4-苄磺酰基-2-丁腈的制作方法
本发明涉及4-苄磺酰基-2-丁腈,或其药学上可接受的盐或溶剂合物。本发明还涉及包含该化合物和药学上可接受的载体的药物组合物。
附图简要说明
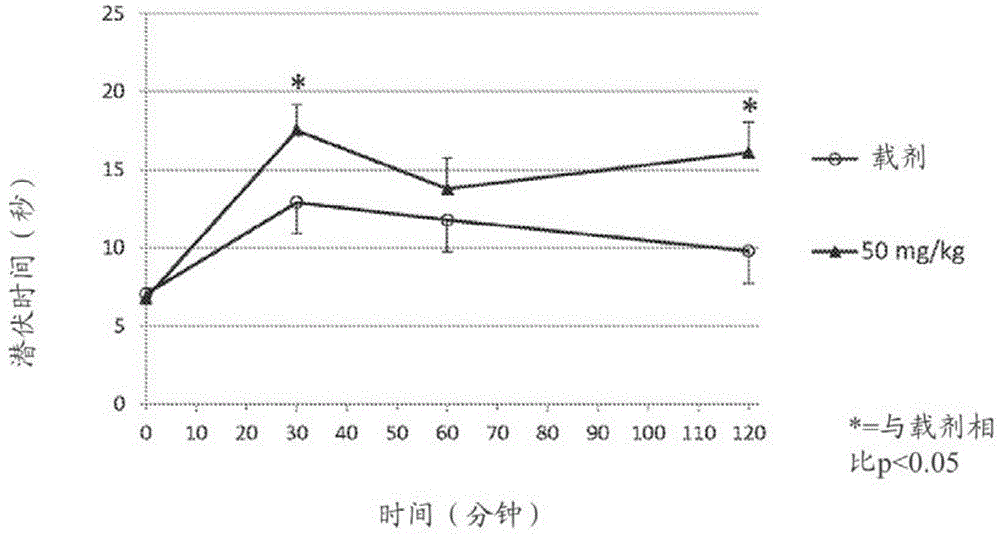
图1显示通过腹膜内施用使用载剂(DMSO)、试验化合物(载剂中50mg/kg)治疗小鼠的甩尾试验结果。以平均值±SEM计算各组的潜伏时间并对时间作图,其中*表示与载剂治疗的小鼠相比p值小于0.05。
发明详述
定义
本文中,“药学上可接受的盐”为保留母体化合物的所需的生物活性并不赋予不需要的毒理学作用的盐。药学上可接受的盐形式包括不同盐的多种结晶多晶型物及无定形形式。药学上可接受的盐可与金属或有机抗衡离子形成并包括,但不限于,碱金属盐例如钠或钾;碱土金属盐例如镁或钙;及铵或四烷基铵盐,即,NX4+(其中X为C1-4)。
本文中,“溶剂合物”为其中化合物以某个固定比例与可接受的共溶剂组合的加成复合物。共溶剂包括但不限于乙酸乙酯、乳酸肉荳蔻基酯、肉荳蔻酸异丙酯、乙醇、1-丙醇、异丙醇、1-丁醇、异丁醇、叔丁醇、丙酮、甲乙酮和二乙醚。
4-苄磺酰基-2-丁腈
发明人合成并鉴定了4-苄磺酰基-2-丁腈。该化合物或其药学上可接受的盐或溶剂合物可有效地治疗炎症、炎症相关病症和疼痛。
4-苄磺酰基-2-丁腈的分子量为221.28,且其反式和顺式结构如下所示。
可通过在压力下加热4-溴-2-丁烯腈及苄基亚磺酸钠(sodium benzylsulfinate)的水溶液来合成4-苄磺酰基-2-丁腈。必需品4-溴-2-丁烯腈可通过烯丙基氰的溴化和随后的碱催化的HBr消除来制备,其生成E-和Z-异构体的约1:1混合物。
药物组合物
本发明提供药物组合物,其包含一种或多种药学上可接受的载体和活性化合物4-苄磺酰基-2-丁腈或其药学上可接受的盐或溶剂合物。该药物组合物可包含顺式或反式异构体之一,或等摩尔或不同量的两种异构体。对于局部制剂而言,药物组合物中活性化合物或其药学上可接受的盐或溶剂合物的量通常为约0.01-20%、或0.05-20%、或0.1-20%、或0.2-15%、或0.5-10%、或1-5%(w/w);对于可注射制剂而言为约0.1-5%,对于贴片制剂而言为0.1-5%,对于片剂制剂而言为约1-90%,且对于胶囊制剂而言为1-100%。
在一个实施方式中,将活性化合物并入任何可接受的载体中,包括霜剂、凝胶剂、洗剂或其它类型的悬浮剂(其可稳定活性化合物)并通过局部施用将其递送到受影响区域。在另一个实施方式中,药物组合物的剂型可以是例如片剂、胶囊剂、颗粒剂、细颗粒剂、粉末剂、糖浆剂、栓剂、可注射溶液剂、贴片等。可利用常规方法制备以上的药物组合物。
药学上可接受的载体,作为无活性成分,可由本领域技术人员利用常规标准选择。药学上可接受的载体包括,但不限于,非水基溶液、悬浮液、乳液、微乳液、胶束溶液、凝胶及软膏。药学上可接受的载体也可包括,但不限于以下的成分:盐水及电解质水溶液;离子及非离子渗透剂例如氯化钠、氯化钾、甘油及右旋糖;pH调节剂及缓冲剂例如氢氧化物、磷酸盐、柠檬酸盐、乙酸盐、硼酸盐的盐;及三乙醇胺;抗氧化剂例如亚硫酸氢盐、亚硫酸盐、偏亚硫酸氢盐、硫代亚硫酸盐、抗坏血酸、乙酰基半胱氨酸、半胱氨酸、谷胱甘肽、丁基化羟基苯甲醚、生育酚及棕榈酸抗坏血酸酯的盐、酸和/或碱;表面活性剂例如卵磷脂、磷脂,包括但不限于磷脂酰胆碱、磷脂酰乙醇胺及磷脂酰基肌醇;泊洛沙姆及保丽视明,聚山梨醇酯例如聚山梨醇酯80、聚山梨醇酯60及聚山梨醇酯20,聚醚例如聚乙二醇及聚丙二醇;聚乙烯例如聚乙烯醇及聚维酮;纤维素衍生物例如甲基纤维素、羟丙基纤维素、羟乙基纤维素、羧甲基纤维素及羟丙基甲基纤维素及其盐;石油衍生物例如矿物油及白矿脂;脂肪例如羊毛脂、花生油、棕榈油、大豆油;单、二及三酸甘油酯;丙烯酸的聚合物例如羧基聚甲烯凝胶及疏水改性的交联的丙烯酸酯共聚物;多糖例如葡聚糖及糖胺聚糖例如透明质酸钠。其他药学上可接受的载体包括黄原胶、卡拉胶、Avicel RC-591(微晶纤维素和的组合)和聚乙二醇。或者,可将活性化合物溶解或悬浮在药学上可接受的脂质制剂中,该脂质制剂是例如Kalepu等(Acta Pharmaceutica Sinica B,3:361-372,2013)描述的那些,例如植物油、椰子油、蓖麻油等。
这类药学上可接受的载体可利用熟知的防腐剂进行防腐对抗细菌污染,这些防腐剂包括,但不限于,苯扎氯铵、乙二胺四乙酸及其盐、苯索氯铵、氯己定、氯丁醇、对羟基苯甲酸甲酯、硫柳汞及苯乙醇,或可配制成单用途或多用途的非防腐的制剂。
例如,活性化合物的片剂制剂或胶囊剂制剂可包含没有生物活性并与活性化合物不发生反应的其它赋形剂。片剂或胶囊剂的赋形剂可包括填充剂、粘合剂、润滑剂及助流剂、崩解剂、湿润剂及释放速率调节剂。粘合剂促进制剂颗粒的粘附并对于片剂制剂而言是重要的。片剂或胶囊剂的赋形剂的示例包括但不限于:羧甲基纤维素、纤维素、乙基纤维素、羟丙基甲基纤维素、甲基纤维素、剌梧桐树胶(karaya gum)、淀粉、黄蓍胶、明胶、硬脂酸镁、二氧化钛、聚(丙烯酸)及聚乙烯吡咯烷酮。例如,片剂制剂可含有无活性成分如胶体二氧化硅、交聚维酮、羟丙甲纤维素、硬脂酸镁、微晶纤维素、聚乙二醇、羧甲淀粉钠和/或二氧化钛。胶囊剂制剂可含有无活性成分如明胶、硬脂酸镁和/或二氧化钛。
例如,活性化合物的贴片制剂可包含一些无活性成分,如1,3-丁二醇、氨基乙酸二羟基铝、乙二胺四乙酸二钠、D-山梨糖醇、明胶、高岭土、对羟基苯甲酸甲酯、聚山梨醇酯80、聚维酮、丙二醇、对羟基苯甲酸丙酯、羧甲基纤维素钠、聚丙烯酸钠、酒石酸、二氧化钛及纯净水。贴片制剂也可含有皮肤渗透性增强剂例如乳酸酯或二乙二醇单乙醚。
包含活性化合物的局部制剂可以是凝胶剂、霜剂、洗剂、液体剂、乳剂、软膏剂、喷雾剂、溶液剂及悬浮剂的形式。局部制剂中的无活性成分例如包括,但不限于,(润肤剂/渗透增强剂)、二乙二醇单乙醚(润肤剂/渗透增强剂)、DMSO(增溶剂)、硅酮弹性体(流变/质地改性剂)、辛/癸酸甘油三酯(润肤剂)、辛水杨酯(润肤剂/UV滤光剂)、硅酮流体(润肤剂/稀释剂)、角鲨烯(润肤剂)、向日葵油(润肤剂),及二氧化硅(增稠剂)。
在一个实施方式中,二乙二醇单乙醚包含在局部凝胶制剂中。
使用方法
炎症是一种由免疫系统先天性及后天性部分活性的激活及延续引起的过程及组织病理学状态。细胞与细胞相互作用中花生四烯酸级联以及细胞因子产生和作用是免疫激活及应答的关键组分,这导致炎症。花生四烯酸残留在许多细胞膜中。当花生四烯酸从膜中裂解时,其可产生多种已知的类花生酸,包括前列腺素及白三烯,这些为已知的促炎性实体。
该活性化合物可在体外有效地抑制人外周血单核细胞中促炎性细胞因子释放(例如IL-1β、IL-6、TNFα、IL-4和IFNγ)。在局部施用于小鼠耳朵肿胀模型时,该活性化合物是抗炎的,其中炎症由花生四烯酸引起。
本发明涉及一种治疗炎症和/或疼痛的方法。4-苄磺酰基-2-丁腈可不加改变使用,或其可以另外含有药学上可接受的载体的药物组合物的形式给予。该方法包括以下步骤:首先确定罹患炎症和/或疼痛的对象,并向该对象给予有效量的活性化合物,所述有效量可治疗炎症和/或疼痛。本文中,“有效量”为通过缓解病理状况或减轻疾病症状来有效地治疗疾病的量。
在一个实施方式中,该方法减轻或缓解与炎症相关的症状。本发明提供一种治疗炎症的局部表现的方法,所述炎症的局部表现的特征在于急性或慢性肿胀、疼痛、发红、温度升高或在一些情况中功能丧失。
在另一个实施方式中,本发明提供一种缓解疼痛症状的方法,其与疼痛的起因无关。可通过本发明的方法治疗的通用术语“疼痛”包括伤害性、神经性及混合型。本发明减轻不同严重程度的疼痛,即轻度、中度和重度疼痛;急性及慢性疼痛。本发明有效地治疗关节疼痛、肌肉疼痛、腱疼痛、烧伤疼痛及炎症(例如风湿性关节炎)引起的疼痛。
在一个实施方式中,本发明可用于治疗肌骨骼系统中相关的炎症和/或疼痛。高度神经支配的肌骨骼系统具有显示疼痛的高能力。此外,肌骨骼系统具有组织肿胀的高能力。在肌骨骼系统中,组织损伤程度通常被放大,其与所致炎症反应不成比例。
本发明提供一种治疗炎症和/或疼痛相关的炎性骨骼或肌肉疾病或病症的方法。该方法包括以下步骤:确定有此需要的对象,并将有效量的活性化合物给予给该对象,所述有效量可治疗炎症和/或疼痛。本文中,
“活性化合物”旨在包括该化合物(顺式异构体、反式异构体,或其混合物)及其药学上可接受的盐或溶剂合物。骨骼或肌肉疾病或病症包括肌骨骼扭伤、肌骨骼过劳、跟腱炎、外周神经根病、骨关节炎、关节退行性疾病、风湿性多肌痛、幼年关节炎、痛风、强直性脊柱炎、牛皮癣关节炎、系统性红斑狼疮、肋软骨炎、腱炎、滑囊炎(例如常见外上髁炎(网球肘)、内上髁炎(投手肘)及大粗隆滑囊炎)、颞下颌关节综合征及纤维肌痛。
在一个实施方式中,本发明涉及一种治疗炎症和/或疼痛相关痛风的方法。痛风是一种慢性炎症疾病,其特征是关节处复发性、突发性和严重的急性炎症攻击(发红和触痛)和疼痛,通常发生于大拇趾基部。痛风由血液中尿酸水平升高所导致。痛风是一种关节炎类型。一些人可发展出慢性痛风,其也称作痛风性关节炎(gouty arthritis)。
本发明的药物组合物可通过局域性给药(local administration)及全身给药加以施用。局域性给药包括局部给药。全身给药包括口服给药、肠胃外(例如静脉内、肌内、皮下或直肠)及其它全身性给药途径。在全身给药中,活性化合物首先到达血浆然后分布到目标组织中。局部给药和口服给药是本发明的优选的给药途径。
组合物的剂量可根据损伤程度及各患者的个体反应变化。对于全身给药,递送的活性化合物的血浆浓度可变化;但一般为1x10-10-1x10-4摩尔/升,且优选1x10-8-1x10-5摩尔/升。
在一个实施方式中,将组合物局部施用于受影响区域上并擦入其中。组合物局部施用的频率为至少每天1或2次,或每天3至4次,这取决于医学问题及疾病病理是慢性还是急性。一般来讲,外用组合物包括约0.01-20%、或0.05-20%、或0.1-20%、或0.2-15%、0.5-10、或1-5%(w/w)的活性化合物。例如,局部组合物包含约1或5%(w/w)的活性化合物。根据受影响区域的大小,将每剂量0.2-85mL(通常0.2-10mL)的局部组合物施用于个体。活性化合物穿过皮肤并被递送到不适部位。
在一个实施方式中,将药物组合物经口服给予对象。口服给药的剂量一般为至少0.1mg/kg/天且小于100mg/kg/天。例如,对于人对象而言,口服给药的剂量是0.1-100或0.5-50mg/kg/天,且优选1-20或1-10mg/kg/天。例如,在人对象中,口服给药的剂量是20-1000mg/天,且优选20-500、20-100、25-200、50-500、50-200、100-600、100-400或200-800kg/天。
在一个实施方式中,将药物组合物经静脉内给予对象。静脉内推注注射(bolus injection)或静脉内输注的剂量通常是0.03-20且优选0.03-10mg/kg/天。
在一个实施方式中,将药物组合物经皮下给予对象。皮下给药的剂量一般为0.3-20,且优选0.3-3mg/kg/天。
本领域技术人员应理解,多种递送机理也适于本发明。
本发明用于治疗哺乳动物对象,例如人、马及狗。本发明特别可以用于治疗人。
以下实施例进一步说明本发明。这些实施例只是用于说明本发明而不应被视为限制。
实施例
实施例1.4-苄磺酰基-2-丁腈的制备
(A)依次使用溴(0.59mole)的叔戊醇(60mL)溶液和乙醇钠的乙醇溶液(345mL,0.60mole)处理烯丙基氰(0.59mole)的叔戊醇(120mL)和石油醚(370mL)溶液。在将反应混合物冷却至室温时,通过真空过滤除去固体并减压浓缩滤液。将残留液体装载至硅胶柱(307.10g)上并使用己烷-乙酸乙酯(19:1;9:1)洗脱。合并适当组分,随后减压浓缩以生成淡黄色液体状的4-溴-2-丁烯腈(47.14g)。
(B)室温下将苄磺酰氯(10.5mmol)一次性加入50mL压力容器内搅拌中的亚硫酸钠(10.5mmol)和碳酸氢钠(20.9mmol)的水(20mL)溶液中。几分钟后,将该容器置于50℃的油浴中,并持续搅拌2小时,之后将容器移出油浴,紧密密封并在室温下储存过夜。
(C)将部分(A)的产物(20.4mmol)添加至含有部分(B)中制备的水溶液的容器中。将该容器紧密密封,置于110℃的油浴中,并持续加热/搅拌12小时。将该容器在室温下放置过夜后,通过真空过滤收集棕色固体,并用水洗涤。空气干燥后,在丙酮(50mL)中溶解固体,并使用活性炭(1g)处理。通过Whatman GF/F玻璃纤维滤器真空过滤混合物并将滤液减压浓缩至较浅棕色固体,将该固体在约80℃下边搅拌边溶解在乙酸(20mL)和水(48mL)的混合物中。将含有溶液的烧瓶密封并在室温下储存过夜。通过真空过滤收集米色(beige-colored)晶状固体,并使用冰冷的乙酸水溶液洗涤,并空气干燥。
产率:1.367g(72%回收率;59%分离产率,基于苄磺酰氯)。mp:89.4-91.4℃;FTIR:2226cm-1(CN);根据C11H11NO2S计算:C,59.71;H,5.01;N,6.33。实测:C,59.64;H,4.87;N,6.35。
实施例2.NMR谱
由光谱数据服务公司(Spectral Data Services,Inc.(SDS))在400MHz下在CDCl3溶液中获取4-苄磺酰基-2-丁腈的1H NMR谱。表1显示NMR结果的化学位移数据。
4-苄磺酰基-2-丁腈的结构包含11个质子。所有质子都具有且存在合理的化学位移。苄基质子的化学位移分配是明确的,如同剩余的次甲基质子那样。5.5-5.6ppm和5.6-5.7ppm处的两对三重峰可通过其耦合常数(J)区分。J~16与E-构型中烯烃质子的预期耦合常数一致,而J~11与Z-构型中烯烃质子的预期耦合常数一致。基于这些信号的RIV值的比较,E/Z比例接近1,且轻微偏向E-异构体。由于6.45-6.6ppm处信号的复杂性,剩余烯烃质子的耦合常数无法准确测量。
TLC(氯仿/乙醇(19:1),在254nm下并使用高锰酸钾试剂检测)显示两个不同且部分重叠的位点,印证了NMR结果,即实施例1的产品是约1比1比例的E-和Z-异构体的混合物。
实施例3.凝胶制剂
表2显示一种凝胶制剂。
表2
实施例4.小鼠中通过腹膜内给药的活性化合物的镇痛活性(甩尾模型)
甩尾试验是动物中疼痛反应的试验。甩尾试验用于基础疼痛研究并旨在测量镇痛剂的有效性,具体方法为观察动物对热的甩尾反应。此试验评价对局部疼痛刺激的伤害性应答,以及药物抑制该应答的能力。
在第一次甩尾测量前30分钟时,使用5mL/kg的体积向小鼠腹膜内给予载剂对照(二甲亚砜,DMSO)和DMSO中的试验化合物4-苄磺酰基-2-丁腈,给药两次。以50mg/kg的剂量给予试验化合物。在第一次甩尾测量前30分钟时,使用8mL/kg的体积通过8mg/kg的皮下注射给予阳性对照化合物吗啡。各组有10只小鼠。
通过测量49℃水浴的甩尾潜伏时间或甩尾时间来评价小鼠对热刺激的应答。简言之,将动物放在限制器中,使其尾巴下垂。将约2英寸的尾部浸入38±1℃的装水烧杯中约30秒,进行两次以使动物适应该过程。
然后,将约2英寸的尾巴浸入49±1℃的装水烧杯中,此时打开计时器。出现第一不适迹象(全身痉挛,尾部蜷曲或快速运动)时停止计时,或如果动物没有应答则在30秒时停止计时,记录潜伏时间,将尾部从水中取出。
在给予试验化合物的剂量后30、60和120分钟,进行甩尾测量。进行ANOVA,如果p<0.05,使用顿式t检验(Dunnett’s t test)以计算载剂对照与试验化合物治疗组之间的差异。使用成对斯氏t检验(Student’s t test)计算吗啡组与对照组之间的差异。
图1显示使用载剂(DMSO)和试验化合物(DMSO中50mg/kg)治疗的小鼠的甩尾试验结果。以平均值±SEM(平均值的标准误)计算各组的潜伏时间并对时间作图,其中*表示与载剂治疗的小鼠相比p值小于0.05。
如图1所示,与时间零处相比,5mL/kg的载剂(DMSO)显示一些镇痛作用。与载剂治疗的小鼠相比,通过50mg/kg腹膜内给药使用试验化合物治疗的小鼠在第30分钟(p值=0.046)和第120分钟(p值=0.020)时显示统计学显著的甩尾潜伏时间。与载剂治疗的小鼠相比,吗啡治疗的小鼠(皮下注射)在第30和60分钟而非第120分钟时显示统计学显著的甩尾潜伏时间。吗啡的数据未包含在图1中。
上述结果提供了以下结论的证据:腹膜内给药时,试验化合物可有效地治疗动物中的伤害性疼痛。小鼠中的腹膜内给药是其他胃肠道外给药途径(例如,静脉内、皮下、肌内)的药代动力学概况的良好代表。因此,这些结果显示将试验化合物经胃肠道外给予对象可有效地减轻伤害性疼痛。
实施例5.小鼠中通过口服给药的活性化合物的镇痛活性,福尔马林模型(预示性实施例)
福尔马林试验为由福尔马林诱导的组织损伤引起的持续疼痛的模型。伤害性及炎症性疼痛是通过将福尔马林稀溶液注射入爪内诱导的,导致包括爪退缩的伤害防御行为。福尔马林模型包括疼痛的炎性、神经性及中枢机理。疼痛的早期(0至约10分钟)是因为伤害性机理而疼痛的晚期(10-40分钟)是因为炎性疼痛和伤害性机理的组合。利用手动舔动测量值评价疼痛行为。研究终点为舔动事件的次数。(Hunskaar等,Pain,30:103-114,1987;Li等,Molecular Pain,6:11,2010)
该研究中使用雄性CD-1小鼠。
在测试前(时间0),将小鼠限制在布中并向左后爪的背侧面皮下注射20μL 5%福尔马林溶液。向小鼠使用10mL/kg的体积经口管饲给予载剂对照(n=10,DMSO)和试验化合物4-苄磺酰基-2-丁腈(n=10,在DMSO中)。试验化合物的量为每剂量50或100mg/kg。
向小鼠使用4mL/kg的体积通过4mg/kg的皮下注射给予阳性载剂吗啡(n=10)。
在福尔马林注射前15分钟,经皮下给予吗啡一次。在时间零处福尔马林注射前60和15分钟时,经口服给予试验化合物和载剂对照两次(BID)。
福尔马林注射后,将动物放入各自笼中,以人工方式观察60分钟。以五分钟的间隔记录舔动事件,总共持续60分钟。计算载剂、阳性对照及试验化合物在0-10分钟与10-40分钟之间的每分钟舔动事件次数。
进行双样本t检验以比较载剂组与试验化合物组。显著性设为P<0.05水平。
实施例6.小鼠中通过局部给药的活性化合物的镇痛活性,福尔马林模型(预示性实施例)
动物及治疗方案与实施例5中所述的那些类似,不同之处如下。
通过将小鼠左后爪浸在各自溶液中约30秒来局部给予试验化合物4-苄磺酰基-2-丁腈(载剂中375mM,n=10)及载剂对照(丙酮:乙醇1:1,n=10)。然后,收回爪并利用纸巾擦拭以避免过度皮肤干燥。
向小鼠使用4mL/kg的体积通过4mg/kg的皮下注射给予阳性对照吗啡(n=10)。
在福尔马林注射前15分钟时,皮下给予吗啡一次。在福尔马林注射前90和15分钟时,局部给予试验化合物和载剂对照两次(BID)。
实施例7.花生四烯酸模型中小鼠中通过局部施用的活性化合物的抗炎活性(预示性实施例)
该实验中使用4-苄磺酰基-2-丁腈。
评价试验化合物、吲哚美辛(阳性对照)和载剂(丙酮:乙醇/1:1)在小鼠的局部花生四烯酸诱导的耳肿胀模型中的抗炎活性。
使用体重22±2g的雄性ICR小鼠并随机分组。将花生四烯酸(20μl丙酮:乙醇/1:1中0.5mg)局部施用到各小鼠右耳的前和后表面。在花生四烯酸施用前30分钟及施用后15分钟,类似地施用试验物质(载剂中375mM)和载剂。测量右耳及左耳的厚度且所计算的差异表明右耳炎症。在花生四烯酸施用后60分钟及90分钟时,利用Dyer型号测微规测量耳肿胀,作为炎症指标。根据下式计算抑制百分比:Ic-It/Ic x 100,其中Ic及It分别指对照及经治疗小鼠的耳朵厚度(mm)的增加。使用ANOVA和顿式t检验来确定载剂对照与治疗组之间的显著差异。显著性定为P<0.05水平。
实施例8.花生四烯酸模型中通过口服施用的小鼠中活性化合物的抗炎活性(预示性实施例)
实验方案与实施例7中描述的那些类似,区别如下。
在载剂(水中1%吐温80悬浮液或脂基药学上可接受的载体)中悬浮活性化合物4-苄磺酰基-2-丁腈至5-15mg/mL。向小鼠经口服给予试验化合物、地塞米松(载剂中的阳性对照)和载剂并评价其在小鼠中局部花生四烯酸诱导的耳肿胀模型中的抗炎活性。
在花生四烯酸前1小时经口管饲给予载剂中的试验化合物(50mg/kg,10mL/kg)和载剂(10mL/kg,),而在花生四烯酸攻击前3小时经口管饲给予地塞米松。
实施例9.通过局部施用的卡拉胶模型中活性化合物的抗炎和镇痛活性(预示性实施例)
评价载剂中的试验材料、吲哚美辛(阳性对照)和载剂(丙酮:乙醇1:1)在大鼠卡拉胶诱导的爪炎症模型中的抗炎和镇痛活性。
该实验中使用大鼠。将卡拉胶(0.1mL的1%悬浮液)皮下注射至左后爪以诱导炎症。在给予卡拉胶后1.5、2.5和3.5小时,将试验材料(1-5%)或载剂以0.05、0.1、0.15或2.0mL的体积局部施用于爪。在给予卡拉胶前1小时以5mg/kg经口服给予吲哚美辛。使用体积描记仪测量爪体积来确定炎症的程度(水肿或肿胀)。通过测量对于使用冯弗雷纤维(von Frey filament)的机械刺激的爪缩回来测定镇痛。在给予卡拉胶后4小时测量炎症和镇痛。试验材料预期具有抗炎和/或镇痛性质,其分别表现为:经测量,与载剂对照相比,爪体积的显著减少和/或引发爪缩回所需的机械压显著增加。
实施例10.CFA诱导的热痛觉过敏中活性化合物的镇痛作用(预示性实施例)
CFA(完全弗氏佐剂)已知诱导炎性疼痛(Walker等,JPET.304:56-62,2003)。
使用重量为180±20g的雄性Sprague-Dawley大鼠。这些动物被分成每组8-10只,在实验前24小时接收针对测试的后爪的CFA(0.1%溶液)的跖下(subplantar)注射(0.1ml)。通过使用IITC Model-336G(美国IITC公司)仪器来测试热痛觉过敏,其中将热调节的玻璃地板设为30℃。将各大鼠置于玻璃地板顶上的塑料盒内。将地板下的光束对准右后爪的跖面。当爪缩回远离热刺激时,自动记录时间。使用约12-14秒的平均组基线潜伏时间(CFA前)和施加的20秒截止潜伏时间来调节光强度。获得各小鼠的缩回潜伏时间并定义为热疼痛阈值。CFA注射后24小时,仅当缩回潜伏时间小于基线的75%时预先选择大鼠进行实验(清楚地存在热痛觉过敏)。
在局部载剂中或口服载剂中制备活性化合物4-苄磺酰基-2-丁腈。
评价局部或口服载剂中的活性化合物、吗啡(阳性对照,p.o.,20mg/kg)、局部载剂(丙酮:乙醇1:1)和口服载剂(DMSO)在福尔马林模型中的镇痛活性。
在再次测量热痛觉过敏水平前60分钟时,将试验物质或载剂口服给予(20-60mg/kg)或局部给予(375mM)后爪的跖面(治疗后)。计算热爪缩回时间的平均值±SEM。应用未配对的斯氏t检验来比较试验物质治疗组和载剂对照组之间的治疗后数值。阳性活性设为P<0.05。
实施例11.慢性缩窄性损伤模型中活性化合物的镇痛活性(预示性实施例)
外周神经伤害可产生综合征,其除自发性疼痛外还包括对轻触的过度应答(触觉异常性疼痛(tactile allodynia))。慢性缩窄性损伤模型是一种神经性疼痛模型。
使用重量为180±20g的雄性Sprague-Dawley大鼠。在戊巴比妥(50mg/kg,5ml/kg,i.p.)麻醉下,在中-股水平下将坐骨神经暴露。将四条结扎线(4-0铬肠线)以约1mm的间隔松弛地扎在神经上。然后,在试验前,将动物单独地放入具有软垫料的笼中7天。坐骨神经的缩窄产生神经损伤及单侧神经性疼痛。
在实验那天,在试验前,动物不能进食过夜。将大鼠放在金属丝网支架上的倒置的树脂玻璃笼中并使之适应20至30分钟。通过Chaplan上/下法利用von Frey细丝对左后爪的跖面评价机械异常疼痛。参见Chaplan等,J.Neuroscience Methods,53:55-63,1994。
仅在以下情况下预先选择大鼠进行实验:神经结扎(治疗前)后7-14天,相对于神经结扎前(结扎前)单独爪的应答减少10克力,即明显存在异常疼痛。
评估局部或口服载剂中的活性化合物、吗啡(阳性对照,p.o.,20mg/kg)、局部载剂(丙酮:乙醇1:1)和口服载剂(DMSO)在福尔马林模型中的镇痛活性。
向左后爪的跖面经口服(20-60mg/kg)或局部(375mM)给予试验物质或载剂。在单剂量的测试物质或载剂之前30分钟(治疗前)和之后1和3小时(治疗后)进行机械异常性疼痛试验。测量对照及试验化合物的爪缩回阈值。
实施例12.关节炎的治疗(预示性实施例)
向小鼠的膝关节直接注射的酵母聚糖引发炎症应答并被用作关节炎的模型(Verschure等,Ann.Rheum Dis.53:455-460,1994)。该模型中测量的终点包括膝关节肿胀评分、滑膜组织中的细胞因子水平和膝的显微病理学。
使用5mL/kg的体积向小鼠经口管饲给予活性化合物4-苄磺酰基-2-丁腈(载剂中50mg/kg)和载剂对照(水悬浮液或其他脂基药学上可接受的载体中的1%吐温80)。
每组5只小鼠,共注射10个膝。在第1天,在第0和12小时时向C57BL6小鼠给予(50mg/kg/剂量)活性化合物或载剂两次。在第2天,在第24小时时向小鼠给予活性化合物或载剂,随后在第25小时时将180μg酵母聚糖(6μL)关节内注射至两个膝关节,并随后在第36小时时第二次给予各活性化合物或载剂。在第3天,在第48小时时再次向小鼠给予活性化合物或载剂。给药后两小时即第50小时时,对膝的水肿进行评分,收集滑膜组织以测量细胞因子水平,并加工膝关节进行组织病理学分析以分析关节中的炎性免疫细胞流入。使用范围为0-3的评分系统在移除皮肤后评价肉眼可见的关节肿胀,其中0表示无肿胀且3表示严重肿胀。从5个膝提取滑膜组织以测量小鼠白介素-1β、白介素-6和白介素-1受体拮抗剂水平。对剩余的5个膝加工进行显微病理学分析以评价炎症位点的细胞流入。
各组的结果表示为平均值±平均值的标准误并进行统计学评估。
关节肿胀减少、细胞因子水平降低且炎症位点的炎性细胞流入减少,这些测量结果显示使用活性化合物进行的治疗预期导致炎症减少。
实施例13.痛风的治疗(预示性实施例)
将尿酸单钠单水合物(MSU)晶体连同游离脂肪酸(FFA)直接注射至小鼠的膝关节引发炎症应答并被用作痛风模型(Joosten等,Arthritis&Rheumatism,62(11):3237-3248,2010)。该模型中测量的终点包括膝关节肿胀评分、滑膜组织中的细胞因子水平和膝的显微病理学。
使用5mL/kg的体积向小鼠经口管饲给予活性化合物4-苄磺酰基-2-丁腈(载剂中50mg/kg)和载剂对照(水悬浮液或脂基药学上可接受的载体中的1%吐温80)。
每组5只小鼠,共注射10个膝。在第1天,在第0和12小时时向C57BL6小鼠给予(50mg/kg/剂量)活性化合物或载剂两次。在第2天,在第24小时时向小鼠给予活性化合物或载剂,随后在第25小时时使用MSU晶体(300μg)和C18:0FFA(200μM,10μL)进行关节内注射。3小时后(第28小时时),对膝的水肿进行评分,收集滑膜组织以测量细胞因子水平,并加工膝关节进行组织病理学分析以分析关节中的炎性免疫细胞流入。使用范围为0-3的评分系统在移除皮肤后评价肉眼可见的关节肿胀,其中0表示无肿胀且3表示严重肿胀。从5个膝提取滑膜组织以测量小鼠白介素-1β、白介素-6和白介素-1受体拮抗剂水平。对剩余的5个膝加工进行显微病理学分析以评价炎症位点的细胞流入。
各组的结果表示为平均值±平均值的标准误并进行统计学评估。
关节肿胀减少、细胞因子水平降低且炎症位点的炎性细胞流入减少,这些测量结果显示使用活性化合物进行的治疗预期导致炎症减少。
实施例14.膝疼痛的治疗(预示性实施例)
目的:为了研究凝胶制剂中或口服制剂中的活性化合物在患有标准NSAID治疗暂时停止后与骨关节炎相关的轻微至中度膝疼痛的人患者中的效力。此研究关注于疼痛性关节炎引起的症状。临床试验将膝关节炎作为其他肌骨骼病症的明确范例。
局部制剂:该实施例使用包含1%和5%的活性化合物4-苄磺酰基-2-丁腈的凝胶制剂(实施例3)。安慰剂含有不具有活性化合物的相同凝胶。
口服制剂:该实施例中使用各自含有100-600mg活性化合物4-苄磺酰基-2-丁腈的胶囊剂或片剂。安慰性胶囊剂或片剂不含活性化合物。
方法:随机、双盲、安慰剂对照、平行治疗多中心临床活性研究。
患者患有疼痛性膝关节炎,被稳定剂量的标准NSAID治疗控制至少2个月,中断NSAID持续7天洗脱期。然后,以1:1:1比率(1%活性凝胶、5%活性凝胶、安慰剂)随机分配患者。招募总共150名患者,治疗14天并在第14、21和28天时随访。
将活性凝胶或安慰剂施用于受影响的膝,一天三次,持续14天,总共进行42次治疗,清醒时每4-6小时给药。
向患者经口服给予胶囊剂或片剂,每天1-4次,持续14天。
治疗患者14天,再随访14天。第14天就诊后,重新开始NSAID。
评价标准:
安全性:
·整个研究中的不良事件。
·招募时(-7天,开始NSAID洗脱期),基线,第14天以及第28天的身体检查。
·招募时(-7天,开始NSAID洗脱期),基线以及第7、14、21、28天时的生命体征。
·基线以及第7、14、21和28天时的临床实验测量。
临床活性:
主要临床活性参数为施用位点处的疼痛测量,其利用移动时疼痛(Pain on Movement)评价(100-mm VAS)和西安大略和麦克马斯特大学(WOMAC)指数(100-mm VAS或5点李克特量表(5-point Likert scale))进行定量。记录治疗对膝肿胀、触痛及炎症的影响,也记录治疗后疼痛减轻或根除的时间。
研究终点:
主要的临床活性终点为:
·在WOMAC功能障碍指数和亚指数中从基线到第14天的变化:
-疼痛(等级0-20)。
-僵硬度(等级0-8)。
-身体功能(等级0-68)。
·从基线(第1天)到第14天移动时疼痛中的变化(1–100mm VAS)。
次要的临床活性终点为:
·从给予剂量前到给予剂量后1小时基线处当前膝疼痛(Current Knee Pain)评分中的变化(100mm VAS)。
·从给予剂量前到给予剂量后2小时基线处当前膝疼痛评分中的变化(100mm VAS)
·全身性疾病评级(Global Rating of Disease)中的变化(5点李克特量表)
·每次施用活性化合物后疼痛减少或消除的时间。
·使用急救药物(APAP)。
经历从基线至第14天移动时疼痛(100-mm VAS)中改善的对象比例等于或大于20%或15mm的最小临床重要改善(MCII)阈值
第14天时移动时疼痛(100-mm VAS)小于40mm的患者可接受症状状况(PASS)阈值的对象比例
有关WOMAC指数,基于OARSI应答者指数是‘应答者’的对象比例。
应理解,上文描述了本发明的优选实施方案,并可进行修改而不背离权利要求书所限定的本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!