同的摩尔消光系数,以分光光度法,通过监测其在280nm的吸光度确定最终ADC浓度:如果ADC浓度低于2mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
当细胞毒性有效荷载缀合于Lys残基(如对于ADC11的制备利用化合物13)时,事先将抗体用SMCC活化。简言之,将70μM(10.5mg/mL)抗体在缓冲液A中的溶液适量的20mM SMCC在DMSO中的溶液混合以保持活化试剂相对抗体8-倍过量。如果需要加入DMSO以达到最终5%(v/v)的DMSO浓度。将混合物在18℃温育和搅拌3h,并且随后使用来自GE Healthcare的PD-10柱通过在Sephadex G-25上的凝胶过滤色谱,去除过量的SMCC。移除小的等份的得到的活化的抗体以计算LAR并且将得到的样品与适当体积的10mM的化合物13在DMSO中的溶液混合,达到化合物相对抗体8-倍过量:考虑到LAR值从不超过8,这保证化合物与可接近反应位点的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v)。将混合物在18℃温育4h并且使用来自GE Healthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,将得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析,使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。使用与母体抗体相同的摩尔消光系数,以分光光度法,通过监测其在280nm的吸光度确定最终ADC浓度:如果ADC浓度低于2mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
实施例16
利用利妥昔单抗和化合物1和12的抗体-药物缀合物ADC 12和13的制备
(a)一般步骤
以分光光度法,通过监测其在280nm的吸光度,使用2.45E5M-1 cm-1的摩尔消光系数和150kDa的分子量检查抗体浓度。用于这些步骤的缓冲液是缓冲液B(50mM磷酸钠pH 8.0)或磷酸盐水缓冲液(“PBS”)。假设药物-接头与游离Cys的缀合反应是定量的,从游离Cys/mol的抗体比例推断药物与抗体比例(“DAR”)。所述测定基于5,5′-二硫双(2-硝基苯甲酸)(“DTNB”)与游离硫醇基形成有色的硫代硝基苯甲酸酯加合物的比色反应。将50μL的测试样品与150μL在缓冲液B中的133μM DTNB混合,在412nm测量吸光度并且将得到的值与从使用已知浓度的Cys的标准曲线的获得的那些相比较。随后将推导的测试样品中游离Cys的浓度相对抗体浓度计算比例。
(b)抗体-药物缀合物的制备
在通过Cys缀合于药物-接头前,将抗体用三(2-羧基乙基)膦盐酸盐(“TCEP”)还原。简言之,将70μM(10.5mg/mL)的抗体在缓冲液B中的溶液与适量的5mM TCEP在水中的溶液混合以保持还原剂相对抗体2.5-倍过量。将混合物在20℃温育和搅拌60min并且之后移除小等份的得到的还原的抗体以计算游离Cys与抗体的比例,同时将剩余的样品与适当体积的10mM药物接头(对于ADC12为化合物1或对于ADC13为化合物12)在DMSO中的溶液混合达到化合物相对抗体6-倍过量:考虑到还原的抗体通常存在少于6个游离Cys/蛋白分子,化合物与可接近游离Cys的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v)并且将混合物在20℃温育30min。之后加入N-乙酰半胱氨酸以猝灭反应,使用适当体积的在水中的10mM溶液以匹配药物-接头的浓度。最后使用来自GEHealthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,从试剂的剩余部分纯化得到的缀合物。通过分析型尺寸排阻层析,使用装有Superdex-10010/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。以分光光度法,通过监测其在280nm的吸光度,使用与母体抗体相同的摩尔消光系数,确定最终ADC浓度:如果ADC浓度低于1mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
实施例17
利用抗CD5和化合物1和12的抗体-药物缀合物ADC 14和15的制备
(a)一般步骤
以分光光度法,通过监测其在280nm的吸光度,使用2.25E5M-1 cm-1的摩尔消光系数和150kDa的分子量检查抗体浓度。用于这些步骤的缓冲液是缓冲液B(50mM磷酸钠pH 8.0)或磷酸盐水缓冲液(“PBS”)。假设药物-接头与游离Cys的缀合反应是定量的,从游离Cys/mol的抗体比例推断药物与抗体比例(“DAR”)。所述测定基于5,5′-二硫双(2-硝基苯甲酸)(“DTNB”)与游离硫醇基形成有色的硫代硝基苯甲酸酯加合物的比色反应。将50μL的测试样品与150μL的在缓冲液B中的133μM DTNB混合,随后测量412nm的吸光度并且将得到的值与从使用已知浓度的Cys的标准曲线获得的那些相比较。随后将推导的测试样品中游离Cys的浓度相对抗体浓度计算比例。
(b)抗体-药物缀合物的制备
在通过Cys缀合于药物-接头之前,将抗体用三(2-羧基乙基)膦盐酸盐(“TCEP”)还原。简言之,将70μM(10.5mg/mL)的抗体在缓冲液B中的溶液与适量的5mM TCEP在水中的溶液混合以保持还原剂相对抗体2.5-倍过量。将混合物在20℃温育和搅拌60min,并且之后移除小等份的得到的还原的抗体,以计算游离Cys与抗体的比例,同时将剩余的样品与适当体积的10mM的药物接头(对于ADC14为化合物1或对于ADC15为化合物12)在DMSO中的溶液混合,达到化合物相对抗体6-倍过量:考虑到还原的抗体通常存在少于6个游离Cys/蛋白分子,化合物与可接近游离Cys的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v),并且将混合物在20℃温育30min。之后加入N-乙酰半胱氨酸以猝灭反应,使用适当体积的在水中的10mM溶液以匹配药物-接头的浓度。最后使用来自GE Healthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,将得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析,使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。以分光光度法,通过监测其在280nm的吸光度,使用与母体抗体相同的摩尔消光系数,确定最终ADC浓度:如果ADC浓度低于1mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
实施例18
利用抗CD4和化合物1和12的抗体-药物缀合物ADC 16和17的制备
(a)一般步骤
以分光光度法,通过监测其在280nm的吸光度,使用2.25E5M-1 cm-1的摩尔消光系数和150kDa的分子量检查抗体浓度。用于这些步骤的缓冲液是缓冲液B(50mM磷酸钠pH 8.0)或磷酸盐水缓冲液(“PBS”)。假设药物-接头与游离Cys的缀合反应是定量的,从游离Cys/mol的抗体比例推断药物与抗体比例(“DAR”)。所述测定基于5,5′-二硫双(2-硝基苯甲酸)(“DTNB”)与游离硫醇基形成有色的硫代硝基苯甲酸酯加合物的比色反应。将50μL的测试样品与150μL的在缓冲液B中的133μM DTNB混合,随后测量412nm的吸光度并且将得到的值与从使用已知浓度的Cys的标准曲线获得的那些相比较。随后将推断的测试样品中游离Cys浓度相对抗体浓度计算比例。
(b)抗体-药物缀合物的制备
在通过Cys缀合于药物-接头之前,将抗体用三(2-羧基乙基)膦盐酸盐(“TCEP”)还原。简言之,将70μM(10.5mg/mL)抗体在缓冲液B中的溶液与适量的5mM TCEP在水中的溶液混合,以保持还原剂相对抗体2.5-倍过量。将混合物在20℃温育和搅拌60min,并且之后移除小等份的得到的还原的抗体以计算游离Cys与抗体的比例,同时将剩余的样品与适当体积的10mM的药物接头(对于ADC16为化合物1或对于ADC17为化合物12)在DMSO中的溶液混合,达到化合物相对抗体6-倍过量:考虑到还原的抗体通常存在少于6个游离Cys/蛋白分子,化合物与可接近游离Cys的摩尔比从不低于1。如果需要加入DMSO以将其浓度保持在5%(v/v),将混合物在20℃温育30min。之后加入N-乙酰半胱氨酸以猝灭反应,使用适当体积的在水中的10mM溶液匹配药物-接头的浓度。最后使用来自GE Healthcare的PD-10柱通过在Sephadex G-25中凝胶过滤,将得到的缀合物从试剂的剩余部分中纯化。通过分析型尺寸排阻层析,使用装有Superdex-100 10/300柱的FPLC系统(运行利用1ml/min的PBS的等梯度方法)检查聚集体的存在:如果对应于聚集体的峰面积超过总峰面积的10%,使用相同色谱系统,利用Superdex 200 16/600制备型柱(运行与上述相同的方法)纯化单体。以分光光度法,通过监测其在280nm的吸光度,使用与母体抗体相同的摩尔消光系数,确定最终ADC浓度:如果ADC浓度低于1mg/mL,使用来自GE Healthcare的Vivaspin装置将其浓缩并且再次如上确定新的浓度。
实施例19式D-X-(AA)w-L1的化合物的合成
化合物28的制备
(a)化合物28的制备
在23℃将DIPEA (10μL,0.06mmol)加入如上述制备实施例中所示制备的化合物9(13mg,0.02mmol)和如上述实施例2中所述制备的化合物8(20mg,0.02mmol)在NMP(6.5mL)中的溶液中。9h后,将反应混合物用H2O稀释并用EtOAc萃取。将合并的有机层经无水Na2SO4干燥,过滤,并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,DCM:CH3OH,100:0至90:10)中纯化,以获得纯的目标化合物28(9mg,38%)。
1H NMR(400MHz,CDCl3/CD3OD):δ9.40(s,1H),8.92(d,J=10.6Hz,1H),7.67(d,J=7.7Hz,1H),7.47(d,J=8.0Hz,2H),7.21(d,J=7.9Hz,2H),7.16(t,J=11.9Hz,1H),6.97(t,J=9.6Hz,2H),6.82(t,J=11.4Hz,1H),6.67-6.64(m,1H),6.64(s,2H),6.08(d,J=11.7Hz,1H),5.68(d,J=11.4Hz,1H),5.62-5.58(m,1H),5.54-5.47(m,1H),5.35-5.29(m,1H),5.20(d,J=9.9Hz,1H),4.94(s,2H),4.77(q,J=8.1Hz,1H),4.54-4.45(m,2H),4.37(d,J=9.2Hz,1H),4.20-4.12(m,1H),4.08(t,J=7.8Hz,1H),3.58(s,3H),3.42(t,J=7.2Hz,2H),3.18-3.00(m,7H),2.81-2.75(m,1H),2.35-2.30(m,3H),2.29-2.25(m,3H),2.17(t,J=7.2Hz,2H),2.14-2.06(m,1H),2.04-1.92(m,1H),1.86-1.74(m,1H),1.76(s,3H),1.61-1.42(m,10H),1.54(d,J=6.3Hz,3H),1.30-1.14(m,4H),1.08(d,J=6.4Hz,3H),0.94(s,9H),0.86(dd,J=6.8,4.3Hz,6H)。
13C NMR(100MHz,CDCl3):δ173.0,172.1,171.0,170.2,168.5,167.1,162.0,161.3,157.2,157.0,145.0,140.2,137.7,137.5,137.1,134.0,132.4,128.8,126.9,124.9,124.4,124.3,124.0,120.5,119.8,108.6,107.3.81.9,74.8,66.1,60.4,58.8,55.4,37.6,37.2,36.0,34.8,31.9,31.6,30.9,30.7,30.0,29.7,29.3,28.1,26.5,26.2,26.1,25.1,19.2,18.3,17.1,16.5,12.9。
ESI-MS m/z:对于C63H90N10O15计算:1226.7。实验值:1267.4(M+H)+。
实施例20式D-X-(AA)w-H的化合物的合成
化合物36的制备
(a)化合物29的制备
在0℃向化合物6(1.01g,1.91mmol)(化合物30b,如WO 2007144423中所述制备,其内容通过引用结合于此)在DCM(40mL)中的溶液中加入吡啶(0.31mL,3.82mmol)和氯甲酸4-硝基苯酯(769.7mg,3.82mmol)。将反应混合物在23℃搅拌1.5h,用10%柠檬酸稀释并用DCM萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的化合物29(783mg,59%)。
1H NMR(400MHz,CDCl3):δ8.26(d,J=9.2Hz,2H),8.02(d,J=10.9Hz,1H),7.43(d,J=9.2Hz,2H),7.22(t,J=9.2Hz,1H),6.92-6.76(m,2H),6.21(d,J=9.3Hz,1H),6.17-6.12(m,1H),5.73-5.64(m,1H),5.65-5.56(m,2H),5.46-5.38(m,1H),5.27(d,J=9.9Hz,1H),4.86(q,J=8.2Hz,1H),4.76(p,J=6.2Hz,1H),4.40(d,J=9.3Hz,1H),4.21(ddd,J=10.7,7.6,4.9Hz,1H),3.66(s,3H),2.89-2.79(m,1H),2.59-2.29(m,6H),1.83(d,J=1.3Hz,3H),1.61(s,3H),1.16(d,J=6.7Hz,3H),1.00(s,9H)。
(b)化合物33的制备
化合物30的制备
向3-溴丙胺氢溴酸盐(1.22g,2.98mmol)在CH2Cl2(30mL)中的溶液中加入4-甲氧基三苯基甲基氯(5.89g,19.1mmol)和DIPEA(6.3,mL,36.38mmol)。将反应混合物在23℃搅拌过夜,用H2O稀释并用CH2Cl2萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物30(8.16g,100%),为白色固体。
1H NMR(400MHz,CDCl3):δ7.50-7.42(m,4H),7.39-7.33(m,2H),7.29-7.22(m,4H),7.22-7.15(m,2H),6.81(d,J=8.9Hz,2H),3.78(s,3H),3.56(t,J=6.8Hz,2H),2.26(t,J=6.7Hz,2H),2.07-1.96(m,2H)。
化合物31的制备
向化合物30(1.49g,3.63mmol)在乙醇(36mL)中的溶液中加入乙基黄原酸钾(1.46g,9.08mmol)。将反应混合物在23℃搅拌过夜并且随后将沉淀的溴化钾从溶液中过滤。在减压下蒸发滤液后,将固体残留物用己烷研碎。将得到的固体通过过滤除去并且蒸发滤液并通过急骤色谱(SiO2,Hex/EtOAc混合物)纯化,以得到化合物31(1.31g,80%)。
1H NMR(400MHz,CDCl3):δ7.50-7.42(m,4H),7.39-7.33(m,2H),7.29-7.22(m,4H),7.22-7.15(m,2H),6.81(d,J=8.9Hz,2H),4.62(q,J=7.1Hz,2H),3.78(s,3H),3.32-3.20(dd,J=7.7,6.9Hz,2H),2.23(t,J=6.7Hz,2H),1.91-1.80(m,2H),1.41(t,J=7.1Hz,3H)。
化合物32的制备
向化合物31(3g,6.64mmol)在DCM(10mL)中的溶液中加入1-丙胺(4,4mL,66.4mmol)。将反应混合物在23℃搅拌10min并在真空下浓缩。无进一步纯化即将获得的残留物用于下一步骤。将其溶解在无水甲醇(50mL)中并且冷却至0℃。加入甲硫代磺酸甲酯(9.7mL,7.97mmol)并且将溶液在23℃搅拌16h。在真空中去除溶剂,将残留的油状物溶解在二氯甲烷中,用pH 7缓冲液和盐水洗涤,干燥并蒸发。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得化合物32(1,6g,59%)。
1H NMR(400MHz,CDCl3):δ7.52-7.44(m,4H),7.38(d,J=8.9Hz,2H),7.32-7.22(m,4H),7.19(d,J=7.3Hz,2H),6.82(d,J=8.9Hz,2H),3.79(s,3H),2.90-2.72(m,2H),2.39(s,3H),2.24(t,J=6.7Hz,2H),1.88(p,J=6.9Hz,2H)。
化合物33的制备
向化合物32(1.22g,2.98mmol)在CH2Cl2(30mL)中的溶液中加入二氯乙酸(0.9mL,10.9mmol)。将反应混合物在23℃搅拌20min并用水稀释。将有机层萃取并且将水相用10%KOH碱化。随后将其用二氯甲烷(3x)完全萃取,并且将合并的有机萃取物经Na2SO4干燥,过滤并浓缩,得到化合物33(409mg,100%)。
1H NMR(400MHz,CDCl3):δ2.83-2.75(m,2H),2.41(s,3H),1.88-1.81(m,2H)。
(c)化合物34的制备
向如上述步骤(a)中所述制备的化合物29(230.2mg,0.33mmol)在CH2Cl2(10mL)中的溶液中加入化合物33(50mg,0.36mmol)和DIPEA(0.06mL,0.36mmol)。将反应混合物在23℃搅拌3h,用H2O稀释并用CH2Cl2萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物34(150mg,66%)。
1H NMR(400MHz,CDCl3):δ8.68(d,J=10.7Hz,1H),7.31(t,J=6.0Hz,1H),6.90(dd,J=12.7,10.2Hz,1H),6.81(dd,J=10.9,8.5Hz,1H),6.38(d,J=9.4Hz,1H),6.16(d,J=11.7Hz,1H),5.69(d,J=11.5Hz,1H),5.65-5.52(m,3H),5.41-5.37(m,1H),5.29-5.56(d,J=8.9Hz,1H),4.83(q,J=8.3Hz,1H),4.54-4.50(m,1H),4.46(d,J=9.4Hz,1H),4.24-4.19(m,1H),3.65(s,3H),3.36-3.16(m,2H),2.86-2.82(m,1H),2.70(t,J=7.2Hz,2H),2.44-2.35(m,5H),2.40(s,3H),2.19-2.04(m,1H),1.98-1.83(m,2H),1.84(s,3H),1.63(d,J=6.6Hz,3H),1.16(d,J=6.6Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C35H53N3O7S2计算:691.33。实验值:692.4(M+H)+。
(d)化合物35的制备
在23℃向如上述步骤(a)中所述制备的化合物29(121.3mg,0.17mmol)在DCM(2.5mL)中的溶液加入3-氨基丙烷-1-硫醇盐酸盐(37.2mg,0.29mmol)在DCM(2.5mL),DIPEA(59μL,0.34mmol)和DMF(0.1mL)中的悬浮液中。将反应混合物在23℃搅拌7h,用H2O稀释并用EtOAc萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物35(40.5mg,37%)。
1H NMR(300MHz,CDCl3):δ8.63(d,J=10.6Hz,1H),7.34-7.21(m,1H),6.88(t,J=11.4Hz,1H),6.76(t,J=9.6Hz,1H),6.68(d,J=9.3Hz,1H),6.13(d,J=11.6Hz,1H),5.7-5.67(m,3H),5.66-5.48(m,3H),4.81(q,J=8.1Hz,1H),4.66-4.50(m,1H),4.46(d,J=9.2Hz,1H),4.25-4.18(m,1H),3.64(s,3H),3.27-3.34(m,1H),2.88-2.77(m,1H),2.66(t,J=7.2Hz,2H),2.42-2.30(m,5H),2.22-2.06(m,2H),1.89-1.74(m,5H),1.62(d,J=6.6Hz,3H),1.15(d,J=6.7Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C68H100N6O14S2计算:1288.67。实验值:1289.4(M+H)+。
(e)化合物36的制备
将化合物35(40.5mg,0.03mmol)在EtOAc(1.5mL)和CH3OH(1.5mL)的混合物中的溶液用二硫苏糖醇(0.36mL,0.36mmol)在含有2mM乙二胺四乙酸(EDTA)的0.05M磷酸钾缓冲液(1.2mL)pH 7.5中的溶液处理。将混合物在23℃搅拌4h。将反应用含有2mM EDTA的0.2M磷酸钾缓冲液pH 6.0的溶液处理并且用EtOAc萃取(x3)。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于HPLC(SiO2,Hex:EtOAc混合物)的系统中纯化,获得化合物36(15mg,38%)。
1H NMR(300MHz,CDCl3):δ8.70(d,J=10.7Hz,1H),7.35-7.21(m,1H),6.89(t,J=11.4Hz,1H),6.80(t,J=9.8Hz,1H),6.42(d,J=9.4Hz,1H),6.16(d,J=11.3Hz,1H),5.75-5.48(m,3H),5.52-5.13(m,3H),4.83(q,J=8.3Hz,1H),4.65-4.38(m,2H),4.32-4.16(m,1H),3.65(s,3H),3.29(q,J=6.6Hz,2H),2.85(dt,J=9.8,6.9Hz,1H),2.54(q,J=7.3Hz,2H),2.48-2.26(m,5H),2.19-2.01(m,1H),1.89-1.74(m,5H),1.63(dd,J=6.8,1.7Hz,3H),1.16(d,J=6.6Hz,3H),1.05(s,9H)。
ESI-MS m/z:对于C34H51N3O7S计算:645.85。实验值:668.4(M+Na)+。
实施例21.化合物14的替代合成
(a)化合物37的制备
向N-Fmoc-1,3-丙烷二胺氢溴酸盐(377mg,1mmol)在CH2Cl2(15mL)中的溶液中加入DIPEA(0.52mL,3mmol)和6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯(323.7mg,1.1mmol)。将反应混合物在23℃搅拌过夜并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物37(430mg,100%),为白色固体。
1H NMR(400MHz,CDCl3):δ7.76(dd,J=7.6,1.0Hz,2H),7.64-7.55(m,2H),7.45-7.37(m,2H),7.31(td,J=7.5,1.2Hz,2H),6.16(bs,1H),5.24(bs,1H),4.42(d,J=6.9Hz,2H),4.21(t,J=6.8Hz,1H),3.27(dq,J=18.3,6.3Hz,4H),2.99(t,J=7.0Hz,2H),2.62(t,J=7.0Hz,2H),2.41(s,3H),1.71-1.59(m,2H)。
(b)化合物38的制备
向如上述步骤(a)中所述制备的37(430mg,1mmol)在CH2Cl2(8mL)中的溶液中加入二乙胺(1.4mL,13.5mmol)。将反应混合物在23℃搅拌6h并在真空下浓缩。将获得的残留物用Et2O研碎并过滤以获得化合物38(148mg,71%),为白色固体。
1H NMR(400MHz,CDCl3):δ3.29(td,J=6.5,2.3Hz,2H),3.01-2.86(m,2H),2.78(t,J=6.6Hz,2H),2.57(t,J=7.1Hz,2H),2.37(s,3H),1.69(p,J=6.6Hz,2H)。
(c)化合物14的制备
向如上述实施例5(a)中所述制备的化合物16(60mg,0.08mmol)在CH2Cl2(2mL)中的溶液中加入如上述步骤(b)中所述制备的化合物38(58mg,0.28mmol)和DIPEA(0.1mL,0.56mmol)在CH2Cl2(2mL)中的溶液。将反应混合物在23℃搅拌3h,用H2O稀释并用CH2Cl2萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物14(60.5mg,95%)。
1H NMR(500MHz,CDCl3):δ8.88(d,J=10.8Hz,1H),7.29-7.24(m,1H),6.90(t,J=11.5Hz,1H),6.82(t J=9.1Hz,1H),6.63(t,J=6.1Hz,1H),6.49(d,J=9.4Hz,1H),6.16(dd,J=11.5,1.5Hz,1H),5.70(d,J=11.5Hz,1H),5.68-5.51(m,3H),5.29(d,J=9.7Hz,1H),4.81(q,J=8.2Hz,1H),4.52(d,J=9.5Hz,1H),4.52-4.43(m,1H),4.24(ddd,J=11.5,7.3,4.3Hz,1H),3.66(s,3H),3.37-3.21(m,3H),3.21-3.12(m,1H),2.97(t,J=7.2Hz,2H),2.90-2.81(m,1H),2.60(t,J=7.2Hz,2H),2.49-2.35(m,3H),2.39(s,3H),2.33(t,J=7.0Hz,2H),2.14-2.07(m,1H),2.07(s,3H),1.84(s,3H),1.73-1.64(m,2H),1.16(d,J=6.7Hz,3H),1.05(s,9H)。
13C NMR(125MHz,CDCl3):δ171.6,168.2,166.4,161.6,157.2,145.2,140.3,137.4,134.2,134.0,131.9,124.4,124.1,122.4,120.7,108.3,105.6,81.8,74.8,60.6,60.4,55.5,37.8,37.2,36.2,35.6,34.7,33.1,31.0,29.8,26.7,26.2,23.0,21.0,17.2,16.6。
实施例22.化合物15的替代合成
(a)化合物39的制备
向如上述实施例5(a)中所述制备的化合物16(178.7mg,0.25mmol)在CH2Cl2(2.5mL)中的溶液中加入如上述实施例20(b)中所述制备的化合物33(120mg,0.88mmol)和DIPEA(0.05mL,0.25mmol)。将反应混合物在23℃搅拌2h,用H2O稀释并用CH2Cl2萃取。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,以获得纯的化合物39(100mg,56%)。
1H NMR(400MHz,CDCl3):δ8.65(d,J=10.7Hz,1H),7.28(t,J=11.6Hz,1H),6.90(t,J=11.5Hz,1H),6.82(t,J=9.6Hz,1H),6.37(d,J=9.4Hz,1H),6.17(d,J=11.7Hz,1H),5.69(d,J=11.4Hz,1H),5.64-5.57(m,2H),5.34(t,J=6.2Hz,1H),5.29(d,J=10.0Hz,1H),4.81(q,J=8.3Hz,1H),4.54-4.49(m,1H),4.45(d,J=9.4Hz,1H),4.28-4.17(m,1H),3.66(s,3H),3.37-3.21(m,2H),2.85(dt,J=9.7,6.9Hz,1H),2.71(t,J=7.2Hz,2H),2.44-2.30(m,5H),2.38(s,3H)2.13-2.06(m,1H),2.07(s,3H),1.93(p,J=6.9Hz,2H),1.84(s,3H),1.16(d,J=6.6Hz,3H),1.06(s,9H)。
ESI-MS m/z:对于C35H52ClN3O7S2计算:725.9。实验值:748.3(M+Na)+。
(b)化合物15的制备
将如上述实施例5(a)中所述制备的化合物39(90mg,0.12mmol)在EtOAc(6.7mL)和CH3OH(6.7mL)的混合物中的溶液用含有2mM乙二胺四乙酸(EDTA)的0.05M磷酸钾缓冲液(6.7mL)pH 7.5中的二硫苏糖醇溶液(0.36mL,0.36mmol)处理。将混合物在23℃搅拌4h。将反应用含有2mM EDTA的0.2M磷酸钾缓冲液pH 6.0的溶液处理,并且用EtOAc萃取(x3)。将合并的有机层经无水Na2SO4干燥,过滤并在真空下浓缩。将获得的残留物在用于急骤色谱的系统(SiO2,Hex:EtOAc混合物)中纯化,获得纯的目标化合物15(40.2mg,46%)。
1H NMR(500MHz,CDCl3):δ8.66(d,J=10.7Hz,1H),7.29(t,J=11.2Hz,1H),6.91(t,J=11.5Hz,1H),6.83(t,J=9.7Hz,1H),6.38(d,J=9.4Hz,1H),6.17(d,J=11.8Hz,1H),5.70(d,J=11.4Hz,1H),5.65-5.51(m,2H),5.34(t,J=6.3Hz,1H),5.29(d,J=10.0Hz,1H),4.82(q,J=8.3Hz,1H),4.56-4.48(m,1H),4.45(d,J=9.3Hz,1H),4.22(ddd,J=11.4,7.5,4.3Hz,1H),3.67(s,3H),3.31(q,J=6.4Hz,2H),2.88-2.83(m,1H),2.55(q,J=7.7Hz,2H),2.47-2.30(m,5H),2.12-2.07(m,1H),2.08(s,3H),1.88-1.76(m,5H),1.17(d,J=6.6Hz,3H),1.06(s,9H)。
13C NMR(125MHz,CDCl3):δ168.2,166.2,161.5,156.7,145.2,140.2,137.3,134.2,134.0,132.0,124.4,124.2,122.3,120.8,108.1,105.5,81.8,74.5,60.6,55.4,39.6,37.3,34.6,33.9,33.3,30.8,26.7,26.3,21.8,21.1,17.2,16.7。
ESI-MS m/z:对于C34H50ClN3O7S计算:679.3。实验值:702.4(M+Na)+。
证明本发明的抗体-药物缀合物的细胞毒性的实施例
用于检测抗肿瘤活性的生物测定
测定的目的是评价测试的样品的体外抑制细胞(使肿瘤细胞生长延迟或停滞的能力)或细胞毒性(杀伤肿瘤细胞的能力)活性。
细胞系和细胞培养
除非另有指示,用于本研究的所有肿瘤细胞系获自美国典型培养物保藏中心(ATCC);BT-474(ATCC HTB-20,乳腺导管癌(Breast Ductal Carcinoma)),SK-BR-3(ATCC HTB-30,乳腺腺癌(Breast Adenocarcinoma))和HCC-1954(ATCC CRL-2338,乳腺导管癌),均HER2+;MDA-MB-231(ATCC HTB-26,乳腺腺癌)和MCF-7(ATCC HTB-22乳腺腺癌,胸腔积液(pleural effusion)),均HER2-;SK-OV-3(ATCC HTB-77,卵巢腺癌(Ovary Adenocarcinoma)),HER2+;NB-4(急性早幼粒细胞白血病(Acute Promyelocytic Leukemia),APL,CD13+,M.Lanotte等人(1991)NB4,a maturation inducible cell line with t(15;17)marker isolated from a human acute promyelocytic leukemia(具有从人急性早幼粒细胞白血病分离的t(15;17)标记物的成熟可诱导细胞系)(M3).Blood 77,1080-1086),(CD13+)和U937(ATCC CRL-1593.2,组织细胞淋巴瘤(Histiocytic Lymphoma),CD13+和CD4+);Raji(ATCC CCL-86,伯基特淋巴瘤(Burkitt’s Lymphoma))(CD13-,CD20+,CD5-,和CD4-);RPMI-8226(ATCC CRM-CCL-155,多发性骨髓瘤(Multiple Myeloma))(CD13-,CD20-,CD5-和CD4-);Karpas-299(DSMZ ACC-31,非霍奇金淋巴瘤(Non-Hodgkin’s Lymphoma))(CD20-,CD5+,和CD4+);MOLT-4(ATCC CRL-1582,急性淋巴细胞白血病(Acute Lymphoblastic Leukemia),CD5+)。此外,用于本研究的两种Raji细胞(ATCC CCL-86,伯基特淋巴瘤)克隆,Raji-克隆#10(高CD5表达)和Raji-克隆18(无CD5表达),由Dr.Juan M.Zapata(Instituto de Investigaciones Biomédicas“Alberto Sols”,CSIC-UAM,Madrid,Spain)提供。将细胞维持在37℃,5%CO2和95%湿度,于Dulbecco’s改良的Eagle’s培养基(DMEM)(用于MCF和MDA-MB-231细胞),RPMI-1640(用于SK-BR-3,HCC-1954,NB-4,U937,Raji,RPMI-8226,Karpas-299,MOLT-4,Raji-克隆#10和Raji-克隆#18细胞),RPMI-1640+1%ITS(用于BT-474细胞)或McCOyS(用于SK-OV-3细胞),所有培养基补充以10%胎牛血清(FCS)和100单位/mL的氨苄青霉素和链霉素。
细胞毒性测定
对于粘附细胞:使用磺酰罗丹明B(SRB)的比色测定适应于细胞生长和细胞毒性的定量测量,如V.Vichai和K.Kirtikara(2006)Sulforhodamine B colorimetric assay for cytotoxicity screening(用于细胞毒性筛选的磺酰罗丹明B比色测定).Nature Protocols,1,1112-1116。简言之,将细胞在96孔微孔板中接种并使得在无药物培养基中静置24小时,之后单独用媒介物或指定的化合物处理72小时。对于定量,将细胞用磷酸盐缓冲盐水(PBS)洗涤两次,在1%戊二醛溶液中固定15min,用PBS冲洗两次,在0.4%SRB-1%乙酸溶液中染色30min,用1%乙酸溶液冲洗数次并空气干燥。随后在10mM氨基丁三醇溶液中萃取SRB并且在微孔板分光光度计中在490nm测量光密度。将细胞存活表达为对照,未处理细胞存活的百分数。
对于悬浮液细胞:使用MTT(3-(4,5-二甲基噻唑-2基-)-2,5-二苯基四唑溴化物)的标准代谢测定适于定量测量细胞生长和细胞毒性,如T.Mosmann(1983)Rapid colorimetric assay for cellular growth and survival:Application to proliferation and cytotoxicity assays(用细胞生长和存活的快速比色测定:对增殖和细胞毒性测定的应用).J.Immunol.Meth.,65,55-63中所述的。简言之,以0.5mg/mL的终浓度将MTT溶液加入细胞培养物中,并在37℃温育1至4小时,直到甲臜晶体形成。从细胞培养物中小心去除培养基,并且将甲臜晶体在100μL DMSO中重悬。混合以保证溶解后,甲臜的定量(大概直接与存活细胞的数量成比例)通过使用平板读取分光光度计在570nm记录吸光度的改变来测量。细胞存活表达为对照,未处理细胞存活的百分数。
IC50值是指与对照细胞存活相比,诱导50%细胞死亡的化合物浓度。
生物活性实施例1-ADC1和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对不同的过表达或不过表达HER2受体的人乳腺癌细胞系,包括BT-474,HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER阴性细胞)评价ADC1连同母体细胞毒性化合物1和4和曲妥珠单抗的体外细胞毒性。SK-OV-3,HER2+卵巢癌细胞系,也包括在本研究中,作为非乳腺组织细胞模型。进行72小时的标准剂量响应(DR)曲线。
曲妥珠单抗的细胞毒性
首先,针对不同肿瘤细胞系测定曲妥珠单抗的体外细胞毒性。在范围从5.0E01至2.6E-03μg/mL(3.4E-07-1.8E-11M)的一式三份的DR曲线中,在两个独立的实验中,曲妥珠单抗为完全失活的,在任何测试的细胞系中未达到IC50,不依赖于其HER2状态(参见表3)。
表3.曲妥珠单抗的体外细胞毒性总结
化合物4的细胞毒性
以DR曲线,使用从1E-01至2.6E-05μg/mL(1.5E-07至3.9E-11M)的十个连续稀释液(1/2.5比例)测定中间体化合物4的细胞毒性。
该化合物的细胞毒性,在两个独立的实验中,在测试的不同细胞系间相对均匀,IC50值在低纳摩尔范围内,从8.9E-05至1.7E-03μg/mL(1.34E-10至2.6E-09M),整个细胞组(panel)中的平均IC50值为5.69E-04μg/mL(8.57E-10M)。此外,化合物4的细胞毒性不依赖于肿瘤细胞系的HER2状态(参见表4)。
表4.化合物4的体外细胞毒性的总结
化合物1的细胞毒性
使用与上述相同的条件,从1E-01至2.6E-05μg/mL(1.1E-07至3.0E-11M)测定母体化合物1的活性。该化合物的细胞毒性,在两个独立的实验中,在测试的不同细胞系中也相对均匀,IC50值在低纳摩尔范围内,从8.9E-04至6.4E-03μg/mL(1.04E-09至7.47E-09M),整个细胞组的平均IC50值为3.41E-03μg/mL(3.98E-09M)。马来酰亚胺接头似乎稍微减小化合物的细胞毒性效应。再次,化合物1的细胞毒性不依赖于肿瘤细胞系的HER2状态(参见表5)。
表5.化合物1的体外细胞毒性的总结
ADC1的细胞毒性
针对不同细胞系测定ADC1的细胞毒性。为了确保适当的浓度范围,以六个不同的,一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1,0.1,0.01和0.001μg/mL开始(相当于3.3E-07,6.6E-08,6.6E-09,6.6E-10,6.6E-11和6.6E-12摩尔浓度),在两个独立的实验中测定缀合物。
代表性的DR曲线显示在图3中。
调节不同DR曲线后,对于ADC1计算的针对不同细胞系的平均IC50值显示在下表6中。ADC1显示与单独母体化合物化合物1相对类似的细胞毒性,重要的是,针对表达HER2+的细胞的明显特异性。因此,我们设想,缀合物实际上通过mAb与肿瘤细胞上的膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中来起作用。在HER2阳性细胞系中,在针对ADC1的敏感性方面存在显著差异。最敏感的细胞系是HCC-1954和SK-BR-3,显示3.88E-02和2.45E-02μg/mL(相当于2.581E-10和1.63E-10M)的IC50,接着是BT-474细胞,其显示显著更高的7.4E-01μg/mL(相当于4.93E-09M)的IC50值。卵巢细胞系SK-OV-3显示甚至更高的7.0E+00μg/mL(相当于4.67E-08M)的IC50值。两种HER阴性细胞显示大约2.0E+01μg/mL(相当于约1.0E-07M)的类似的敏感性(参见表6)。
表6.ADC1的体外细胞毒性的总结
因此,最具响应性的HER2阳性细胞系比HER2阴性细胞系敏感约300-800倍,表明缀合物针对表达HER2的细胞的特异性。
为了通过图表比较单独mAb曲妥珠单抗的细胞毒性与缀合物ADC1的细胞毒性,显示单独用mAb(10μg/mL)或10或1μg/mL的ADC处理不同细胞系后的细胞存活的百分数的直方图,显示在图4中。如可以从图4看出的,在相等的10μg/mL的浓度,单独mAb曲妥珠单抗针对任何测试的细胞系显示很小的或不显示细胞毒性(<20%máx),不依赖于其HER2状态。相比之下,ADC1显示针对表达HER2的细胞,HCC-1954和SK-BR-3的有效细胞毒性,至更小的程度,BT-474和SK-OV-3,与对照细胞相比,分别诱导88%,82%,52%和47%的平均细胞存活抑制。在该浓度,ADC1显示一些针对HER阴性细胞MCF-7和MDA-MB-231的细胞毒性,分别为38%和32%的平均细胞存活抑制百分数。在1μg/mL的浓度,ADC1缀合物显示与在10μg/mL观察到的稍微类似的针对HER2阳性细胞的细胞毒性,但在此种情况下没有可检测的对HER2阴性细胞的效应(图4)。
这些结果明确表明ADC1缀合物针对表达HER2的人肿瘤细胞的显著体外细胞毒性和特异性。
生物活性实施例2-ADC2和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对不同的表达或不表达HER2受体的人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER阴性细胞)评价ADC2,曲妥珠单抗化合物5ADC,连同母体细胞毒性化合物5和8以及mAb曲妥珠单抗的体外细胞毒性。进行72小时的标准剂量响应(DR)曲线。
化合物8的细胞毒性
以DR曲线,使用从01E+00至2.6E-04μg/mL(1.6E-06至4.0E-10M)的十个连续稀释液(1/2.5比例)测定中间体化合物8的细胞毒性。
该化合物的细胞毒性,在两个独立的实验中,在不同的测试的细胞系间相对均匀,IC50值在纳摩尔范围内,从3.40E-03至6.75E-03μg/mL(5.4E-09至1.0E-08M),整个细胞组的平均IC50值是5.53E-03μg/mL(相当于8.79E-09M)。此外,化合物8的细胞毒性不依赖于肿瘤细胞系的HER2状态(表7)。
表7.化合物8的体外细胞毒性的总结数据
化合物5的细胞毒性
在与上述相同的条件中,从01E+00至2.6E-04μg/mL(1.2E-06至3.1E-10M),测定化合物5,携带马来酰亚胺接头的修饰的化合物8的活性。
该化合物的细胞毒性,在两个独立的实验中,测试的不同细胞系中也相对均匀,IC50值在纳摩尔范围内,从1.9E-02至7.7E-02μg/mL(2.32E-08至9.41E-08M),整个细胞组的平均IC50值为4.33E-02μg/mL(5.26E-08M)。与化合物8相比,化合物5中马来酰亚胺接头的存在稍微减小化合物的细胞毒性。此外,化合物5的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表8)。
表8.化合物5的体外细胞毒性的总结数据
ADC2的细胞毒性
针对不同细胞系测定ADC2的细胞毒活性。仅为了保证适当的浓度范围,在五个不同浓度范围中,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1,0.1,和0.01μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线显示在图5中。调节所有的不同DR曲线后,对于ADC2针对不同细胞系计算的平均IC50值显示在表9中。缀合物ADC2显示针对表达HER2+的细胞,HCC-1954和SK-BR-3的特异性,其中该化合物显示与母体化合物5类似的细胞毒性,分别为5.8E+00和2.2E-01μg/mL(相当于4.0E-08和1.5E-09M)的平均IC50值。两个HER-细胞系,MCF-7和MDA-MB-231,在测试的浓度范围内几乎不响应于ADC2,未达到IC50值(>5.0E+01μg/mL)(参见图5和表9)。
因此,我们猜想,缀合物通过mAb与肿瘤细胞上的膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表9.ADC2(曲妥珠单抗化合物5ADC)的体外细胞毒性的总结数据
为了通过图表比较单独曲妥珠单抗的细胞毒性与缀合物ADC2的细胞毒性,显示用单独曲妥珠单抗(10μg/mL)或10或1ug/mL的ADC处理不同细胞系后的细胞存活百分数的直方图显示在图6中。在10μg/mL的浓度,单独mAb曲妥珠单抗不显示针对任何测试的细胞系的细胞毒性,不依赖于其HER2状态。相比之下,与对照细胞相比,ADC2缀合物显示针对表达HER2的细胞HCC-1954和SK-BR-3的显著和特异的细胞毒性,分别诱导57%和78%的平均细胞存活抑制。
在1μg/mL的浓度,ADC2缀合物显示与在10μg/mL观察到的稍微类似的针对HER2阳性细胞的细胞毒性,同样,无可检测的对HER2阴性细胞的效应(图6)。
这些结果明确表明ADC2缀合物针对表达HER2的人肿瘤细胞的显著的体外细胞毒性和特异性。
生物活性实施例3-ADC3和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对不同的表达或不表达HER2受体的人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER阴性细胞评价)ADC3,连同母体细胞毒性化合物12和4以及mAb曲妥珠单抗的体外细胞毒性。进行72小时的标准剂量响应(DR)曲线。
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-07至3.9E-12M)的十个连续稀释液(1/2.5比例)测定母体化合物4的细胞毒性。
化合物4的细胞毒性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在皮摩尔范围内,从1.16E-04至2.80E-04μg/mL(1.75E-10至4.23E-10M),整个细胞组的平均IC50值是1.97E-04μg/mL(相当于2.96E-10M)。此外,化合物4的细胞毒性不依赖于肿瘤细胞系的HER2状态(表10)。
表10.化合物4的体外细胞毒性的总结数据
在上述相同的实验条件下,在01E+01至2.6E-03μg/mL(7.9E-06至2.0E-09M)的浓度范围内,测定化合物12,携带可切割肽接头的修饰的化合物4的活性。
化合物12的细胞毒性,在两个独立的实验中,在测试的不同细胞系中也相对均匀,IC50值在低纳摩尔范围内,从7.60E-03至3.05E-02μg/mL(6.02E-09至2.42E-08M),整个细胞组的平均IC50值为1.63E-02μg/mL(1.29E-08M)(表11)。与化合物4相比,化合物12中肽接头的存在对化合物的细胞毒性具有消极作用。化合物12的细胞毒性相当不依赖于肿瘤细胞系的HER2状态。
表11.化合物12的体外细胞毒性的总结数据
ADC 3的细胞毒性
最后,针对不同细胞系测定ADC3的细胞毒性。为了确保适当的浓度范围,在六个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1,0.1,0.01和0.001μg/mL(相当于3.33E-07,6.64E-08,6.64E-09,6.64E-10,6.64E-11和6.64E-12摩尔浓度)开始,在两个独立的实验中测定缀合物。代表性的DR曲线显示在图7中。对于ADC3针对测试的不同细胞系计算的平均IC50值显示在表12中。
缀合物ADC3明确显示显著的针对表达HER2+的细胞的特异性,其中该化合物显示与母体化合物4(比携带肽接头的中间体化合物12多约1log活性)类似的有效细胞毒性。两种HER2+细胞系,HCC-1954和SK-BR-3,均显示相当的针对ADC3的敏感性,平均IC50值分别为8.83E-02和6.77E-02μg/mL(相当于5.86E-10和4.49E-10M)。两种HER阴性细胞系,MCF-7和MDA-MB-231,显示显著更低的针对ADC3的敏感性,平均IC50值分别为9.40E+00和>5.0E+01μg/mL(相当于约6.24E-08M和>3.32E-07M)。
图8是ADC3针对HER2阳性和阴性乳腺癌细胞的细胞毒性的图。发现HER2+细胞系(平均IC507.80E-02μg/mL)比HER2阴性MCF-7细胞(平均IC509.40E+00μg/mL)对ADC3敏感至少>120倍并且远比MDA-MB-231细胞敏感,明确显示ADC3针对表达HER2的细胞的特异性(图7和表12)。因此,我们猜想,缀合物实际上通过mAb与肿瘤细胞上膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表12.ADC3(曲妥珠单抗化合物12)的体外细胞毒性的总结数据。
为了通过图表比较单独mAb曲妥珠单抗的细胞毒性与缀合物ADC3的细胞毒性,显示利用单独mAb(10μg/mL)或10或1μg/mL的ADC3处理不同细胞系后的细胞存活的百分数的直方图,显示在图8中,其显示曲妥珠单抗相对ADC3针对不同人乳腺癌细胞系的细胞毒活性。
在10μg/mL的相等浓度,单独曲妥珠单抗,不显示针对任何测试的细胞系的细胞毒性,不依赖于其HER2状态。相比之下,ADC3缀合物显示针对表达HER2的细胞,HCC-1954和SK-BR-3的有效细胞毒性。与对照细胞相比,在这些细胞系中,ADC3分别发挥83%和84%的细胞存活抑制。在该浓度,ADC3还对HER2阴性细胞,MCF-7和MDA-MB-231具有一些效应,分别产生33%和20%的略微细胞存活抑制。在1μg/mL的浓度,ADC3缀合物显示与在10μg/mL观察到的类似的针对HER2阳性细胞的细胞毒性,但无可检测的对HER2阴性细胞的效应(图8)。这些结果明确表明ADC3针对表达HER2的人肿瘤细胞的显著的体外细胞毒性和特异性。
生物活性实施例4-ADC4和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对表达或不表达HER2受体的不同人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER2阴性细胞)评价ADC4,连同母体细胞毒性化合物13和4以及mAb曲妥珠单抗的体外细胞毒性。进行72小时的标准剂量响应(DR)曲线。
化合物4的细胞毒性
以DR曲线,使用从1E-01至2.6E-05μg/mL(1.5E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定母体化合物化合物4的细胞毒性。
化合物4的细胞毒性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在低纳摩尔范围内,从2.43E-04至4.45E-04μg/mL(3.6E-10至6.7E-10M),整个细胞组的平均IC50值为3.3E-04μg/mL(相当于4.98E-10M)。因此,化合物4的细胞毒性不依赖于肿瘤细胞系的HER2状态(表13)。
表13.化合物4的体外细胞毒性的总结数据
化合物13的细胞毒性
在与上述相同的条件下,从1E-01至2.6E-05μg/mL(1.3E-07至2.0E-11M)测定化合物13,携带含硫醇基的修饰的化合物4的活性。
化合物13的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中也是相对均匀的,IC50值在低纳摩尔范围内,从7.95E-04至2.63E-03μg/mL(1.0E-09至3.5E-09M),整个细胞组的平均IC50值为1.83E-03μg/mL(2.44E-09M)(表14)。与化合物4相比,化合物13中含硫醇基尾的存在稍微减少(约5倍)化合物的细胞毒活性。此外,化合物13的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表14)。
表14.化合物13的体外细胞毒性的总结数据
ADC4的细胞毒性
针对不同细胞系测定ADC4的细胞毒性。仅为了保证适当的浓度范围,以四个不同浓度范围,每个以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线显示在图9中。调节所有不同DR曲线后,对于ADC4针对测试的不同细胞系计算的平均IC50值显示在表15中。
缀合物ADC4显示针对表达HER2+的细胞,HCC-1954和SK-BR-3的特异性,其中该化合物显示与母体化合物4和13类似的有效细胞毒性,平均IC50值分别为1.17E-01和4.80E-02μg/mL。两种HER阴性细胞系,MCF-7和MDA-MB-231,分别显示显著更低的针对ADC4的敏感性,平均IC50值为5.35E+00和6.50E+00μg/mL。似乎缀合物优选通过mAb与肿瘤细胞上膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表15.ADC4的体外细胞毒性的总结数据。
为了通过图表比较单独mAb曲妥珠单抗的细胞毒性与缀合物ADC4的细胞毒性,显示用单独mAb(10μg/mL)或10或1μg/mL ADC4处理不同细胞系后细胞存活的百分数的直方图显示在图10中。在10μg/mL的浓度,单独mAb曲妥珠单抗,不显示针对任何测试的细胞系的细胞毒性,不依赖于其HER2状态。相比之下,ADC4缀合物显示显著和特异的针对表达HER2的细胞,HCC-1954和SK-BR-3的细胞毒性,与对照细胞相比,分别诱导80%和75%的平均细胞存活抑制。在该浓度,ADC4还具有对HER2阴性细胞,MCF-7和MDA-MB-231的效应,分别产生53%和40%的细胞存活抑制。在1μg/mL的浓度,ADC4缀合物显示与在10μg/mL观察到的稍微类似的对HER2阳性细胞的细胞毒性针,但无可检测的对HER2阴性细胞的效应(图10)。这些结果明确表明ADC4缀合物针对表达HER2的人肿瘤细胞的显著的体外细胞毒性和特异性。
生物活性实施例5-ADC5和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性。
针对表达或不表达HER2受体的不同人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER阴性细胞)评价ADC5连同母体细胞毒性化合物15和40的体外细胞毒性。
化合物40的细胞毒性
化合物40如WO2007144423中所述制备(该专利申请中的化合物1),其内容通过引用结合于此。
以DR曲线,使用从1E-02至2.6E-06μg/mL(1.65E-08至4.29E-12M)的十个连续稀释液(1/2.5比例)测定母体化合物40的细胞毒性。
化合物40的细胞毒性,在两个独立的实验中,在测试的不同细胞系中非常均匀,IC50值在低纳摩尔范围内,从4.90E-05至1.73E-04μg/mL(8.10E-11至2.84E-10M),整个细胞组的平均IC50值为1.06E-04μg/mL(相当于1.75E-10M)。因此化合物40的细胞毒性不依赖于肿瘤细胞系的HER2状态(表16)。
表16.化合物40的体外细胞毒性的总结数据。
化合物15的细胞毒性
在与上述相同的条件下,从1E-01至2.6E-05μg/mL(1.47E-07至3.82E-11M)测定化合物15,携带含硫醇基的修饰的化合物40的活性。
化合物15的细胞毒性,在两个独立的实验中,在测试的不同细胞系中也非常均匀,IC50值在纳摩尔范围内,从4.80E-04至1.49E-03μg/mL(7.06E-10至2.19E-09M),整个细胞组的平均IC50值为1.03E-03μg/mL(1.51E-09M)。与化合物40相比,化合物15中含硫醇基尾的存在稍微减少(约8倍)化合物的细胞毒活性。此外,化合物15的细胞毒性不依赖于肿瘤细胞系的HER2状态。(表17)
表17化合物15的体外细胞毒性的总结数据
ADC 5的细胞毒性
针对不同细胞系测定ADC5的细胞毒性。仅为了保证适当的浓度范围,以四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图11中。在调节所有不同DR曲线后,对于ADC5针对测试的不同细胞系计算的平均IC50值显示在表18中。
缀合物ADC5显示针对表达的HER2+的细胞,HCC-1954和SK-BR-3的特异性,其中化合物显示与母体化合物化合物40和化合物15类似的细胞毒活性,平均IC50值分别为1.13E-01和4.61E-02μg/mL。两个HER阴性细胞系,MCF-7和MDA-MB-231,都显示显著更低的针对ADC5的敏感性,平均IC50值分别为1.23E+00和1.45E+00μg/mL。
似乎缀合物ADC5优选通过mAb与肿瘤细胞上膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表18.ADC5的体外细胞毒性的总结数据。
为了通过图表比较单独mAb曲妥珠单抗细胞毒活性与缀合物ADC5的细胞毒活性,显示用单独mAb(10μg/mL)或10μg/mL或1μg/mL的ADC5处理不同细胞系后的细胞存活百分数的直方图,显示在图12中。在10μg/mL的浓度,单独mAb曲妥珠单抗不显示针对任何测试的细胞系的细胞毒活性,不依赖于其HER2状态。相比之下,ADC5缀合物显示针对表达HER2的细胞的显著和特异的细胞毒性,HCC-1954和SK-BR-3,与对照细胞相比,分别诱导82%和72%的平均细胞存活抑制。在该浓度,ADC5还对HER2阴性细胞,MCF-7和MDA-MB-231具有作用,分别产生58%和54%的细胞存活抑制。在1μg/mL的浓度,ADC5缀合物显示与在10μg/mL观察到的相对类似的针对HER2阳性细胞的细胞毒活性(分别为77%和75%),但远小的针对HER2阴性细胞的活性,分别为24%和15%的细胞存活抑制(图12)。这些结果表明ADC5缀合物针对表达HER2的人肿瘤细胞的显著的体外细胞毒活性和相对特异性。
生物活性实施例6-ADC6和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对表达或不表达HER2受体的不同人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER2阴性细胞)评价ADC6连同母体细胞毒性化合物18和8的体外细胞毒性。进行标准剂量响应(DR)曲线达数小时。
化合物8的细胞毒性
以DR曲线,使用从1E+00至2.6E-04μg/mL(1.59E-06至4.13E-10M)的十个连续稀释液(1/2.5比例)测定母体化合物8的细胞毒性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中相对均匀(针对SK-BR-3细胞稍微更具活性),IC50值在低纳摩尔范围内,从1.85E-03至9.50E-03μg/mL(2.94E-09至1.51E-08M),整个细胞组的平均IC50值为5.45E-03μg/mL(相当于8.67E-09M)。因此,化合物8的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表19)。
表19.化合物8的体外细胞毒性的总结数据
化合物18的细胞毒性
在与上述相同的条件下,从1E+00至2.6E-04μg/mL(1.39E-06至3.63E-10M)测定化合物18,携带含硫醇基的修饰的化合物8的活性。该化合物的细胞毒性,在两个独立的实验中,在测试不同细胞系中也非常均匀,IC50值在纳摩尔范围内,从4.40E-03至1.85E-02μg/mL(6.14E-09至2.58E-08M),整个细胞的组平均IC50值为1.06E-02μg/mL(1.48E-08M)。与化合物8相比,化合物18中含硫醇基尾的存在对化合物的活性具有很小的影响。此外,化合物18的细胞毒性似乎相当不依赖于肿瘤细胞系的HER2状态(表20)。
表20.化合物18的体外细胞毒性的总结数据
ADC6的细胞毒性
针对不同细胞系测定ADC6的细胞毒性。仅为了保证适当的浓度范围,以四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR显示在图13中。在调节所有不同DR曲线后,对于ADC6针对测试的不同细胞系计算的平均IC50值显示在表21中。
缀合物ADC6,尽管有限,显示一些针对表达HER2+的细胞,HCC-1954和SK-BR-3的特异性。在这些细胞系中,该缀合物比母体化合物8和18单独细胞毒性稍低(分别为5.6和3.2倍),平均IC50值分别为1.04E+01和3.80E+00μg/mL。两个HER阴性细胞系,MCF-7和MDA-MB-231,显示稍微更低的针对ADC6(5倍低于)的敏感性,平均IC50值分别为3.50E+01和4.40E+01μg/mL。似乎缀合物ADC6对对表达HER2的细胞更优先,通过mAb与肿瘤细胞上膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表21.ADC6的体外细胞毒性的总结数据。
为了通过图表比较单独mAb曲妥珠单抗的细胞毒活性与缀合物ADC6的细胞毒活性,显示用单独mAb(10μg/mL)或10或1μg/mL的ADC6处理不同细胞系后细胞存活百分数的直方图,显示在图14中。在10μg/mL的浓度,单独mAb曲妥珠单抗不显示针对任何测试的细胞系的细胞毒活性,不依赖于其HER2状态。相比之下,ADC6缀合物显示针对表达HER2的细胞,HCC-1954和SK-BR-3的特异性细胞毒性,与对照细胞相比,分别诱导57%和70%的平均细胞存活抑制。在该浓度,ADC6对HER2阴性细胞,MCF-7和MDA-MB-231也具有剩余效应,分别产生9%和7%的细胞存活抑制。在1μg/mL的浓度,ADC6缀合物仍然具有针对HER2阳性细胞的细胞毒活性,尽管低于在10μg/mL观察到的(分别为19%和38%)。在该浓度,ADC6针对HER2阴性细胞是完全失活的(图14)。这些结果表明ADC6缀合物针对表达HER2的人肿瘤细胞的优选的体外细胞毒活性。
生物活性实施例7-ADC7和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对表达或不表达HER2受体的不同人乳腺癌细胞系评价,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER2阴性细胞)ADC7连同母体细胞毒性化合物24,25和41的体外细胞毒性。进行72小时的标准剂量响应(DR)曲线。
化合物41的细胞毒性
化合物41如WO 2009/080761中所述制备(该专利申请中的化合物72),其内容通过引用结合于此。
以DR曲线,使用从1E-02至2.6E-06μg/mL(1.8E-08至4.6E-12M)的十个连续稀释液(1/2.5比例)测定母体化合物41的细胞毒性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在低纳摩尔范围内,从1.0E-04至2.6E-04μg/mL(1.8E-10至4.6E-10M),整个细胞组的平均IC50值为1.6E-04μg/mL(相当于2.9E-10M)。因此,化合物41的细胞毒性不依赖于肿瘤细胞系的HER2状态(表22)
表22.化合物41的体外细胞毒性的总结数据。
化合物24的细胞毒性
在与上述相同的条件下,从1E+00至2.6E-04μg/mL(1.6E-06至4.1E-10M)测定化合物24的活性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在纳摩尔范围内,从9.0E-03至1.8E-02μg/mL(1.4E-08至2.8E-08M),整个细胞组的平均IC50值为1.5E-02μg/mL(2.4E-08M)。与化合物41相比,化合物24中1,3-亚丙基二胺基团的存在显著减小(约2logs)化合物的细胞毒活性。此外,化合物24的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表23)。
表23.化合物24的体外细胞毒性的总结数据。
化合物25的细胞毒性
在与上述相同的条件下,从1E-01至2.6E-05μg/mL(1.2E-07至3.2E-11M)测定化合物25,携带MC接头的修饰的化合物24的活性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中均匀,IC50值在纳摩尔范围内,从2.5E-02至5.3E-02μg/mL(3.1E-08至6.5E-08M),整个细胞组的平均IC50值为4.1E-02μg/mL(4.9E-08M)。与化合物24相比,化合物25中MC接头的存在非常稍微地减少化合物的细胞毒活性,尤其是在MDA-MB-231细胞中。化合物25的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表24)。
表24.化合物25的体外细胞毒性的总结数据
ADC7的细胞毒性
针对不同细胞系测定ADC7的细胞毒性。仅为了保证适当的浓度范围,以四种不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(起始浓度10μg/mL)显示在图15中。在调节所有不同DR曲线后,对于ADC7针对测试的不同细胞系计算的平均IC50值显示在中表25中。
缀合物ADC7显示针对表达HER2+的细胞,HCC-1954和SK-BR-3的特异性,其中化合物显示与母体化合物41几乎类似的细胞毒活性,平均IC50值分别为3.7E-01和8.9E-02μg/mL。两个HER阴性细胞系,MCF-7和MDA-MB-231,实质上不响应于ADC7。该缀合物似乎通过mAb与阳性肿瘤细胞上的膜相关HER2受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表25.ADC7的体外细胞毒性的总结数据
为了通过图表比较单独mAb曲妥珠单抗的细胞毒活性与缀合物ADC7的细胞毒活性,显示用mAb单独(50μg/mL)或50或1μg/mL的ADC7处理不同细胞系后细胞存活百分数的直方图,显示在图16中。在50μg/mL的浓度,mAb曲妥珠单抗单独,不显示针对任何测试的细胞系的显著细胞毒活性,不依赖于其HER2状态。相比之下,ADC7缀合物显示显著和特异针对表达HER2的细胞,HCC-1954和SK-BR-3的细胞毒性,与对照细胞相比,分别诱导77%和76%的平均细胞存活抑制。在该浓度,ADC7仅对HER2阴性细胞,MCF-7和MDA-MB-231具有剩余效应,分别产生13%和15%的细胞存活抑制。以更低的浓度的ADC7(低至1μg/mL)在HCC-1954和SK-BR-3细胞中检测类似的活性和特异性,分别产生68%和79%的细胞存活抑制(图16)。总之,这些结果明确表明ADC7缀合物针对表达HER2的人肿瘤细胞的显著的体外细胞毒活性和特异性。
生物活性实施例8-ADC8和相关试剂针对HER2阳性和阴性乳腺癌细胞的细胞毒性
针对表达或不表达HER2受体的不同人乳腺癌细胞系,包括HCC-1954和SK-BR-3(HER2阳性细胞)以及MDA-MB-231和MCF-7(HER阴性细胞)评价ADC8连同母体细胞毒性化合物24,27和41的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物41的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.8E-08至4.6E-12M)的十个连续稀释液(1/2.5比例)测定母体化合物41的细胞毒活性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中均匀,IC50值在低纳摩尔范围内,从7.5E-05至1.4E-04μg/mL(1.3E-10至2.4E-10M),整个细胞组的平均IC50值是1.1E-04μg/mL(相当于1.9E-10M)。因此,化合物41的细胞毒性不依赖于肿瘤细胞系的HER2状态(表26)。
表26.化合物41的体外细胞毒性的总结数据
化合物24的细胞毒性
在与上述相同的条件下,从01E+00至2.6E-04μg/mL(1.6E-06至4.1E-10M)测定化合物24的活性。该化合物的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在纳摩尔范围内,从9.0E-03至1.8E-02μg/mL(1.4E-08至2.9E-08M),整个细胞组的平均IC50值为1.5E-02μg/mL(2.4E-08M)。与化合物41相比,化合物24中1,3-亚丙基二胺基团的存在显著减小(约2logs)化合物的细胞毒活性。化合物24的细胞毒性似乎不依赖于肿瘤细胞系的HER2状态(表27)。
表27.化合物24的体外细胞毒性的总结数据
化合物27的细胞毒性
以DR曲线,使用从01E+00至2.6E-04μg/mL(1.4E-06至3.6E-10M)的十个连续稀释液(1/2.5比例)测定化合物27的活性。化合物27的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在微摩尔范围内,从1.05E-02至3.9E-02μg/mL(1.5E-08至5.5E-08M),整个细胞组的平均IC50值2.5E-02μg/mL(3.5E-08M)。与化合物24相比,化合物27中MPA接头的存在对化合物的细胞毒活性无显著影响。化合物27的活性不依赖于肿瘤细胞系的HER2状态(表28)。
表28.化合物27的体外细胞毒性的总结数据
ADC8的细胞毒性
针对不同细胞系测定ADC8的细胞毒活性。以四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(10μg/mL的最大浓度)显示在图17中。ADC8显示针对表达HER2+的细胞,特别是SK-BR-3(最敏感的细胞系)的一些特异性。除了这些细胞(其大约比HER2阴性细胞(IC502.3E-09M)敏感4倍),ADC8显示,与母体化合物27类似的不依赖于HER2的细胞毒活性,IC50值在纳摩尔范围内(表29)。
表29.ADC8的体外细胞毒性的总结数据
为了通过图表比较单独mAb曲妥珠单抗的细胞毒活性与缀合物ADC8的细胞毒活性,显示用mAb单独(50μg/mL)或ADC8(50或10μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图18中。在50μg/mL,单独mAb在任何测试的细胞系中都不具备活性,除了SK-BR-3,其中,其产生小于20%的细胞存活抑制。ADC8,相应,不显示对于HER2+细胞系的显著特异性,在所有分析的细胞中产生多于60%的强烈细胞存活抑制。在10μg/mL的浓度,ADC8显示一些,但很少的,针对HER2+细胞的特异性,在HCC-1954和SK-BR-3细胞中(二者都是HER2阳性)产生78%的细胞存活抑制,同时对HER2阴性细胞具有较小的效应,在MCF7和MDA-MB-231中分别为59%和41%。HER2阳性细胞比HER2阴性细胞对ADC8大约更敏感1.5和2倍之间。
生物活性实施例9-ADC9和相关试剂针对CD13阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD13受体的不同人肿瘤细胞系,包括NB4和U937(CD13阳性细胞)以及Raji和RPMI-8226(CD13阴性细胞)评价ADC9连同母体细胞毒性化合物1和4的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
抗CD13小鼠单克隆抗体的细胞毒性
首先,针对不同肿瘤细胞系测定单独抗CD13小鼠mAb的体外细胞毒活性。在范围从5.0E+01至1.3E-02μg/mL(3.3E-07-8.7E-11M)的一式三份的DR曲线,在两个独立的实验中,抗体实质上是失活的,在任何测试的细胞系中均未达到不依赖于其CD13状态的IC50(表30)。
表30.抗CD13小鼠mAb的体外细胞毒活性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是非常均匀的,IC50值在低纳摩尔范围内,从7.9E-05至2.65E-03μg/mL(1.2E-10至4.0E-09M),整个细胞组的平均IC50值为8.4E-04μg/mL(相当于1.2E-09M)。因此,化合物4的细胞毒性非常不依赖于肿瘤细胞系的CD13状态(表31)。
表31.化合物4的体外细胞毒性的总结数据
化合物1的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.1E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定化合物1的活性。化合物1的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中略微均匀,IC50值在纳摩尔范围内,从8.0E-04至6.3E-03μg/mL(9.4E-10至7.3E-09M),整个细胞组的平均IC50值是2.8E-03μg/mL(3.3E-09M)。与化合物4相比,化合物1中马来酰亚胺接头的存在不非常显著地改变化合物的细胞毒活性。此外,化合物的细胞毒性与肿瘤细胞系的CD13状态无关(表32)。
表32.化合物1的体外细胞毒性的总结数据
ADC9的细胞毒性
针对不同细胞系测定ADC9的细胞毒活性。在四个不同浓度范围内,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(0.1μg/mL的最大浓度)显示在图19中。
缀合物ADC9显示针对表达CD13+的细胞的显著特异性,其中该化合物表现出与母体化合物4和1相似或比其稍高的细胞毒活性。CD13+细胞系,NB4和U937二者,都显示相当的针对ADC9的敏感性,平均IC50值分别为8.7E-03和2.4E-02μg/mL。两个CD13阴性细胞系,Raji和RPMI-8226,显示显著更低的针对ADC9的敏感性,平均IC50值分别为1.6E+00和5.9E-01μg/mL。平均,CD13+细胞系(平均IC501.66E-02μg/mL)比CD13-细胞(平均IC501.08E+00μg/mL)对ADC9更敏感大约65倍。比较ADC9在NB4细胞(最敏感的)相对Raji细胞(最不敏感)中的活性;发现大约180倍的差异。这些结果明确显示缀合物针对表达CD13的细胞的特异性(表33)。因此,我们猜想,ADC9,至少部分,通过mAb与肿瘤细胞上膜相关CD13受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表33.ADC9的体外细胞毒性的总结数据
为了通过图表比较单独mAb的细胞毒活性与缀合物ADC9的细胞毒活性,显示用单独mAb(50μg/mL)或50或0.1μg/mL的ADC处理不同细胞系后细胞存活百分数的直方图显示在图20中。在50μg/mL的相等浓度,抗CD13抗体单独,不显示针对任何测试的细胞系细胞毒活性,不依赖于其CD13状态。相比之下,ADC9缀合物显示针对所有细胞系的有效细胞毒活性,诱导大于80%的细胞存活抑制。在0.1μg/mL的浓度,缀合物ADC9显示与在50μg/mL观察到的类似的针对CD13阳性细胞的细胞毒活性,但对CD13阴性细胞没有任何可检测的效应。这些结果进一步表明ADC9针对表达CD13的人肿瘤细胞显著的体外细胞毒活性和特异性。
生物活性实施例10-ADC10和相关试剂针对CD13阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD13受体的不同人肿瘤细胞系,包括NB4和U937(CD13阳性细胞)以及Raji和RPMI-8226(CD13阴性细胞)评价ADC10连同母体细胞毒性化合物12和4的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中非常均匀,IC50值在低纳摩尔范围内,从7.9E-05至2.65E-03μg/mL(1.2E-10至4.0E-09M),整个细胞组的平均IC50值是8.4E-04μg/mL(相当于1.2E-09M)。因此,化合物4的细胞毒性非常不依赖于肿瘤细胞系的CD13状态(表34)。
表34.化合物4的体外细胞毒性的总结数据
化合物12的细胞毒性
以DR曲线,使用从01E+00至2.6E-04μg/mL(7.9E-07至2.0E-10M)的十个连续稀释液(1/2.5比例)测定化合物12的活性。化合物12的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中稍微均匀,IC50值在纳摩尔范围内,从4.4E-03至4.8E-02μg/mL(3.5E-09至3.8E-08M),整个细胞组的平均IC50值是2.0E-02μg/mL(1.6E-08M)。与化合物4相比,化合物12中长接头的存在减小(大约1log)化合物的细胞毒活性。此外,化合物的细胞毒性与肿瘤细胞系的CD13状态不相关(表35)。
表35.化合物12的体外细胞毒性的总结数据
ADC10的细胞毒性
针对不同细胞系测定ADC10的细胞毒活性。在四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(1μg/mL的最大浓度)显示在图21中。
缀合物ADC10显示针对表达CD13+的细胞的显著特异性,其中该化合物表现出与母体化合物4和12相似或比其稍高的细胞毒活性。CD13+细胞系,NB4和U937二者均显示相当的针对ADC10的敏感性,平均IC50值分别为7.2E-03和9.8E-03μg/mL。两个CD13阴性细胞系,Raji和RPMI-8226,显示显著更低的针对ADC10的敏感性,平均IC50值分别为1.0E+01和5.3E+00μg/mL。平均,CD13+细胞系(平均IC508.50E-03μg/mL)比CD13-细胞(平均IC507.83E+00μg/mL)对ADC10大约更敏感900倍。比较ADC10在NB4细胞(最敏感的)相对Raji细胞(最不敏感)的活性;发现约1440倍的差异。这些结果明确显示ADC10针对表达CD13的细胞的特异性(表36)。因此,我们猜想,ADC10至少部分通过mAb与肿瘤细胞上膜相关CD13受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表36.ADC10的体外细胞毒性的总结数据
为了通过图表比较单独mAb的细胞毒活性与缀合物ADC10的细胞毒活性,显示用单独mAb(50μg/mL)或50或1μg/mL的ADC处理不同细胞系后的细胞存活百分数的直方图,显示在图22中。在50μg/mL的相等浓度,单独抗CD13抗体,不显示针对任何测试的细胞系细胞毒活性,不依赖于其CD13状态。相比之下,ADC10缀合物显示针对所有细胞系的有效细胞毒活性,诱导大于80%的细胞存活抑制,除了Raji细胞,其中其产生较低的,但仍然重要的约70%的抑制。在1μg/mL的浓度,ADC10显示与在50μg/mL观察到的类似的针对CD13阳性细胞的细胞毒活性,但对CD13阴性细胞无任何可检测效应。这些结果进一步表明ADC10针对表达CD13的人肿瘤细胞的显著的体外细胞毒活性和特异性。
生物活性实施例11-ADC11和相关试剂针对CD13阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD13受体的不同人肿瘤细胞系,包括NB4和U937(CD13阳性细胞)以及Raji和RPMI-8226(CD13阴性细胞)评价ADC11连同母体细胞毒性化合物13和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定化合物40的细胞毒活性。化合物40的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是均匀的,IC50值在低皮摩尔范围内,从3.1E-05至1.7E-04μg/mL(5.2E-11至2.8E-10M),整个细胞组的平均IC50值是8.6E-05μg/mL(相当于1.4E-10M)。的细胞毒性化合物40不依赖于肿瘤细胞系上CD13表达水平(表37)。
表37.化合物40的体外细胞毒性的总结数据
化合物13的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.3E-07至3.4E-11M)的十个连续稀释液(1/2.5比例)测定化合物13的活性。化合物13的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是稍微均匀的,IC50值在纳摩尔范围内,从1.7E-03至1.0E-02μg/mL(2.7E-09至1.4E-08M),整个细胞组的平均IC50值是4.3E-03μg/mL(5.7E-09M)。与化合物40相比,化合物13中含硫醇基尾的存在减小(小于1log)化合物的细胞毒活性。化合物的细胞毒性非常不依赖于肿瘤细胞系的CD13状态(表38)。
表38.化合物的体外细胞毒性的总结数据13
ADC11的细胞毒性
针对不同细胞系测定ADC11的细胞毒活性。在四个不同的浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,在两个独立的实验中测定缀合物。代表性的DR曲线(1μg/mL的最大浓度)显示在图23中。ADC11显示一些,但很少,针对表达CD13的细胞的特异性。缀合物在所有测试的细胞系中具有相当类似的细胞毒活性,除了Raji细胞(其稍不敏感些)。ADC11的活性与母体化合物13的相当,IC50值在低纳摩尔范围内(表39)。
表39.ADC11的体外细胞毒性的总结数据
为了通过图表比较单独抗CD13mAb的细胞毒活性与缀合物ADC11的细胞毒活性,显示单独mAb(50μg/mL)或ADC11(50或1μg/mL)处理不同细胞系后细胞存活百分数的直方图显示在图24中。在50μg/mL的浓度,单独抗CD13抗体,显示一些针对CD13+细胞系的细胞毒活性,产生约30%的细胞存活抑制。在CD13-细胞中,抗体实质上失活。在相同的浓度,ADC11缀合物显示针对所有细胞系的有效细胞毒活性,一些,但非常少的,针对表达CD13的细胞的特异性,其中其诱导几乎100%的细胞存活减少。在CD13-细胞中,ADC11诱导大于80%的细胞存活减少。在1μg/mL的浓度,ADC11显示更多的针对CD13+细胞,NB-4和U937的特异性,其中其分别诱导99和85%的细胞存活减少。在CD13-细胞,Raji和RPMI8226中,缀合物分别诱导38和60%的细胞存活减少。
生物活性实施例12-ADC12和相关试剂针对CD20阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD20抗原的不同人癌细胞系,包括Raji(CD20阳性细胞);RPMI-8226和Karpas-299(CD20阴性细胞)测定ADC12连同母体细胞毒性化合物1,4,以及40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
利妥昔单抗的细胞毒性
首先,针对表达或不表达CD20抗原的不同人癌细胞系,包括Raji(CD20阳性细胞);RPMI-8226和Karpas-299(CD20阴性细胞)测定单独利妥昔单抗的体外细胞毒活性。在范围从5.0E+01至2.62E-05μg/mL(3.4E-07至1.7E-13M)的一式三份的DR曲线中,抗体相当钝化,在任何测试的细胞系中均未达到IC50,不依赖于其CD20状态(表40)。
表40.利妥昔单抗的体外细胞毒性的总结数据
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物化合物40的细胞毒活性。化合物40的细胞毒活性在测试的不同细胞系中是非常均匀的,IC50值在低亚纳摩尔范围内,从8.6E-05至1.1E-04μg/mL(1.4E-10至1.9E-10M),整个细胞组的平均IC50值为9.6E-05μg/mL(相当于1.6E-10M)。化合物40的细胞毒性不依赖于肿瘤细胞系的CD20状态(表41)。
表41.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在测试的不同细胞系中是均匀的,IC50值在纳摩尔范围内,从5.7E-04至1.4E-03μg/mL(8.6E-10至2.1E-09M),整个细胞组的平均IC50值是9.7E-04μg/mL(1.5E-09M)。与化合物40相比,化合物4中含胺基团的存在稍微减小化合物的细胞毒活性。化合物4的细胞毒活性也不依赖于肿瘤细胞系的CD20状态(表42)。
表42.化合物4的体外细胞毒性的总结数据
化合物1的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.2E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定化合物1的活性。化合物1的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是非常均匀的,IC50值在纳摩尔范围内,从1.6E-03至2.8E-03μg/mL(1.9E-09至3.3E-09M),整个细胞组的平均IC50值是2.1E-03μg/mL(2.5E-09M)。与化合物4相比,化合物1中马来酰亚胺接头的存在不显著改变化合物的细胞毒活性。此外,化合物的细胞毒性也不依赖于肿瘤细胞系的CD20状态(表43)。
表43.化合物的体外细胞毒性的总结数据1
ADC12的细胞毒性
针对不同肿瘤细胞系测定ADC12的细胞毒活性。在四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,分别测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图25中。尽管在CD20阳性Raji细胞中较高,ADC12在所有测试的细胞系中显示相对类似的细胞毒活性,在纳摩尔范围内。Raji细胞(CD20+),显示9.5E-02μg/mL的平均IC50值,而RPMI-8226和Karpas-299细胞(均为CD20-)各自的值,分别为4.0E-01和4.1E-01μg/mL(表44)。因此,CD20阳性细胞比CD20阴性细胞对ADC12稍微更不敏感(4倍)。
表44.ADC12的体外细胞毒性的总结数据
为了通过图表比较单独mAb利妥昔单抗的细胞毒活性与缀合物ADC12的细胞毒活性,显示用单独mAb(50μg/mL)或ADC12(1和0.1μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图26中。单独利妥昔单抗在50μg/mL的浓度,在所有测试的细胞系中实质上失活,不依赖于其CD20状态。相比之下,ADC12,在1μg/mL的浓度,在所有测试的细胞系中显示有效细胞毒活性,处理72小时候引起大于70%的细胞存活减少。在更低的浓度,0.1μg/mL,ADC12缀合物显示一些特异性,在CD20阳性细胞(Raji)中诱导约60%的细胞存活减少,而在CD20阴性细胞(RPMI-8226和Karpas-299)中实质上失活。
生物活性实施例13-ADC13和相关试剂针对CD20阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD20抗原的不同人癌细胞系,包括Raji(CD20阳性细胞);RPMI-8226和Karpas-299(CD20阴性细胞)评价ADC13连同母体细胞毒性化合物1,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物化合物40的细胞毒活性。化合物40的细胞毒活性在测试的不同细胞系中非常均匀,IC50值在低亚纳摩尔范围内,从8.6E-05至1.1E-04μg/mL(1.4E-10至1.9E-10M),整个细胞组的平均IC50值是9.6E-05μg/mL(相当于1.6E-10M)。化合物40的细胞毒性不依赖于肿瘤细胞系的CD20状态(表45)。
表45.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在测试的不同细胞系中是均匀的,IC50值在纳摩尔范围内,从5.7E-04至1.4E-03μg/mL(8.6E-10至2.1E-09M),整个细胞组的平均IC50值是9.7E-04μg/mL(1.5E-09M)。与化合物40相比,化合物4中含胺基团的存在稍微减小化合物的细胞毒活性。化合物4的细胞毒活性也不依赖于肿瘤细胞系的CD20状态(表46)。
表46.化合物的体外细胞毒性的总结数据4
化合物12的细胞毒性
以DR曲线,使用从01E+00至2.6E-04μg/mL(7.9E-07至2.1E-10M)的十个连续稀释液(1/2.5比例)测定化合物12的活性。化合物12的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中是非常均匀的,IC50值在纳摩尔范围内,从2.2E-02至6.7E-02μg/mL(1.8E-08至5.5E-08M),整个细胞组的平均IC50值是3.9E-02μg/mL(3.1E-08M)。与化合物4和化合物40相比,化合物12中马来酰亚胺接头的存在减少化合物的细胞毒活性。此外,化合物的细胞毒性也不依赖于肿瘤细胞系的CD20状态(表47)。
表47.化合物的体外细胞毒性的总结数据12
ADC13的细胞毒性
针对不同肿瘤细胞系测定ADC13的细胞毒活性。在四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,分别测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图27中。尽管在CD20阳性Raji细胞中较高,ADC13在所有测试的细胞系显示类似的细胞毒活性,在纳摩尔范围内。Raji细胞(CD20+),显示2.5E-01μg/mL的平均IC50值,而RPMI-8226和Karpas-299细胞(均为CD20-)各自的值是1.1E+00μg/mL(表48)。因此,CD20阳性细胞比CD20阴性细胞对ADC13稍微更敏感(约5倍)。
表48.ADC13的体外细胞毒性的总结数据
为了通过图表比较单独mAb利妥昔单抗的细胞毒活性与缀合物ADC13的细胞毒活性,显示用单独mAb(50μg/mL)或ADC13(1和0.1μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图28中。单独利妥昔单抗,在50μg/mL的浓度,在所有测试的细胞系中实质上失活,不依赖于其CD20状态。相比之下,在1和0.1μg/mL ADC13显示细胞毒活性,对表达CD20的Raji细胞具有一些特异性。在1μg/mL,处理72小时后,ADC13引起,大于65%的Raji细胞(CD20+)的细胞存活的减小,而在RPMI-8226和Karpas-299细胞(CD20-)中分别诱导35-45%。在0.1μg/mL,ADC13缀合物显示更明确的特异性,在CD20+细胞中诱导约50%的细胞存活减少,而在CD20-细胞中失活。
生物活性实施例14-ADC14和相关试剂针对CD5阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD5抗原的不同人癌细胞系,包括Karpas-299和MOLT-4(二者均为CD5+);Raji和RPMI-8226(二者均为CD5-)评价ADC14连同母体细胞毒性化合物1,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
抗CD5mAb的细胞毒性
首先,针对表达或不表达CD5抗原的不同人癌细胞系,包括Karpas-299和MOLT-4(二者均为CD5+);Raji和RPMI-8226(二者均为CD5-)测定单独抗CD5小鼠mAb的体外细胞毒活性。在范围从5.0E+01至1.3E-02μg/mL(3.3E-07至8.7E-11M)的一式三份的DR曲线中,在两个独立的实验中,抗体实质上失活,在任何测试的细胞系中均未达到IC50,不依赖于其CD5状态(表49)。
表49.抗CD5mAb的体外细胞毒性的总结数据
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物40的细胞毒活性。化合物40的细胞毒活性在测试的不同细胞系中非查功能均匀,IC50值在低的纳摩尔范围内,从7.5E-05至3.6E-04μg/mL(1.2E-10至5.9E-10M),整个细胞组的平均IC50值是1.6E-04μg/mL(相当于2.6E-10M)。化合物40的细胞毒性不依赖于肿瘤细胞系中CD5表达水平(表50)。
表50.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在测试的不同细胞系中是均匀的,IC50值在纳摩尔范围内,从6.3E-04至2.7E-03μg/mL(9.5E-10至4.1E-09M),整个细胞组的平均IC50值1.3E-03μg/mL(1.9E-09M)。与化合物40相比,化合物4中含胺基团的存在减少化合物的细胞毒活性(约1log),化合物4的细胞毒活性也不依赖于肿瘤细胞系的CD5表达水平(表51)。
表51.化合物4的体外细胞毒性的总结数据
化合物1的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.2E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定化合物1的活性。化合物1的细胞毒活性在测试的不同细胞系中几乎均匀,除了Raji细胞,其中化合物活性较低,IC50值在纳摩尔范围内,从1.8E-03至1.1E-02μg/mL(2.2E-09至1.3E-09M),整个细胞组的平均IC50值是4.6E-03μg/mL(5.3E-09M)。与化合物4相比,化合物1中含有马来酰亚胺的接头的存在不改变化合物的细胞毒活性。化合物的细胞毒性也不依赖于肿瘤细胞系中CD5表达水平(表52)。
表52.化合物的体外细胞毒性的总结数据1
ADC14的细胞毒性
针对不同肿瘤细胞系测定ADC14的细胞毒活性。在四个不同浓度范围内,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,分别测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图29中。ADC14显示一些针对CD5阳性细胞的选择性的趋势,尽管中纳摩尔范围内的平均IC50值对于所有测试的细胞系相对类似,独立地于CD5状态(表53)。
表53.ADC14的体外细胞毒性的总结数据
为了通过图表比较单独抗CD5mAb的细胞毒活性与缀合物ADC14的细胞毒活性,显示单独mAb(50μg/mL)或ADC14(1μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图30中。单独抗CD5mAb,在50μg/mL的浓度,在所有测试的细胞系中实质上失活。ADC14,在1μg/mL的浓度,显示针对CD5阳性细胞,Karpas-299和MOLT-4的特异细胞毒活性,分别诱导约84%和70%的细胞存活抑制,而针对CD5阴性细胞实质上失活(图30)。
生物活性实施例15-ADC16和相关试剂针对CD4阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD4抗原的不同人癌细胞系,包括Karpas-299和U937(二者均为CD4+);Raji和RPMI-8226(二者均为CD4-)评价ADC16连同母体细胞毒性化合物1,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
抗CD4mAb的细胞毒性
首先,针对表达或不表达CD4抗原的不同人癌细胞系,包括Karpas-299和U937(二者均为CD4+);Raji和RPMI-8226(二者均为CD4-)测定单独抗CD4小鼠mAb的体外细胞毒活性。在范围从5.0E+01至1.3E-02μg/mL(3.3E-07至8.7E-11M)的一式三份的DR曲线,在两个独立的实验中,抗体实质上失活,在任何测试的细胞系中未达到IC50,不依赖于其CD4状态(表54)。
表54.抗CD4mAb的体外细胞毒性的总结数据
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物化合物40的细胞毒活性。化合物40的细胞毒活性在测试的不同细胞系中是均匀的,IC50值在低亚纳摩尔范围内,从7.9E-05至2.8E-04μg/mL(1.3E-10至4.7E-10M),整个细胞组的平均IC50值是1.5E-04μg/mL(相当于2.5E-10M)。化合物40的细胞毒性不依赖于肿瘤细胞系的CD4表达水平(表55)。
表55.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在测试的不同细胞系中也是均匀的,IC50值在纳摩尔范围内,从6.1E-04至2.7E-03μg/mL(9.2E-10至4.1E-09M),整个细胞组的平均IC50值是1.2E-03μg/mL(1.8E-09M)。与化合物40相比,化合物4中含胺基团的存在减少化合物的细胞毒活性。化合物4的细胞毒活性也不依赖于肿瘤细胞系中CD4表达水平(表56)。
表56.化合物4的体外细胞毒性的总结数据
化合物1的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.2E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定化合物1的活性。化合物1的细胞毒活性在测试的不同细胞系中也是均匀的,IC50值在纳摩尔范围内,从1.5E-03至7.3E-03μg/mL(1.7E-09至8.6E-09M),整个细胞组的平均IC50值3.4E-03μg/mL(3.9E-09M)。与化合物4相比,化合物1中含马来酰亚胺的接头的存在不改变化合物的细胞毒活性。化合物的细胞毒性也不依赖于肿瘤细胞系中CD4表达水平(表57)。
表57.化合物的体外细胞毒性的总结数据1
ADC16的细胞毒性
针对不同肿瘤细胞系测定ADC16的细胞毒活性。在四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,分别测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图31中。ADC16针对CD4阳性细胞存在一些特异性,尽管对CD4阴性细胞的敏感性的平均差异相对低,大约7倍(是较不敏感和最敏感的细胞系,Raji和Karpas-299之间的最大差异,分别为约14倍)(表58)。尽管显示小的治疗窗口,可能的是,观察到的ADC16的至少部分细胞毒性由mAb和肿瘤细胞的细胞膜的CD4糖蛋白的相互作用介导。
表58.ADC16的体外细胞毒性的总结数据
为了通过图表比较单独抗CD4mAb的细胞毒活性与缀合物ADC16的细胞毒活性,显示mAb单独(50μg/mL)或ADC16(1和0.1μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图32中。单独抗CD4mAb,在50μg/mL的浓度,在所有测试的细胞系中实质上失活。相比之下,ADC16,在1μg/mL的浓度,在所有测试的细胞系中显示有效细胞毒活性,不依赖于其CD4状态,处理72小时后引起大于60%的细胞存活的减少(范围为60-90%)。甚至在0.1μg/mL的浓度,ADC16显示针对CD4阳性细胞,Karpas-299和U937的特异细胞毒活性,分别引起约80%和70%的细胞存活抑制,而针对CD4阴性细胞失活(图32)。
生物活性实施例16-ADC17和相关试剂针对CD4阳性和阴性人肿瘤细胞的细胞毒性
针对表达或不表达CD4抗原的不同人癌细胞系,包括Karpas-299和U937(二者均为CD4+);Raji和RPMI-8226(二者均为CD4-)评价ADC17连同母体细胞毒性化合物12,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物40的细胞毒活性。化合物40的细胞毒活性在测试的不同细胞系中是均匀的,IC50值在低亚纳摩尔范围内,从7.9E-05至2.8E-04μg/mL(1.3E-10至4.7E-10M),整个细胞组的平均IC50值是1.5E-04μg/mL(相当于2.5E-10M)。化合物40的细胞毒性不依赖于肿瘤细胞系中CD4表达水平(表59)。
表59.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在测试的不同细胞系中也是均匀的,IC50值在纳摩尔范围内,从6.1E-04至2.7E-03μg/mL(9.2E-10至4.1E-09M),整个细胞组的平均IC50值是1.2E-03μg/mL(1.8E-09M)。与化合物40相比,化合物4中含胺基团的存在减少化合物的细胞毒活性。化合物4的细胞毒活性也不依赖于肿瘤细胞系中CD4表达水平(表60)。
表60.化合物的体外细胞毒性的总结数据4
化合物12的细胞毒性
以DR曲线,使用从01E+00至2.6E-04μg/mL(7.9E-07至2.1E-10M)的十个连续稀释液(1/2.5比例)测定化合物12的活性。化合物12的细胞毒活性,在两个独立的实验中,在测试的不同细胞系中相对均匀,IC50值在纳摩尔范围内,从7.3E-02至4.1E-01μg/mL(5.8E-08至3.2E-07M),整个细胞组的平均IC50值是1.7E-01μg/mL(1.3E-07M)。与化合物40(几乎3log)和化合物4(几乎2log)相比,化合物12中长的含马来酰亚胺接头的存在强烈减少化合物的细胞毒活性。此外,化合物的细胞毒性也不依赖于肿瘤细胞系的CD4表达水平(表61)。
表61.化合物的体外细胞毒性的总结数据12
ADC17的细胞毒性
针对不同肿瘤细胞系测定ADC17的细胞毒活性。在四个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从50,10,1和0.1μg/mL开始,分别测定缀合物。代表性的DR曲线(起始浓度1μg/mL)显示在图33中。ADC17显示针对CD4阳性细胞的特异性,关于CD4阴性细胞的敏感性的平均差异为约40倍(11-64倍之间的范围)(表62)。可能观察到的ADC17的细胞毒活性的主要部分由mAb和肿瘤细胞细胞膜中的CD4糖蛋白的相互作用介导。
表62.ADC17的体外细胞毒性的总结数据
为了通过图表比较单独抗CD4mAb的细胞毒活性与缀合物ADC17的细胞毒活性,显示用mAb单独(50μg/mL)或ADC17(1和0.1μg/mL)处理不同细胞系后的细胞存活百分数的直方图,显示在图34中。单独抗CD4mAb,在50μg/mL的浓度,在所有测试的细胞系中实质上失活。相比之下,ADC17,在1ug/mL的浓度,在测试的四个细胞系的三个中(除了Raji细胞)显示有效细胞毒活性,在处理72小时后,引起大于70%的细胞存活的减少(范围为72-95%)。甚至在0.1μg/mL的浓度,ADC17显示针对CD4阳性细胞的特异性细胞毒活性,Karpas-299和U937,分别诱导约70%和75%的细胞存活抑制,而针对CD4阴性细胞非常钝化(图34)。
生物活性实施例17-ADC14和相关试剂针对具有高或无CD5表达的Raji细胞克隆的细胞毒性
针对表达或不表达CD5抗原的Raji细胞克隆评价ADC14连同母体细胞毒性化合物1,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
抗CD5mAb的细胞毒性
针对表达(C#10)或不表达(C#18)CD5抗原的Raji细胞克隆测定单独抗CD5小鼠mAb的体外细胞毒活性。在范围从5.0E+01至1.3E-02μg/mL(3.3E-07-8.7E-11M)的一式三份的DR曲线中,在两个独立的实验中,抗体实质上失活,在任何测试的细胞系中均未达到IC50,不依赖于其CD5状态(表63)。
表63.抗CD5mAb的体外细胞毒性的总结数据
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物40的细胞毒活性。化合物40的细胞毒活性在表达CD5的(克隆#10)和不表达CD5(克隆#18)的Raji细胞之间相对类似,平均IC50值在亚纳摩尔范围内,分别为4.95E-04和8.90E-04μg/mL(相当于8.17E-10和1.47E-09M)。尽管在CD5阳性细胞中稍高,化合物40的细胞毒性似乎非常不依赖于肿瘤细胞系中的CD5表达水平(表64)
表64.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR响应曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在表达CD5的(克隆#10)和不表达CD5(克隆#18)的Raji细胞之间相对类似,尽管在零表达(null)细胞中,化合物未达到IC50值。在CD5阳性细胞中,该化合物显示9.9E-03μg/mL(相当于1.57E-08M)的平均IC50值。尽管在CD5阳性细胞中稍高,化合物4的细胞毒性似乎相当不依赖于肿瘤细胞系中的CD5表达水平(参见作为参考的表65中的IC20值)。
表65.化合物4的体外细胞毒性的总结数据
化合物1的细胞毒性
以DR曲线,使用从01E-01至2.6E-05μg/mL(1.2E-07至3.0E-11M)的十个连续稀释液(1/2.5比例)测定化合物1的活性。在两个Raji细胞克隆之间化合物1的细胞毒活性不同,在CD5过表达的细胞(克隆#10)中比在无CD5的细胞(克隆#18)中更有活性(约1log),平均IC50值分别为2.9E-03和3.8E-02μg/mL(相当于3.4E-09和4.4E-08M)(表66)。在该情况下化合物1的细胞毒性似乎不依赖于肿瘤细胞系的CD5状态。
表66.化合物1的体外细胞毒性的总结数据
ADC14的细胞毒性
针对两个Raji克隆测定ADC14的细胞毒活性。在三个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从10,1和0.1μg/mL开始测定缀合物。代表性的DR曲线(起始浓度10μg/mL)显示在图35中。ADC 14显示针对过表达CD5的细胞(克隆#10)的特异性,其中该化合物表现出与母体化合物1,4和40类似的或甚至更高的细胞毒活性。在表达CD5的Raji细胞中,该缀合物显示1.6E-01μg/mL的平均IC50值。在无CD5的细胞中,缀合物比在CD5阳性细胞中活性低50倍,显示9.0E+00μg/mL的平均IC50值。尽管有一些保留,由于在两个Raji细胞克隆之间观察到的针对一些母体化合物的一些差异的敏感性,这些结果表明,ADC14针对表达CD5的细胞具有特异性(表67)。因此,我们猜想,ADC14至少部分通过mAb与肿瘤细胞上的膜相关CD5受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表67.ADC14的体外细胞毒性的总结数据
为了通过图表比较单独抗CD5mAb的细胞毒活性与缀合物ADC14的细胞毒活性,显示用mAb单独(50μg/mL)或ADC14(10μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图36中。单独抗CD5mAb,在50μg/mL的浓度,针对两个Raji细胞克隆无活性,不依赖于其CD5状态。相反,ADC14,在10μg/mL的浓度,显示有效和稍微选择性的针对CD5阳性Raji细胞(克隆#10)的细胞毒活性,在处理72小时后引起几乎90%的其细胞存活的减少。在相同条件下,ADC14引起30%的无CD5细胞(克隆#18)的细胞存活的减少(图36)。
生物活性实施例18-ADC15和相关试剂针对具有高或无CD5表达的Raji细胞克隆的细胞毒性
针对表达或不表达CD5抗原Raji细胞克隆评价ADC15连同母体细胞毒性化合物12,4,和40的体外细胞毒活性。进行72小时的标准剂量响应(DR)曲线。
化合物40的细胞毒性
以DR曲线,使用从01E-03至2.6E-07μg/mL(1.7E-09至4.3E-13M)的十个连续稀释液(1/2.5比例)测定母体化合物40的细胞毒活性。化合物40的细胞毒活性在表达CD5的(克隆#10)和不表达CD5(克隆#18)的Raji细胞之间相对类似,平均IC50值在亚纳摩尔范围内,分别为4.95E-04和8.90E-04μg/mL(相当于8.17E-10和1.47E-09M)。尽管在CD5阳性细胞中稍高,化合物40的细胞毒性似乎相当不依赖于肿瘤细胞系的CD5表达水平(表68)。
表68.化合物40的体外细胞毒性的总结数据
化合物4的细胞毒性
以DR响应曲线,使用从01E-02至2.6E-06μg/mL(1.5E-08至4.0E-12M)的十个连续稀释液(1/2.5比例)测定化合物4的细胞毒活性。化合物4的细胞毒活性在表达CD5的(克隆#10)和不表达CD5(克隆#18)的Raji细胞之间相对类似,尽管在零表达细胞中该化合物未达到IC50值。在CD5阳性细胞中,该化合物显示9.9E-03μg/mL(相当于1.57E-08M)的平均IC50值。尽管在CD5阳性细胞中稍高,化合物4的细胞毒性似乎相当不依赖于肿瘤细胞系中的CD5表达水平(参见作为参考表69中的IC20值)。
表69.化合物的体外细胞毒性的总结数据4
化合物12的细胞毒性
以DR曲线,使用从01E+00至2.6E-4μg/mL(7.9E-07至2.1E-10M)的十个连续稀释液(1/2.5比例)测定化合物12的活性。化合物12的细胞毒活性在表达CD5的(克隆#10)和不表达CD5的(克隆#18)Raji细胞之间相对类似,尽管在零表达细胞中,该化合物未达到IC50值。在CD5阳性细胞中,该化合物显示2.7E-01μg/mL(相当于2.15E-07M)的平均IC50值。尽管在CD5阳性细胞中稍高,化合物12的细胞毒性似乎相当不依赖于肿瘤细胞系中CD5表达水平(参见作为参考的表70中的IC20值)。
表70.化合物的体外细胞毒性的总结数据12
ADC15的细胞毒性
针对两个Raji克隆测定ADC15的细胞毒活性。在三个不同浓度范围,各自以一式三份的DR曲线(十个连续稀释液,1/2.5比例),从10,1和0.1μg/mL开始测定缀合物。代表性的DR曲线(起始浓度10μg/mL)显示在图37中。ADC15显示针对过表达CD5的细胞(克隆#10)的显著特异性,其中该化合物显示出与母体化合物40类似并且甚至比化合物4和12高的细胞毒活性。在表达CD5的Raji细胞中,该缀合物显示9.3E-01μg/mL的平均IC50值。在无CD5的细胞中,该缀合物比在CD5阳性细胞中活性小远大于10倍,未达到IC50值。尽管有所保留,由于在两个Raji细胞克隆之间观察到的针对母体化合物4和12的潜在敏感性,这些结果表示,ADC15针对表达CD5的细胞具有特异性(表71)。我们可以假想ADC15至少部分通过mAb与肿瘤细胞上的膜相关CD5受体的相互作用,和随后细胞内递送细胞毒性药物到靶组织中起作用。
表71.ADC15的体外细胞毒性的总结数据
为了通过图表比较单独抗CD5mAb的细胞毒活性与缀合物ADC15的细胞毒活性,显示用单独mAb(50μg/mL)或ADC15(10μg/mL)处理不同细胞系后细胞存活百分数的直方图,显示在图38中。单独抗CD5mAb,在50μg/mL的浓度,针对两种Raji细胞克隆无活性,不依赖于其CD5状态。相反,ADC15在10μg/mL的浓度显示有效和选择性的针对CD5阳性Raji细胞(克隆#10)的细胞毒活性,在处理72小时后引起细胞存活的80%的减少。在相同条件下,ADC15在无CD5细胞(克隆#18)上无活性(图38)。
- 还没有人留言评论。精彩留言会获得点赞!