一种磁性石墨烯载药体系的制备及应用的制作方法
本发明涉及纳米材料领域和生物技术领域,具体涉及一种具有肿瘤磁靶向、乳腺癌治疗的磁性石墨烯载药体系制备方法及其应用。
背景技术:
乳腺癌是女性最常见的恶性肿瘤之一,在我国乳腺癌占全身各种恶性肿瘤的7%~10%,仅次于子宫颈癌,发病率约为18.7人/10万人,近年来增长趋势明显。虽然在我国乳腺癌的早期诊断与治疗已取得长足进步,但仍有八成乳腺癌患者被发现时已经是中晚期,尽管手术能切除大部分肿瘤,但多数手术已不能达到根治,肿瘤最终会复发和转移,治疗效果不佳。因此,关注乳腺癌,关注女性健康,越来越成为我国医学研究和临床的发展所趋。
紫杉醇(Paclitaxel)是从短叶红豆杉树皮中分离得到的一种二萜类化合物,水溶性较差,属疏水性药物。它是一种新型的微管稳定剂,它通过诱导和促进微管蛋白聚合,稳定微管而阻止肿瘤细胞的繁殖,临床研究显示紫杉醇治疗乳腺癌具有较好的疗效。
石墨烯是2004年被发现的一种新型二维平面纳米材料,其特殊的单原子层结构决定了它具有丰富而新奇的物理性质。由于石墨烯具有单原子层结构,其比表面积很大,非常适合用作药物载体。但结构完整的石墨烯化学稳定性高,其表面呈惰性状态,必修对其进行有效的功能化才能进一步应用。目前,利用磁性纳米颗粒装载到石墨烯表面制备磁性石墨烯纳米复合材料,在磁靶向载药、磁共振成像及污水处理等领域表现出极大的应用潜力。
CN201510805317.7公布了一种荧光标记的磁性氧化石墨烯复合药物载体的制备方法,该药物载体的磁性颗粒粒径在10-60nm,量子点粒径在1.5-10nm,该复合纳米药物载体同时具备量子点的荧光性能、磁性纳米颗粒的磁性能以及石墨烯的高比表面积,可用作生物体内靶向定位和荧光成像。
CN201510242930.2公布了一种对宫颈癌能够实现诊断与治疗的多功能磁性氧化石墨烯。首先将羧基化的Fe3O4粒子水溶液与氨基化PEG修饰的氧化石墨烯GO水溶液混合,在EDC缩合条件下,制备出磁性氧化石墨烯纳米材料。并通过π-π作用将阿霉素DOX负载到磁性氧化石墨烯上,并研究其对宫颈癌HELA细胞的毒性作用。
目前,尚未有磁性石墨烯负载紫杉醇并用于乳腺癌治疗中的研究或报道。本发明开发了一种具有良好稳定性和生物相容性、对疏水性药物紫杉醇负载率高的磁性石墨烯载药体系。
技术实现要素:
本发明的目的是提供一种具有肿瘤磁靶向、乳腺癌治疗的磁性石墨烯载药体系的制备方 法。该方法制备的复合纳米药物载体具有分散性好且均匀、磁响应性强、生物相容性好、药物负载量高等优点,在纳米药物载体领域具有广泛的应用。
为达到本发明的目的,本发明的技术方案包括以下步骤:①首先采用两种表面活性剂十六烷基三甲基溴化铵和聚苯乙烯磺酸钠依次改性氧化石墨烯,并通过水热合成法合成磁性氧化石墨烯;②然后用两亲性聚合物PF127修饰,并用水合肼将磁性氧化石墨烯还原为具有良好生物相容性的磁性石墨烯;③通过π-π作用将疏水性抗癌药物紫杉醇(PTX)负载到磁性石墨烯上,制备出抗肿瘤和生物相容性于一体的磁性石墨烯载药体系;④将磁性石墨烯载药体系与MCF-7乳腺癌细胞共培育,用MTT法检测细胞毒性。⑤采用荧光显微镜进行观察活细胞形态。
具体步骤为:
步骤1:采用两种表面活性剂改性氧化石墨烯,得到功能化的氧化石墨烯;并通过水热法合成磁性氧化石墨烯。
A、将0.11~0.13g氧化石墨烯GO加入到80~120mL,0.5~1.5%质量比的十六烷基三甲基溴化铵CTAB水溶液中,在冰浴中超声分散20~40min后,搅拌5~7h,于10000r min-1下离心分离,并用蒸馏水多次离心洗涤。将产物与0.23~0.25g聚苯乙烯磺酸钠PSS溶液混合,超声分散20~40min,搅拌5~7h,继续用蒸馏水离心清洗,于30~50℃真空干燥,所得样品即功能化的氧化石墨烯f-GO。
B、然后将0.11~0.13g f-GO和0.5~1.5g铁盐添加到40~60mL多元醇中,冰浴中超声搅拌1~3h使Fe3+附着在GO上,然后离心去除多余的Fe3+。然后将0.5~1.5g PEG和3.4~3.8g NaAc添加到上述悬浮液中,超声处理20~40min,然后再搅拌11~13h,得到水热前驱体,在聚四氟乙烯为内衬的反应釜中190~210℃下反应11~13h小时,待反应结束后,自然冷却至室温,并将所得样品用无水乙醇反复清洗并用磁铁收集Fe3O4/f-GO,于30~50℃下真空干燥。
步骤2:采用两亲性聚合物PF127修饰,并用水合肼将磁性氧化石墨烯还原为具有良好生物相容性的磁性石墨烯。
将300~400mg PF127加入到10~20mL,0.5~1.5mg/mL Fe3O4/f-GO水溶液中,室温搅拌下加入300~500μL水合肼,于30~50℃下超声搅拌反应12~36h,将氧化石墨烯(GO)还原为石墨烯(GN)。反应结束后将混合液进行压滤即得PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127)。
步骤3:通过π-π作用将疏水性抗癌药物紫杉醇(PTX)负载到磁性石墨烯上。
将0.008~0.012g Fe3O4/f-GN/PF127加入到20~40mL,0.8~1.2mg/mLPTX的PBS缓 冲液中,25℃下避光搅拌14~18h,离心并洗涤,冷冻干燥得到载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
步骤4:本发明对磁性石墨烯和载药的磁性石墨烯都进行了细胞毒性实验,将其与乳腺癌MCF-7细胞共培育,用MTT法检测细胞毒性。
(1)在装有磁性石墨烯载药体系(PTX-Fe3O4/f-GN/PF127)和磁性石墨烯载体(Fe3O4/f-GN/PF127)的EP管中,加入100μL75%乙醇消毒,12000rpm/min,离心5min,随后用PBS清洗,离心,重复2次,备用。
(2)将MCF-7细胞培养于DMEM高糖培养基中,37℃、5%CO2恒温培养箱中贴壁培养,每2~3天换液体一次,每3~5天传代一次。当细胞贴壁生长密度达到80%~90%后,弃去细胞培养基,加入2mL PBS洗涤2次,弃去PBS,加入2mL含有0.25%的胰蛋白酶与0.02%EDTA的消化液消化,消化30~60s后弃去大部分消化液,剩下的消化液再继续消化大约l~2min;镜下观察,细胞完全从细胞壁上分离并尽可能吹打成单个细胞,将细胞的密度调到1×104个/mL,然后加入细胞瓶或者培养板中传代培养。
(3)称取25mg MTT(3-(4,5-二甲基噻唑)-2,5-二苯基四氮唑溴盐,噻唑蓝,Sigma公司)溶于5mL PBS,配成浓度为5mg/mL的MTT溶液。用0.22μm多孔滤膜过滤灭菌,用前现配。将对数期生长的MCF-7细胞接种于96孔板,利用DMEM高糖完全培养基进行培养,培养24h后,分别加入不同浓度的磁性石墨烯载药体系和磁性石墨烯载体,同时设置只加入DMEM高糖完全培养基作为对照组,未接种细胞作为空白组,每组设置6个复孔,分别继续培养24h,每孔加入20μL、5mg/mL的MTT溶液,培养箱内继续培养4h后,弃去孔内的培养基,然后每孔加入200μL二甲基亚砜(DMSO),置摇床避光低速振荡10min,使结晶物充分溶解。利用酶标仪在490nm波长下测定吸光值(OD)。将对照组的细胞存活率定义为100%,存活率按公式计算:
细胞存活率=(试验组OD值/对照组OD值)×l00%
步骤5:取25mg荧光素二乙酸酯(Fluorescein Diacetate,FDA)溶于5mL丙酮中配制成母液,使用时将母液按1:1000的体积比稀释于PBS中。MCF-7细胞用5μg/mL的FDA/PBS溶液孵育10min,在这个过程中,FDA进入活细胞膜,在488nm的激发下发出荧光。培养3天后的活细胞通过荧光显微镜(Zeiss Axovert 200)观察。
所述步骤1A中,所述的超声时间优选30min,搅拌时间优选6h。
所述步骤1B中,所述的铁盐可选自水合氯化铁、水合硝酸铁、水合硫酸铁中的一种。
所述步骤1B中,多元醇选自乙二醇、1-2-丙二醇、二缩二乙二醇、三缩三乙二醇等。
所述步骤2中的反应温度优选40℃;反应时间优选24h。
所述步骤3中PTX的浓度优选1mg/mL,搅拌时间优选16h。
本发明所取得的有益效果:
(1)本发明通过多步合成法,合成磁性石墨烯载药体系,制备方法操作简单,实验条件温和。
(2)本发明合成的磁性石墨烯药物载体具有分散性好且均匀、磁响应性强好、生物相容性好、药物负载量高的优点。
(3)本发明首次采用磁性石墨烯作为药物载体,负载疏水性抗癌药物紫杉醇,尚未见到有探讨对乳腺癌MCF-7细胞毒性的研究报道。本发明合成的磁性石墨烯载药体系在用量较少的情况下200μg/mL就表现出对MCF-7乳腺癌细胞较好的抑制作用。
附图说明
图1为具体实施例所制备的样品的FTIR图谱:a.氧化石墨烯(GO);b.磁性石墨烯(Fe3O4/f-GO);c.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
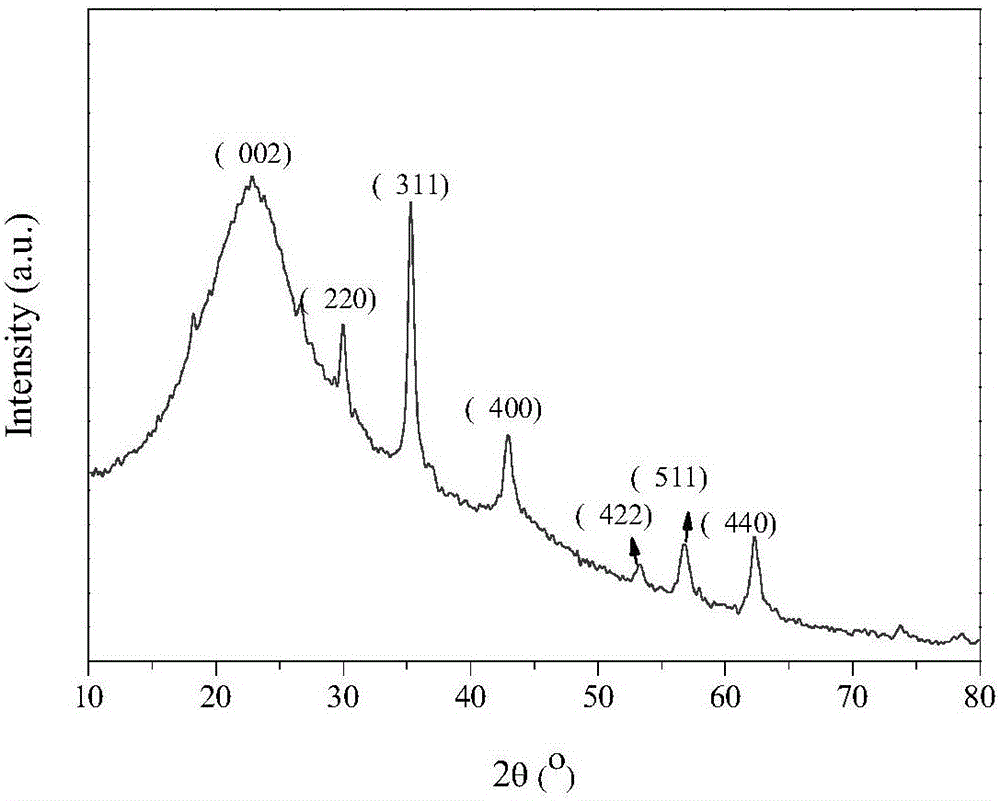
图2为具体实施例所制备磁性石墨烯Fe3O4/f-GO的XRD图谱。
图3为具体实施例所制备样品的SEM图:a.氧化石墨烯(GO);b.改性氧化石墨烯(f-GO);c.磁性石墨烯(Fe3O4/f-GO);d.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
图4所示为具体实施例所制备的样品的TEM图:a.改性氧化石墨烯(f-GO);b.磁性石墨烯(Fe3O4/f-GO);c.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
图5为具体实施例所制备磁性石墨烯载药前后的VSM图:a.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);b.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
图6为具体实施例所制备的磁性石墨烯载药体系PTX-Fe3O4/f-GN/PF127在37℃,pH=7.4条件下的PBS缓冲溶液所测的药物释放曲线。
图7为MCF-7细胞加入样品后的细胞毒性图:a.PF127修饰的磁性石(Fe3O4/f-GN/PF127);b.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
图8为MCF-7细胞加入不同浓度的磁性石墨烯载体Fe3O4/f-GN/PF127后的荧光显微镜图片:a.0μg/mL;b.1μg/mL;c.10μg/mL;d.100μg/mL.
图9为MCF-7细胞加入不同浓度的磁性石墨烯载药体系PTX-Fe3O4/f-GN/PF127后的荧光显微镜图片:a.0μg/mL;b.100μg/mL;c.200μg/mL;d.500μg/mL.
具体实施方式
以下结合实施例对本发明做进一步说明,但下述实施例对本发明的保护范围并无明确限制。
实施例
将0.12g的GO加入到100mL1%的CTAB蒸馏水溶液中,在冰浴中超声分散30min后,搅拌6h,于10000r min-1下离心分离,并用蒸馏水多次离心洗涤。将产物与0.24g的PSS溶液200mL混合,超声分散30min,搅拌6h,继续用蒸馏水离心清洗2~3次,于40℃真空干燥,所得样品即为表面带负电荷的改性氧化石墨烯f-GO。
将制备好的0.12g f-GO和1g FeCl3·6H2O添加到50mL乙二醇中,冰浴超声搅拌2h使Fe3+附着在GO上,然后离心除去多余的Fe3+。超声分散在乙二醇中30min,形成均匀的悬浮液。然后将1g PEG和3.6g NaAc添加到上述悬浮液中,超声处理30min,再搅拌12h,得到水热前驱体,在聚四氟乙烯为内衬的反应釜中200℃下反应12h。待反应结束后,自然冷却至室温,并将所得样品用无水乙醇反复清洗并用磁铁收集得到磁性石墨烯Fe3O4/f-GO,于40℃下真空干燥。
将400mg的PF127加入到15mL,1.0mg/mL的Fe3O4/f-GO水溶液中,室温搅拌下加入400μL的水合肼,于40℃下超声搅拌反应24h,将氧化石墨烯(GO)还原为石墨烯(GN)。反应结束后将混合液进行压滤即得Fe3O4/f-GN/PF127。将0.01gFe3O4/f-GN/PF127加入到30mL,1mg/mL PTX的PBS缓冲液中,25℃下避光搅拌16h,离心并洗涤,冷冻干燥得到载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
对制备过程中的中间产物及本发明最终产物进行性能分析。
图1为具体实施例所制备的样品的FTIR图谱:a.氧化石墨烯(GO);b.磁性石墨烯(Fe3O4/f-GO);c.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。
通过与文献谱图对照,图1a中,1725cm-1处为羧基上的C=O的伸缩振动峰,3430cm-1处为羟基中O-H键的伸缩振动,1617cm-1处为C=C键的伸缩振动吸收峰。与图1a相比,图1b中在700cm-1处出现了一个峰,此峰对应于Fe-O的伸缩振动,表明成功得到了磁性氧化石墨烯。对比图1b,图1c中在2975cm-1,2890cm-1出现了新峰,分别对应于-CH3和-CH2的伸缩振动峰,表明PF127成功负载到石墨烯上。图1d中,1722cm-1为紫杉醇中酯基中C=O键的伸缩振动峰,1072cm-1为酯基中C-O键的伸缩振动峰,1247cm-1为-COO的伸缩振动峰,1652cm-1处为酰胺中C=O键的伸缩振动峰,707cm-1对应于芳香环中C-H键,由此表明磁性石墨烯成功负载了药物紫杉醇。
图2所示为具体实施例所制备磁性石墨烯Fe3O4/f-GO的XRD图谱。在2θ=22.9°对应于 GO石墨结构的(002)晶面。经与JCPDS标准图谱(JCPDS:19-0629)对照,Fe3O4/f-GO中的Fe3O4属于立方晶系。图中2θ=30.1°,35.4°,43.1°,53.5°,56.9°,62.5°的衍射峰分别对应于立方相Fe3O4纳米晶的(220),(311),(400),(422),(511),(440)晶面。由此证明成功制备了磁性氧化石墨烯。
图3为具体实施例所制备样品的SEM图:a.氧化石墨烯(GO);b.改性氧化石墨烯(f-GO);c.磁性石墨烯(Fe3O4/f-GO);d.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。从图3a中可看出改性前GO有褶皱片层结构。从图3b中可明显看出改性后氧化石墨烯具有类似的层状结构,分布较均匀,无明显团聚现象。图3c为Fe3O4/f-GO的SEM图,图中可明显看出在改性后氧化石墨烯表面布满了大量Fe3O4粒子,且分布较均匀,无明显团聚现象。图3d为磁性石墨烯被还原后,采用两亲性聚合物PF127修饰的产物照片,Fe3O4仍很好地分布在还原的磁性石墨烯表面。图3e为负载紫杉醇的磁性石墨烯的SEM图,从图中可看出,载药后磁性石墨烯仍保持很好的形貌。
图4所示为具体实施例所制备的样品的TEM图:a.改性氧化石墨烯(f-GO);b.磁性石墨烯(Fe3O4/f-GO);c.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);d.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。图4a为f-GO的TEM图,从图中可以清晰地看到氧化石墨烯的褶皱片层结构,而且具有良好的分散性。从图4b中可以清晰地看到黑色小颗粒均匀分布在氧化石墨烯的褶皱片层上。从图4c中可看出PF127修饰过后,GN与Fe3O4的边缘变得模糊,这是由于两亲性物质PF127的疏水端作用于石墨烯,亲水端分散于水中。图4d为载了疏水性紫杉醇的磁性石墨烯的TEM图,从图中可以看出,载药后整体形貌变得模糊。
图5所示为具体实施例所制备磁性石墨烯载药前后的VSM图:a.PF127修饰的磁性石墨烯(Fe3O4/f-GN/PF127);b.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。由图5a可知,Fe3O4/f-GO的饱和磁化强度(Ms)和剩磁(Mr)分别为9.39emu/g和0.82emu/g,矫顽力(Hc)为35.53Oe,在室温下没有磁滞环现象,剩磁基本为零,表现了较好的超顺磁性。从图5b中可以看出在室温下没有磁滞环现象,剩磁基本为零,饱和磁化强度(Ms)为5.23emu/g,保持了较好的超顺磁性。
紫杉醇标准曲线的绘制:
精确称量紫杉醇5.00mg置于l00mL容量瓶中,用含0.1%(w/v)Tween 80的PBS溶解,并稀释成一系列已知浓度的溶液,用紫外-可见分光光度计测定这些溶液在230nm处的吸光度,以吸光度对药物浓度作图,得到紫杉醇的吸光度-浓度标准曲线,该标准曲线的方程为:Y=16.032X+0.0766,(相关系数R2=0.981)。借助载药率衡量体系的载药能力,载药率=(药物的质量/载药体系的质量)×100%,计算出紫杉醇的载药率为67.9%。
将10.0mg负载紫杉醇的载药体系分散于含0.1%(w/v)Tween 80的PBS溶液中,置于50mL的PBS溶液中缓释,在集热式磁力搅拌器中恒温37℃并以100rpm速度振荡,在特定的时间间隔里取3.0mL的释放介质作为样品,使用紫外分光光度计测定波长为230nm时的吸光度,并补加3.0mL的PBS溶液,得到不同缓释时间下的吸光度。通过线性回归方程得到各个时间点样品的浓度,根据以下公式计算药物累积释放率并得到其关于时间的曲线。Wn=(50Cn+3×∑Cn-1)/m0×100%,其中Wn表示第n次药物的累积释放率,Cn是第n次取样的质量浓度,m0是载药体系中紫杉醇的质量。
图6为具体实施例所制备的磁性石墨烯载药体系PTX-Fe3O4/f-GN/PF127在37℃,pH=7.4条件下的PBS缓冲溶液所测的药物释放曲线。如图6所示,紫杉醇从一开始就有释放,0-60h为突释阶段,释放了所负载药物的85%,60h以后为缓释部分,缓释阶段可以保持到100h。
细胞毒性实验:
将对数期生长的MCF-7细胞接种于96孔板,利用DMEM高糖完全培养基进行培养,每孔200μL(细胞的密度1×104个/mL),培养24h后,分别加入不同浓度的磁性石墨烯载药体系PTX-Fe3O4/f-GN/PF127(1μg/mL、10μg/mL、100μg/mL、200μg/mL、500μg/mL)和磁性石墨烯Fe3O4/f-GN/PF127(0.5μg/mL、1μg/mL、10μg/mL、50μg/mL、100μg/mL),同时设置只加入DMEM高糖完全培养基的MCF-7作为对照组,未接种细胞作为空白组,每组设置6个复孔,分别继续培养24h,每孔加入20μlMTT溶液(5mg/mL),培养箱内继续培养4h后,弃去孔内的培养基,然后每孔加入200μ1的二甲基亚砜(DMSO),置摇床避光低速振荡10min,使结晶物充分溶解。利用酶标仪在490nm波长下测定吸光值(OD)。将对照组的细胞存活率定义为100%,存活率按公式计算:细胞存活率=(试验组OD值/对照组OD值)×l00%。
图7为MCF-7细胞加入样品后的细胞毒性图:a.PF127修饰的磁性石(Fe3O4/f-GN/PF127);b.载药的磁性石墨烯(PTX-Fe3O4/f-GN/PF127)。如图7a所示,与对照组相比,各浓度的Fe3O4/f-GN/PF127载体对MCF-7的细胞活性没有显著影响。如图7b所示,与对照组相比,在200μg/mL-500μg/mL范围内,不同浓度载药磁性石墨烯对MCF-7细胞存活率影响很大。当载药磁性石墨烯浓度为200μg/mL时,细胞活性降低为31%;当浓度增大到500μg/mL时,细胞活性为11%;在1μg/mL~500μg/mL范围内,随着浓度的升高,细胞活性变化逐渐减低。
细胞形态观察实验:
将对数期生长的MCF-7细胞接种于96孔板,每孔200μL(细胞的密度1×104个/mL),培养24h后,分别加入不同浓度的磁性石墨烯载药体系,同时设置只加入DMEM高糖完全培养基的MCF-7细胞作为对照组,每组三个复孔,培养24h后,用移液器每孔弃去100μL的培养基,各 孔加入10μg/mL的FDA/PBS溶液100μL,使FDA的终浓度为5μg/mL,孵育10min,在这个过程中,FDA进入活细胞膜,10分钟后弃去孔中的所有液体,各孔再加入PBS缓冲液200μL,在488nm的激发下发出荧光,荧光显微镜拍照。
图8为MCF-7细胞加入不同浓度的磁性石墨烯载体Fe3O4/f-GN/PF127后的细胞毒性图:a.0μg/mL;b.1μg/mL;c.10μg/mL;d.100μg/mL.如图8所示,加入磁性石墨烯载体后,与图7a的MTT结果一致,对于MCF-7细胞活性没有明显的影响。
图9为MCF-7细胞加入不同浓度的磁性石墨烯载药体系PTX-Fe3O4/f-GN/PF127后的荧光显微镜图片:a.0μg/mL;b.100μg/mL;c.200μg/mL;d.500μg/mL.如图9a所示,未加入磁性石墨烯载药体系,控制组具有最多的活细胞数目。如图9b-d所示,与图7b的MTT结果一致,在100μg/mL-500μg/ml范围内,磁性石墨烯载药体系对MCF-7细胞存活率影响很大,随着浓度的升高,细胞活性降低。
综上所述,所合成的磁性石墨烯载药体系表现出对乳腺癌MCF-7细胞较好的抑制作用,在生物医药领域有广阔的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!
- 氨基硅烷化磁性氧化石墨烯纳米粒子共固定化漆酶和介体系统及其制备方法与制造工艺
- 一种耐热纳米碳溶胶、纳米石墨辐照接枝改性锦纶单丝滤布及其制备方法与制造工艺
- 一种高韧性纳米碳溶胶、纳米石墨辐照接枝改性锦纶单丝滤布及其制备方法与制造工艺
- 一种耐磨纳米碳溶胶、纳米石墨辐照接枝改性锦纶单丝滤布及其制备方法与制造工艺
- 磁性四氧化三铁/石墨烯复合材料的制备及其在制备磁性油漆中的应用的制造方法与工艺
- 一种石墨烯/碳点/磁性复合材料的制备方法与制造工艺
- 荧光磁性氧化石墨烯基4?氯酚分子印迹聚合物的制备和应用
- 一种用于吸附染料的磁性氧化石墨烯吸附剂的制备方法
- 一种磁性石墨烯3d打印耗材的制备方法
- 一种磁性纳米颗粒/SiO<sub>2</sub>气凝胶及其制备方法以及处理高放废液的方法