用于治疗癌症的组合疗法的制作方法
本申请要求于2015年4月20日提交的美国临时申请号62/150,185的优先权和权益,将其内容通过引用以其全文结合在此。
技术领域
本披露涉及包含人类组蛋白甲基转移酶EZH2(催化组蛋白H3上的赖氨酸27(H3-K27)的单至三甲基化的PRC2复合物的催化亚基)的抑制剂和一种或多种其他治疗剂(特别是抗癌剂)的组合物,并且涉及用于治疗癌症的组合疗法的方法。
背景技术:
组合疗法治疗癌症已变得更加常见,部分原因是由于通过多途径攻击疾病的已知优势。虽然在过去的几十年中已经确定了许多有效的组合疗法治疗,鉴于每年由癌症造成的持续大量的死亡,仍然对确定用于抗癌治疗的有效治疗方案存在持续的需要。
技术实现要素:
本披露部分地基于这样的发现:EZH2抑制增加了对抗VEGF剂(诸如舒尼替尼)的敏感性。相应地,本披露的一个方面涉及一种用于通过给予治疗有效量的EZH2抑制剂和一种或多种酪氨酸激酶抑制剂或VEGF/VEGFR抑制剂来治疗对其有需要的受试者(例如,患者)中的癌症的方法。例如,该癌症抵抗酪氨酸激酶抑制剂治疗或抵抗VEGF/VEGFR抑制剂治疗。例如,该癌症是肾细胞癌(例如,晚期透明细胞肾细胞癌)。
本披露部分地基于这样的发现:EZH2抑制剂(诸如化合物44)和糖皮质激素受体激动剂(GRag)(诸如强的松、泼尼松龙或地塞米松)配合使用显著地提高癌症治疗效果。化合物44和泼尼松龙的组合延伸了对EZH2抑制敏感的细胞的范围,从仅携带突变的细胞延伸至所有GCB NHL细胞。
本披露涉及用于通过给予治疗有效量的EZH2抑制剂和选自由糖皮质激素受体激动剂(GRag)、CHOP和BCL2抑制剂组成的组的一种或多种治疗剂来治疗对其有需要的患者中的癌症的方法。例如,该BCL2抑制剂是纳成托克斯(navitoclax)。
在一些实施例中,该癌症是生发中心B亚型的NHL。
在一些实施例中,该癌症是EZH2突变体癌症。可替代地,该癌症是EZH2野生型癌症。
在一些实施例中,该癌症是EZH2抑制剂抗性或难治性癌症。
EZH2抑制剂和GRag可同时或顺序地给予。例如,EZH2抑制剂在给予GRag之前给予。
在本披露的一个方面,本文所述的组合疗法诱导调节特定基因(例如,糖皮质激素靶基因)的表达。如本文所用,糖皮质激素靶基因是指由糖皮质激素直接或间接调节的基因。在某些实施例中,基于本文所述的组合疗法,基因被上调。在某些实施例中,该基因是瑟斯崔因(Sestrin)、TNF和/或GILZ。在某些实施例中,这些基因可以用作生物标记物,即该基因表达谱可以用于识别适用于本文所述的组合疗法的患者。在某些实施例中,该基因表达可以用于监测或评估本文所述的组合疗法的剂量或功效。
在一个实施例中,该癌症是血液癌症。在某个实施例中,该癌症是淋巴瘤。在某个实施例中,该癌症是生发中心B亚型(GCB)的非霍奇金淋巴瘤(NHL)或弥漫性大B细胞淋巴瘤(DLBCL)。
在一个实施例中,EZH2抑制剂的治疗效果由GRag在携带EZH2突变体的细胞中增强。
在本披露的一个方面,EZH2抑制剂增强WT EZH2细胞中的GRag治疗效果。在一个实施例中,这些细胞是WT GCB EZH2细胞。在本披露的某个实施例中,EZH2抑制剂是化合物44,并且GRag是强的松。
本披露的一个方面基于这样的发现:EZH2抑制剂和GRag的组合逆转了在EZH2抑制剂抗性或难治性突变体细胞(包括携带EZH2突变体的细胞)中的不敏感性。
在突变体EZH2GCB淋巴瘤细胞中,还观察到CHOP化学疗法方案的所有单一组分的组合益处。此外,在两种不同的EZH2突变体异种移植模型中,与CHOP具有强的组合益处,并且此效果在其中化学疗法方案省略了阿霉素的第三种EZH2突变体异种移植模型的研究中被保留。
本披露是基于这样的发现:EZH2组蛋白甲基转移酶抑制剂和其他抗癌剂可以组合用于治疗某些肿瘤,并且具有比那些通过单独使用EZH2组蛋白甲基转移酶抑制剂和抗癌剂治疗肿瘤取得更优的结果。相应地,本披露提供一种包含EZH2组蛋白甲基转移酶抑制剂和一种或多种其他治疗剂的组合物,以及它们的使用来治疗疾病(例如癌症)的方法,其中该过程可以受调节组蛋白或其他蛋白质的甲基化状态的影响。在某个实施例中,本披露的特征在于一种包含化合物44和强的松的组合物。在某些实施例中,本披露的特征在于一种包含化合物44和纳成托克斯的组合疗法。本披露还包括用于组合疗法的方法,这些方法包括将EZH2组蛋白甲基转移酶抑制剂和一种或多种治疗剂(诸如化合物44和强的松)用于治疗癌症,例如,GCB淋巴瘤、滤泡性淋巴瘤(FL)和弥漫性细胞大B细胞淋巴瘤(DCLBL)。具体而言,本披露的方法对于治疗或预防癌症或抑制癌细胞增生是有用的。
在一个方面,本披露的特征在于一种包含具有以下化学式(VIa)的化合物和一种或多种其他治疗剂(诸如酪氨酸激酶抑制剂)或其药学上可接受的盐或酯的组合物。
具有化学式(VIa)的化合物可以包括一个或多个以下特征:
Ra和Rb各自独立地是H或C1-C6烷基。
Ra和Rb与其所附接的N原子一起是具有0或1个另外的杂原子的4至7元杂环烷基环、任选地经一个或多个-Q3-T3取代的C1-C6烷基和4至12元(例如,4至7元)杂环烷基环。
Q3是键或未经取代或经取代的C1-C3烷基接头。
T3是H、卤素、4至7元杂环烷基、C1-C3烷基、ORd、COORd、-S(O)2Rd、或-NRdRe,Rd和Re各自独立地是H或C1-C6烷基。
R7是C1-C6烷基、C3-C8环烷基或4至12元(例如,4至7元)杂环烷基,这些基团各自任选地经一个或多个-Q5-T5取代。例如,R7不是H。
R7是任选地经一个或多个-Q5-T5取代的4至7元杂环烷基。
R7是哌啶基、四氢吡喃、环戊基、或环己基,这些基团各自任选地经一个Q5-T5取代。
T5是H、卤素、C1-C6烷基、C1-C6烷氧基、C3-C8环烷基、C6-C10芳基、或4至12元(例如,4至7元)杂环烷基。
Q5是键,并且T5是C1-C6烷基、C3-C8环烷基、或4至12元(例如,4至7元)杂环烷基。
Q5是CO、S(O)2、或NHC(O);并且T5是C1-C6烷基、C1-C6烷氧基、C3-C8环烷基、或4至12元(例如,4至7元)杂环烷基。
Q5是C1-C3烷基接头,并且T5是H或C6-C10芳基。
Q5是C1-C3烷基接头,并且T5是C3-C8环烷基、4至7元杂环烷基、或S(O)qRq。
R7是环戊基或环己基,这些基团各自任选地经一个Q5-T5取代。
Q5是NHC(O),并且T5是C1-C6烷基或C1-C6烷氧基。
R7是异丙基。
R2和R4各自独立地是H或任选地经氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、或C6-C10芳基取代的C1-C6烷基。
R8是H、甲基、或乙基。
R8是甲基。
R8是乙基。
R8是4至7-杂环烷基,例如,四氢吡喃。
本披露的特征在于一种包含选自表1的化合物或其药学上可接受的盐或酯和一种或多种其他治疗剂的组合物。
例如,EZH2抑制剂是具有以下化学式的化合物44(也被称为EPZ-6438、E7438):
或其药学上可接受的盐或溶剂化物和一种或多种其他治疗剂。
例如,EZH2抑制剂是具有以下化学式的化合物A:
或其立体异构体或其药学上可接受的盐或溶剂化物。
例如,EZH2抑制剂是具有以下化学式的化合物B(也被称为EPZ011989):
或其立体异构体或其药学上可接受的盐或溶剂化物。
例如,EZH2抑制剂是具有以下化学式的化合物C:或其立体异构体或其药学上可接受的盐或溶剂化物。
例如,EZH2抑制剂是具有以下化学式的GSK-126:或其立体异构体或其药学上可接受的盐或溶剂化物。
在本披露的这个和其他方面,在一个实施例中,其他治疗剂是抗癌剂。
在本披露的这个和其他方面,在一个实施例中,其他治疗剂是酪氨酸激酶抑制剂。例如,EZH2抑制剂和一种或多种酪氨酸激酶抑制剂同时或顺序地给予。例如,EZH2抑制剂在给予一种或多种酪氨酸激酶抑制剂之前给予。
在本披露的这个和其他方面,在一个实施例中,其他治疗剂是VEGF/VEGFR抑制剂。例如,EZH2抑制剂和一种或多种VEGF/VEGFR抑制剂同时或顺序地给予。例如,EZH2抑制剂在给予一种或多种VEGF/VEGFR抑制剂之前给予。
在本披露的这个和其他方面,在一个实施例中,其他治疗剂是糖皮质激素类。
在本披露的这个和其他方面,在一个实施例中,其他治疗剂选自强的松、泼尼松龙、环磷酰胺、长春新碱、阿霉素、马磷酰胺、顺铂、阿糖胞苷、依维莫司、地西他滨、地塞米松、及其类似物、衍生物、或组合。
在本披露的这个和其他方面,在一个实施例中,其他治疗药剂是强的松、或其类似物或衍生物。
本披露还提供了包含选自具有本文所披露化学式(I)-(VIa)的那些的化合物或其药学上可接受的盐和一种或多种治疗剂、以及药学上可接受的载体的药物组合物。
本披露还提供了包含选自表I的化合物、一种或多种其他治疗剂或其药学上可接受的盐、以及药学上可接受的载体的药物组合物。
本披露还提供了包含选自具有本文所披露化学式(I)-(VIa)的那些化合物的化合物或其药学上可接受的盐、一种或多种其他治疗剂、以及药学上可接受的载体的药物组合物。
本披露的另一个方面是一种用于通过向对其有需要的受试者给予治疗有效量的组合物来治疗或预防疾病的方法,该组合物包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种另外的治疗剂。本披露的疾病可以通过调节组蛋白或其他蛋白质的甲基化状态来影响、治疗、或预防。例如,该疾病是癌症、癌前病状、或神经系统疾病。优选地,该淋巴瘤是非霍奇金淋巴瘤、滤泡性淋巴瘤或弥散性大B细胞淋巴瘤。可替代地,白血病是慢性骨髓性白血病(CML)。该癌前病状是例如,骨髓增生异常综合征(MDS,以前称为白血病前期)。
本披露的受试者包括已经被诊断为癌症或癌前病状、具有癌症或癌前病状的症状、或具有发展癌症或癌前病状的风险的任何人类受试者。本披露的受试者包括表达突变体EZH2的任何人类受试者。例如,突变体EZH2包括一个或多个突变,其中该突变是取代、点突变、无义突变、错义突变、缺失、或插入。本披露的突变体EZH2可在如SEQ ID NO:6所定义的底物口袋结构域中包括突变。突变体EZH2可在氨基酸Y641处具有取代。优选地,突变体EZH2具有以下突变中的一个:在SEQ ID NO:1的氨基酸位置641处的苯丙氨酸(F)取代野生型残基酪氨酸(Y)(Y641F);在SEQ ID NO:1的氨基酸位置641处的组氨酸(H)取代野生型残基酪氨酸(Y)(Y641H);在SEQ ID NO:1的氨基酸位置641处的天冬酰胺(N)取代野生型残基酪氨酸(Y)(Y641N);在SEQ ID NO:1的氨基酸位置641处的丝氨酸(S)取代野生型残基酪氨酸(Y)(Y641S);以及在SEQ ID NO:1的氨基酸位置641处的半胱氨酸(C)取代野生型残基酪氨酸(Y)(Y641C)。
EZH2的其他突变可包括但不限于:在SEQ ID NO:1的氨基酸位置677处的甘氨酸(G)取代野生型残基丙氨酸(A)(A677G);在SEQ ID NO:1的氨基酸位置687处的缬氨酸(V)取代野生型残基丙氨酸(A)(A687V);在SEQ ID NO:1的氨基酸位置674处的甲硫氨酸(M)取代野生型残基缬氨酸(V)(V674M);在SEQ ID NO:1的氨基酸位置685处的组氨酸(H)取代野生型残基精氨酸(R)(R685H);在SEQ ID NO:1的氨基酸位置685处的半胱氨酸(C)取代野生型残基精氨酸(R)(R685C);在SEQ ID NO:3的氨基酸位置322处的丝氨酸(S)取代野生型残基天冬酰胺(N)(N322S)、在SEQ ID NO:3的氨基酸位置288处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R288Q)、在SEQ ID NO:3的氨基酸位置573处的异亮氨酸(I)取代野生型残基苏氨酸(T)(T573I)、在SEQ ID NO:3的氨基酸位置664处的谷氨酸(E)取代野生型残基天冬氨酸(D)(D664E)、在SEQ ID NO:5的氨基酸位置458处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R458Q)、在SEQ ID NO:3的氨基酸位置249处的赖氨酸(K)取代野生型残基谷氨酸(E)(E249K)、在SEQ ID NO:3的氨基酸位置684处的半胱氨酸(C)取代野生型残基精氨酸(R)(R684C)、在SEQ ID NO:21的氨基酸位置628处的组氨酸(H)取代野生型残基精氨酸(R)(R628H)、在SEQ ID NO:5的氨基酸位置501处的组氨酸(H)取代野生型残基谷氨酰胺(Q)(Q501H)、在SEQ ID NO:3的氨基酸位置192处的天冬酰胺(N)取代野生型残基天冬氨酸(D)(D192N)、在SEQ ID NO:3的氨基酸位置664处的缬氨酸(V)取代野生型残基天冬氨酸(D)(D664V)、在SEQ ID NO:3的氨基酸位置704处的亮氨酸(L)取代野生型残基缬氨酸(V)(V704L)、在SEQ ID NO:3的氨基酸位置132处的丝氨酸(S)取代野生型残基脯氨酸(P)(P132S)、在SEQ ID NO:21的氨基酸位置669处的赖氨酸(K)取代野生型残基谷氨酸(E)(E669K)、在SEQ ID NO:3的氨基酸位置255处的苏氨酸(T)取代野生型残基丙氨酸(A)(A255T)、在SEQ ID NO:3的氨基酸位置726处的缬氨酸(V)取代野生型残基谷氨酸(E)(E726V)、在SEQ ID NO:3的氨基酸位置571处的酪氨酸(Y)取代野生型残基半胱氨酸(C)(C571Y)、在SEQ ID NO:3的氨基酸位置145处的半胱氨酸(C)取代野生型残基苯丙氨酸(F)(F145C)、在SEQ ID NO:3的氨基酸位置693处的苏氨酸(T)取代野生型残基天冬酰胺(N)(N693T)、在SEQ ID NO:3的氨基酸位置145处的丝氨酸(S)取代野生型残基苯丙氨酸(F)(F145S)、在SEQ ID NO:21的氨基酸位置109处的组氨酸(H)取代野生型残基谷氨酰胺(Q)(Q109H)、在SEQ ID NO:21的氨基酸位置622处的半胱氨酸(C)取代野生型残基苯丙氨酸(F)(F622C)、在SEQ ID NO:3的氨基酸位置135处的精氨酸(R)取代野生型残基甘氨酸(G)(G135R)、在SEQ ID NO:5的氨基酸位置168处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R168Q)、在SEQ ID NO:3的氨基酸位置159处的精氨酸(R)取代野生型残基甘氨酸(G)(G159R)、在SEQ ID NO:5的氨基酸位置310处的半胱氨酸(C)取代野生型残基精氨酸(R)(R310C)、在SEQ ID NO:3的氨基酸位置561处的组氨酸(H)取代野生型残基精氨酸(R)(R561H)、在SEQ ID NO:21的氨基酸位置634处的组氨酸(H)取代野生型残基精氨酸(R)(R634H)、在SEQ ID NO:3的氨基酸位置660处的精氨酸(R)取代野生型残基甘氨酸(G)(G660R)、在SEQ ID NO:3的氨基酸位置181处的半胱氨酸(C)取代野生型残基酪氨酸(Y)(Y181C)、在SEQ ID NO:3的氨基酸位置297处的精氨酸(R)取代野生型残基组氨酸(H)(H297R)、在SEQ ID NO:21的氨基酸位置612处的丝氨酸(S)取代野生型残基半胱氨酸(C)(C612S)、在SEQ ID NO:3的氨基酸位置694处的酪氨酸(Y)取代野生型残基组氨酸(H)(H694Y)、在SEQ ID NO:3的氨基酸位置664处的丙氨酸(A)取代野生型残基天冬氨酸(D)(D664A)、在SEQ ID NO:3的氨基酸位置150处的苏氨酸(T)取代野生型残基异亮氨酸(I)(I150T)、在SEQ ID NO:3的氨基酸位置264处的精氨酸(R)取代野生型残基异亮氨酸(I)(I264R)、在SEQ ID NO:3的氨基酸位置636处的亮氨酸(L)取代野生型残基脯氨酸(P)(P636L)、在SEQ ID NO:3的氨基酸位置713处的苏氨酸(T)取代野生型残基异亮氨酸(I)(I713T)、在SEQ ID NO:5的氨基酸位置501处的脯氨酸(P)取代野生型残基谷氨酰胺(Q)(Q501P)、在SEQ ID NO:3的氨基酸位置243处的谷氨酰胺(Q)取代野生型残基赖氨酸(K)(K243Q)、在SEQ ID NO:5的氨基酸位置130处的天冬氨酸(D)取代野生型残基谷氨酸(E)(E130D)、在SEQ ID NO:3的氨基酸位置509处的甘氨酸(G)取代野生型残基精氨酸(R)(R509G)、在SEQ ID NO:3的氨基酸位置566处的组氨酸(H)取代野生型残基精氨酸(R)(R566H)、在SEQ ID NO:3的氨基酸位置677处的组氨酸(H)取代野生型残基天冬氨酸(D)(D677H)、在SEQ ID NO:5的氨基酸位置466处的天冬酰胺(N)取代野生型残基赖氨酸(K)(K466N)、在SEQ ID NO:3的氨基酸位置78处的组氨酸(H)取代野生型残基精氨酸(R)(R78H)、在SEQ ID NO:6的氨基酸位置1处的蛋氨酸(M)取代野生型残基赖氨酸(K)(K6M)、在SEQ ID NO:3的氨基酸位置538处的亮氨酸(L)取代野生型残基丝氨酸(S)(S538L)、在SEQ ID NO:3的氨基酸位置149处的谷氨酰胺(Q)取代野生型残基亮氨酸(L)(L149Q)、在SEQ ID NO:3的氨基酸位置252处的缬氨酸(V)取代野生型残基亮氨酸(L)(L252V)、在SEQ ID NO:3的氨基酸位置674处的缬氨酸(V)取代野生型残基亮氨酸(L)(L674V)、在SEQ ID NO:3的氨基酸位置656处的缬氨酸(V)取代野生型残基丙氨酸(A)(A656V)、在SEQ ID NO:3的氨基酸位置731处的天冬氨酸(D)取代野生型残基丙氨酸(A)(Y731D)、在SEQ ID NO:3的氨基酸位置345处的苏氨酸(T)取代野生型残基丙氨酸(A)(A345T)、在SEQ ID NO:3的氨基酸位置244处的天冬氨酸(D)取代野生型残基丙氨酸(A)(Y244D)、在SEQ ID NO:3的氨基酸位置576处的色氨酸(W)取代野生型残基半胱氨酸(C)(C576W)、在SEQ ID NO:3的氨基酸位置640处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N640K)、在SEQ ID NO:3的氨基酸位置675处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N675K)、在SEQ ID NO:21的氨基酸位置579处的酪氨酸(Y)取代野生型残基天冬氨酸(D)(D579Y)、在SEQ ID NO:3的氨基酸位置693处的异亮氨酸(I)取代野生型残基天冬酰胺(N)(N693I)、以及在SEQ ID NO:3的氨基酸位置693处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N693K)。
EZH2的其他突变可以包括:在SEQ ID NO:3、5或21的氨基酸位置730、391、461、441、235、254、564、662、715、405、685、64、73、656、718、374、592、505、730、或363或者编码SEQ ID NO:3、5、或2的核酸序列的相应核苷酸位置处的移码;在SEQ ID NO:3、5或21的氨基酸位置148和149处的谷氨酸(E)和亮氨酸(L)缺失,或者在SEQ ID NO:3,5或21的氨基酸位置733、25、317、62、553、328、58、207、123、63、137、或60处的无义突变。
本披露的受试者可具有对任何一种或多种其他治疗剂或本文所述的任何化合物的抗性。例如,该受试者可具有对EZH抑制剂或强的松的抗性。
本披露的特征在于一种用于抑制癌细胞增生的方法,该方法包括使所述癌细胞与包含具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐和一种或多种另外的治疗药剂的组合物接触。抑制癌细胞增生包括延缓癌细胞生长、诱导细胞死亡、降低癌细胞活力、抑制或延缓肿瘤生长、或减小肿瘤大小。
本披露的特征在于组合疗法的方法,其中具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐和一种或多种其他治疗剂同时或顺序地给予。例如,具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐可在给予一种或多种其他治疗剂之前给予。例如,具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐可在给予包含具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐和一种或多种其他治疗剂的组合物之前给予。同时或顺序地给予具有化学式(I)-(VIa)的任何化合物和/或每一治疗剂,可以通过任何适当的途径来实现,包括但不限于口服途径、静脉途径、肌内途径、并且通过粘膜组织直接吸收。可以通过相同途径或通过不同途径给予这些治疗剂。
本披露所展示的组合疗法的方法可导致协同效应,其中化合物或其他治疗剂的组合的效果大于任何化合物或其他治疗剂作为单一药剂给予时所产生的效果的总和。协同效应还可以是任何化合物或其他治疗剂作为单一药剂给予时所不能达到的效果。协同效应可以包括但不限于通过减小肿瘤大小、抑制肿瘤生长或增加受试者存活来治疗癌症的效果。协同效应还可以包括降低癌症细胞活力、诱发癌症细胞死亡、和抑制或延迟癌症细胞生长。
本披露的组合物可以每天0.01mg/kg至约1000mg/kg的剂量给予。具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐可以每天0.01mg/kg至约1000mg/kg的剂量给予。任何其他治疗剂可以每天0.01mg/kg至约1000mg/kg的剂量给予。
除非另有定义,否则本文中使用的所有技术和科学术语都具有与本发明所属领域的普通技术人员一般所理解的相同的含义。在说明书中,除非上下文明确指示其他情况,否则单数形式也包含复数含义。尽管在本发明的实践或测试中可以使用与本文所述的方法和材料相似或等效的方法和材料,但是下文描述了适合的方法和材料。本文提及的所有公开案、专利申请、专利以及其他参考文献均通过引用结合在此。不承认本文所引用的参考文献是所要求的发明的现有技术。在发生冲突的情况下,以包括定义的本说明书为准。另外,材料、方法以及实例仅是说明性的并不意在是限制性的。
根据以下详细描述和权利要求书,本发明的其他特征和优点将变得显而易见。
附图说明
图1是示出体外组合测定的设计的方案。
预处理模型A:在烧瓶中用7个剂量的化合物44(也称为EZH-6438)或DMSO将淋巴瘤细胞预处理4天。使用HP D300数字分配器将细胞归一化并且用化合物44和感兴趣的化合物共处理(以恒定比率的两种化合物的组合在基质中的3个重复板)。在共处理3天后,使用经由ATP含量测量细胞活力
预处理模型B、C:用1个剂量的感兴趣的化合物(B)或7个剂量的化合物44(C)和DMSO将淋巴瘤细胞预处理4天。然后将细胞归一化并且用1个剂量的感兴趣的化合物和7个剂量的化合物44和DMSO共处理3天。使用Guava试剂测定活力。
共处理模型:用7个剂量的化合物44和1个剂量的感兴趣的化合物将淋巴瘤细胞处理4或7天。使用Guava试剂测定活力。
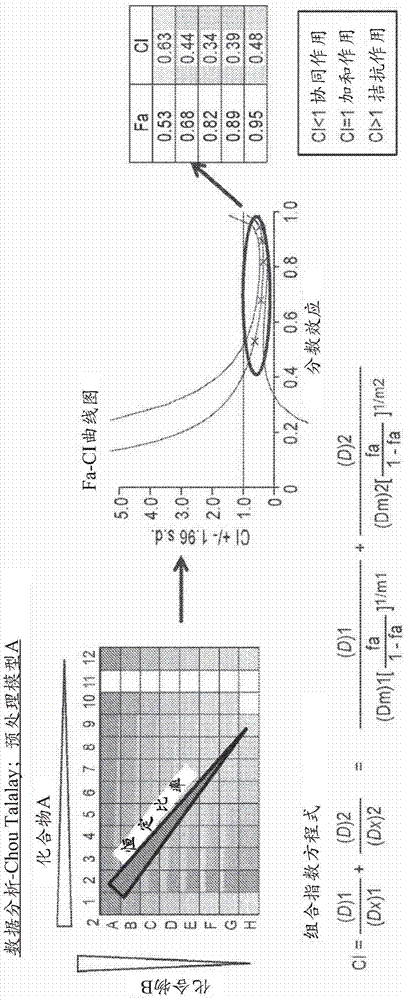
图2是显示使用Chou-Talalay方法的数据分析的方案。使用Chou-Talalay方法3对药物组合进行了协同作用定量。组合指数(CI)方程式提供了加和作用(CI=1)、协同作用(CI<1)和拮抗作用(CI>1)的数量定义。此公式采用来自恒定比率的药物组合的Fa值来确定CI值。所得的曲线图(Fa-CI曲线图)示出了由95%置信区间括号内的所得CI值。这些Fa-CI曲线图是使用软件Calcusyn产生的。协同作用的统计学上显著的CI值例如CI值<1的那些,其中置信区间线也低于1。
图3A-3F是一系列Fa-Cl曲线图,这些曲线图显示了CHOP组分和化合物44在突变体EZH2生发中心B细胞淋巴瘤细胞系中的组合益处。化合物44和阿霉素在WSU-DLCL2细胞(A)中协同作用,并且在SU-DHL-10细胞(D)中产生加和效应。在WSU-DLCL2细胞(C)和SU-DHL-10细胞(F)中观察到与马磷酰胺的组合益处。在EZH2Y646突变体细胞系:WSU-DLCL2细胞(B)和SU-DHL-10细胞(E)中还观察到与长春新碱的组合益处。在WSU-DLCL2中,阿霉素的剂量范围是0.16-20nM,长春新碱是0.04-5nM,马磷酰胺是0.156-10μM,并且化合物44是15-1000nM。在SU-DHL-10细胞中,阿霉素的剂量范围是0.5-60nM,长春新碱是0.016-2nM,马磷酰胺是0.156-10μM,并且化合物44是1.56-100nM。根据预处理模型A处理细胞,并用该Calcusyn软件进行数据分析。
图4A-4D是一系列图,这些图显示了糖皮质激素受体激动剂增强了化合物44在EZH2突变体淋巴瘤系中的效力。当与糖皮质激素受体激动剂组合时,化合物44的效力显著地增加了。根据预处理模型A,添加泼尼松龙(A、C)或地塞米松(B、D)在2个EZH2Y646F突变体DLBCL系中产生在化合物44的IC50的剂量依赖性转变。在两种细胞系中,泼尼松龙的剂量范围是15nM-1000nM,并且地塞米松的剂量范围是1.5nM-100nM。化合物44的剂量范围在WSU-DLCL2细胞中是15-1000nM,并且在SU-DHL-10细胞中是1.5-100nM。
图5A-5D是一系列图,这些图显示了化合物44与糖皮质激素受体激动剂在EZH2 WT生发中心(而不是活化的B细胞淋巴瘤系)中的组合益处。根据预处理模型A,在用化合物44和泼尼松龙(图5A)或地塞米松(图5B)处理后,在DOHH2 EZH2野生型GCB细胞中观察到组合益处。相比之下,在Toledo细胞(一种EZH2野生型ABC淋巴瘤系)中没有观察到组合益处(图5C和5D分别为Cpd44+泼尼松龙和Cpd44+地塞米松)。在两种细胞系中,泼尼松龙的剂量范围是从15nM-1000nM,并且地塞米松的剂量范围是从1.5nM-100nM。化合物44在DOHH2细胞中是从0.16-10nM,并且在Toledo细胞中是15.6-1000nM。
图6是一个汇总表,该表显示化合物44/糖皮质激素受体激动剂组合克服了抵抗EZH2抑制剂的细胞系中的EZH2抑制剂(EZH2i)不敏感性。总的来说,泼尼松和化合物44的组合导致在所有测试的GCB细胞系(不仅是EZH2i敏感型细胞系)中的更高敏感性。除了RL细胞,其中药剂加入的顺序是至关重要的,因为用泼尼松龙预温育,接着加入化合物44是有无效的。
图7A和7B是两个曲线图,这些曲线图显示在EZH2突变体淋巴瘤细胞系中使用化合物44和其他靶向疗法的组合观察到非常强的协同作用。当化合物44与BCL2抑制剂纳成托克斯(在图7A中)以及与mTOR抑制剂依维莫司(在图7B中)组合时,观察到非常强的协同作用。纳威托克斯的剂量范围是0.16-10μM,依维莫司是0.04-5nM,并且化合物44是31-2000nM。在预处理模型A中产生这些数据,并且用Calcusyn软件分析数据。
图8是与化合物44的组合的总结表。在EZH2突变体淋巴瘤系中测试的所有药剂与化合物44实现了组合益处。糖皮质激素受体激动剂显示对EZH2WT和突变体GCB淋巴瘤系(而不是在ABC淋巴瘤细胞系中)的组合益处。
图9A-9C是一系列图,这些图显示了化合物44-CHOP组合在几个EZH2突变体淋巴瘤异种移植模型中显示出相比单一药剂增强的抗肿瘤活性。(9A)。如在这些方法中指定的,使用化合物44、CHOP、或其组合处理WSU-DLCL2(EZH2 Y646F)异种移植物28天。绘制了平均肿瘤体积+/-SEM图。150mg/kg TID和225mg/kg BID剂量的化合物44在肿瘤生长抑制中比单独运载体在统计学上更有意义(*p值<0.05)。相比单独使用任何单一的药剂,使用225mg/kg BID的化合物44加上CHOP处理导致更大的肿瘤消退(相对于运载体,***p值<0.001)。通过重复测量ANOVA计算统计。(9B)。如在这些方法中指定的,使用化合物44、CHOP、或其组合处理SU-DHL6(EZH2 Y646N)异种移植物28天。在顶部图中绘制了平均肿瘤体积+/-SEM图。在28天的处理期中,CHOP或单一药剂化合物44单独对肿瘤生长没有效果,但是使用225mg/kg BID化合物44加上CHOP处理导致肿瘤消退,同时在给药停止32天后还保持肿瘤生长延迟(*p值<0.0001)。直至60天的存活曲线(底部图)显示使用化合物44+CHOP治疗动物出现的显著的肿瘤生长延迟(**p值<0.05)。使用双尾t检验进行统计。(9C)。如在这些方法中指定的,使用化合物44、COP(没有阿霉素成分的SOC)、或其组合处理SUDHL-10(EZH2 Y646F)异种移植物28天。在顶部图中绘制了平均肿瘤体积+/-SEM图。在中部图中绘制了在肿瘤生长延迟研究中直至60天的百分比存活图(注意:500mg/kg和250m g/kg+COP存活曲线是重叠的)。在底部图中比较了平均肿瘤重量,显示出组之间肿瘤重量显著的不同(*p值<0.05,**p值<0.01,****p值<0.0001)。
图10A-10C是显示在EZH2突变体细胞系中糖皮质激素靶基因被泼尼松龙+化合物44组合上调的图。(A)的表达水平。瑟斯崔因;(B)。将TNF和(C)GILZ对使用所指示的单一试剂或组合的每个细胞系的DMSO对照进行归一化。使用ΔΔCt方法和RPLPO作为管家基因来定量倍数变化值。
图11A和11B是显示整体的H3K27乙酰化和三甲基化不受泼尼松龙或组合治疗影响的图。使用增加剂量的泼尼松龙、化合物44、或组合化合物44+恒定剂量的泼尼松龙处理细胞4天。酸提取的组蛋白通过ELISA分析H3K27me3水平(图11A)(泼尼松龙,左图;化合物44和组合,右图,其中IC50值作为每个图的插图)或通过蛋白质印迹分析H3K27ac水平(图11B)。对于泼尼松龙的治疗,因为使用该化合物没有观察到剂量依赖性变化,H3K27me3数值被表示为柱状图。
图12是蛋白质印迹,该印迹显示使用化合物44或泼尼松龙单一药剂治疗对SMARCBl蛋白质水平没有影响。
图13是显示EZH2参与抗VEGF疗法的抗药性的方案,该方案在患有转移性肾细胞癌的患者中仍旧是一项挑战,因为最初对治疗起反应的患者最终产生抗性和肿瘤进展。
图14是显示体内组合测定的设计的方案。
图15A-15E是一系列图,这些图显示了EZH2的抑制在ccRCC细胞系中增加了对舒尼替尼的敏感性。图15A-B:用单一药剂和组合进行细胞抑制测定。图15C:指示舒尼替尼和GSK126组合的浓度与协同作用的组合指数值。图15D-E:用舒尼替尼、GSK126或两者处理48小时的ccRCC细胞系。条形图指示,与单独的单一药剂相比,组合治疗组中的细胞活力显著降低。
图16A-16B是一系列图,这些图显示了EZH2的敲低在ccRCC细胞系中增加了对舒尼替尼的敏感性。图16A:蛋白质印迹分析显示786-0细胞系中EZH2敲低的效率。图16B:与乱序模板对照786-0_shRNA相比,786-0细胞中EZH2的特异性敲低对舒尼替尼更敏感。
图17A-17C是一系列图,这些图显示了EZH2抑制在RP-R-02LM ccRCC PDX模型中使肿瘤对舒尼替尼敏感。肿瘤生长曲线显示,与单独的舒尼替尼相比,组合组中的肿瘤生长显著减少(图17A和17B)。随着时间变化的体重曲线指示小鼠对两种药物和给药方案耐受良好(图17C)。
图18A-18B是一系列图,这些图显示了与单独的药剂相比,舒尼替尼和化合物B的组合显示增加的抗转移效应。图18A包括肺组织的代表性图片,并且H&E染色指示在舒尼替尼治疗组中减少的转移在组合组中是增加的。肺中存在的肿瘤细胞中Ki67和CD31水平的表达在治疗组之间没有差异。图18B是进行不同治疗的肺中的肿瘤结节数量的图。
图19A-19B是一系列图,这些图显示了舒尼替尼和化合物B的组合在ccRCC PDX模型中降低了细胞增生。如Ki67染色(图19A)和定量(图19B)所指示,在肿瘤中主要位点处观察到组合治疗和化合物B治疗的肿瘤中的细胞增生减少。*P<0.05,**p,0.001,ns=不显著
图20A-20B是一系列图,这些图显示了舒尼替尼和化合物B的组合在ccRCC PDX模型中降低了血管密度。如mCD31染色(图20A)和定量(图20B)所指示,在舒尼替尼和组合治疗组中,在肿瘤中初始位点处观察到血管密度显著降低。*P<0.05,**p<0.001,***p<0.0001,ns=不显著
具体实施方式
本披露部分地基于这样的发现:EZH2组蛋白甲基转移酶抑制剂和其他抗癌剂可以组合用于治疗某些肿瘤,并且具有比那些通过单独使用EZH2组蛋白甲基转移酶抑制剂和抗癌剂治疗肿瘤取得更优的结果。相应地,本披露提供一种包含EZH2组蛋白甲基转移酶抑制剂和一种或多种其他治疗剂的组合物,以及它们的使用来治疗疾病(例如癌症)的方法,其中该过程可以受调节组蛋白或其他蛋白质的甲基化状态的影响。在某个实施例中,本披露的特征在于一种包含具有化学式(I)-(VIa)的化合物和酪氨酸激酶抑制剂(诸如舒尼替尼)的组合物。在某个实施例中,本披露的特征在于一种包含具有化学式(I)-(VIa)的化合物和抗VEGF剂(诸如舒尼替尼)的组合物。在某个实施例中,本披露的特征在于一种包含具有化学式(I)-(VIa)的化合物和强的松的组合物。本披露还包括用于组合疗法的方法,这些方法包括将EZH2组蛋白甲基转移酶抑制剂和一种或多种治疗剂,诸如具有化学式(I)-(VIa)的化合物和舒尼替尼或强的松,用于治疗癌症,例如,肾细胞癌、滤泡性淋巴瘤(FL)和弥漫性细胞大B细胞淋巴瘤(DCLBL)。具体而言,本披露的方法对于治疗或预防癌症或抑制癌细胞增生是有用的。
EZH2是组蛋白甲基转移酶,它是PRC2复合物的催化亚基,该PRC2复合物催化组蛋白H3上的赖氨酸27(H3-K27)的单至三甲基化。组蛋白H3-K27三甲基化是抑制接近组蛋白修饰位点的特定基因的转录的机制。已知这种三甲基化是癌症(诸如前列腺癌)中表达改变的癌症标记物(参见例如,美国专利申请公开号2003/0175736;通过引用以其全文结合在此)。其他研究为EZH2表达调节异常、转录抑制和致瘤性转化间的功能联系提供了证据。Varambally等人(2002)Nature[自然]419(6907):624-9Kleer等人(2003)Proc NatlAcadSci USA[美国国家科学院院刊]100(20):11606-11。
对于抗VEGF疗法的响应在患有转移性肾细胞癌的患者中仍旧是一项挑战,因为最初对治疗起反应的患者最终产生抗性和进展。表观遗传改变(诸如作为组蛋白甲基转移酶的zeste同系物的增强子(EZH2)的过表达)已经显示在人类恶性肿瘤中频繁地过表达,涉及许多基因的表观遗传沉默和调节肿瘤血管发生。最近据报道,EZH2涉及抗药性。参见例如,Herranz、Nicolás等人“Polycomb complex 2is required for E-cadherin repression by the Snaill transcription factor[多梳蛋白(polycomb)复合物2是通过Snaill转录因子的E-钙粘蛋白抑制所需要的]”Molecular and cellular biology[分子与细胞生物学]28.15(2008):4772-4781;和Adelaiye、Remi等人“Sunitinib dose-escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications[舒尼替尼剂量递增克服了透明细胞肾细胞癌的短暂抗性并且与表观遗传修饰相关]”Molecular cancer therapeutics[分子癌症治疗学](2014):molcanther-0208。相应地,EZH2的抑制可以使肿瘤对抗VEGF剂重新敏化。
本披露的一个方面涉及用于通过向表达突变体EZH2的受试者给予治疗有效量的EZH2抑制剂和一种或多种其他治疗剂(诸如酪氨酸激酶抑制剂或抗VEGF剂)来治疗或缓解受试者的癌症或癌前病状的症状的方法。本披露的突变体EZH2是指突变体EZH2多肽或编码突变体EZH2多肽的核酸序列。在某些实施例中,突变体EZH2在如SEQ ID NO:6所定义的它的底物口袋结构域中包括一个或多个突变。例如,该突变体可以是取代、点突变、无义突变、错义突变、缺失、或插入。
人类EZH2核酸和多肽已经在先前描述。参见例如,Chen等人(1996)Genomics[基因组学]38:30-7[746氨基酸];Swiss-Prot登录号Q15910[746个氨基酸];GenBank登录号NM_004456和NP_004447(同种型a[751个氨基酸]);以及GenBank登录号NM_152998和NP_694543(同种型b[707个氨基酸]),这些参考文献中的每一个通过引用以其全文结合在此。
以下提供了基于核受体结合SET结构域蛋白1(NSD1)的A链的部分EZH2蛋白的结构模型。此模型对应于SEQ ID NO:1的EZH2序列的氨基酸残基533-732。
以下提供了此结构模型的相应氨基酸序列。底物口袋结构域中的残基加下划线。SET结构域中的残基以斜体显示。
据信EZH2的催化位点位于被称为SET结构域的蛋白质的保守结构域中。EZH2的SET结构域的氨基酸序列由跨越Swiss-Prot登录号Q15910(SEQ ID NO:1)的氨基酸残基613-726的以下部分序列提供:
HLLLAPSDVAGWGIFIKDPVQKNEFISEYCGEIISQDEADRRGKVYDKYMCSFLFNLNNDFVVDATRKGNKIRFANHSVNPNCYAKVMMVNGDHRIGIFAKRAIQTGEELFFDY(SEQ ID NO:7)。
SEQ ID NO:7中加下划线显示的酪氨酸(Y)残基是Swiss-Prot登录号Q15910(SEQ ID NO:1)中的Tyr641(Y641)。
GenBank登录号NP_004447(SEQ ID NO:3)的SET结构域跨越氨基酸残基618-731并且与SEQ ID NO:6相同。对应于在SEQ ID NO:7中加下划线显示的Swiss-Prot登录号Q15910中的Y641的酪氨酸残基是GenBank登录号NP_004447(SEQ ID NO:3)中的Tyr646(Y646)。
GenBank登录号NP_694543(SEQ ID NO:5)的SET结构域跨越氨基酸残基574-687并且与SEQ ID NO:7相同。对应于在SEQ ID NO:7中加下划线显示的Swiss-Prot登录号Q15910中的Y641的酪氨酸残基是GenBank登录号NP_694543(SEQ ID NO:5)中的Tyr602(Y602)。
编码GenBank登录号NP_004447的SET结构域的核苷酸序列是catctattgctggcaccatctgacgtggcaggctgggggatttttatcaaagatcctgtgcagaaaaatgaattcatctcagaatactgtggagagattatttctcaagatgaagctgacagaagagggaaagtgtatgataaatacatgtgcagctttctgttcaacttgaacaatgattttgtggtggatgcaacccgcaagggtaacaaaattcgttttgcaaatcattcggtaaatccaaactgctatgcaaaagttatgatggttaacggtgatcacaggataggtatttttgccaagagagccatccagactggcgaagagctgttttttgattac
(SEQ ID NO:8),
其中编码Y641的密码子加下划线显示。
出于本申请的目的,人类EZH2的氨基酸残基Y641应被理解为是指在Swiss-Prot登录号Q15910中是或对应于Y641的酪氨酸残基。
还出于本申请的目的,人类EZH2的Y641突变体,和相等地,EZH2的Y641突变体应被理解为是指人类EZH2中对应于野生型人类EZH2的Y641的氨基酸残基被除酪氨酸外的氨基酸残基取代。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被除酪氨酸外的氨基酸残基取代。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被苯基丙氨酸(F)取代。对应于这个实施例的EZH2的Y641突变体此处被称为Y641F突变体,或等同地,Y641F。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被组氨酸(H)取代。对应于这个实施例的EZH2的Y641突变体此处被称为Y641H突变体,或等同地,Y641H。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被天冬酰胺(N)取代。对应于这个实施例的EZH2的Y641突变体此处被称为Y641N突变体,或等同地,Y641N。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被丝氨酸(S)取代。对应于这个实施例的EZH2的Y641突变体此处被称为Y641S突变体,或等同地,Y641S。
在一个实施例中,EZH2的Y641突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的Y641的单个氨基酸残基被半胱氨酸(C)取代。对应于这个实施例的EZH2的Y641突变体此处被称为Y641C突变体,或等同地,Y641C。
在一个实施例中,EZH2的A677突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的A677的单个氨基酸残基被非丙氨酸的氨基酸,优选是甘氨酸(G)取代。对应于这个实施例的EZH2的A677突变体此处被称为A677突变体,并且优选是A677G突变体,或等同地,A677G。
在一个实施例中,EZH2的A687突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的A687的单个氨基酸残基被非丙氨酸的氨基酸,优选是缬氨酸(V)取代。对应于这个实施例的EZH2的A687突变体此处被称为A687突变体,并且优选是A687V突变体,或等同地,A687V。
在一个实施例中,EZH2的R685突变体的氨基酸序列不同于野生型人类EZH2的氨基酸序列,不同之处仅是相应于野生型人类EZH2的R685的单个氨基酸残基被非丙氨酸的氨基酸,优选是组氨酸(H)或半胱氨酸(C)取代。对应于这个实施例的EZH2的R685突变体此处被称为R685突变体,并且优选是R685C突变体或R685H突变体,或等同地,R685H或R685C。
在一个实施例中,EZH2的突变体的氨基酸序列在如SEQ ID NO:6所定义的它的底物口袋结构域中的一个或多个氨基酸残基不同于野生型人类EZH2的氨基酸序列。对应于这个实施例的EZH2的突变体此处被称为EZH2突变体。
其他示例性取代氨基酸突变包括在SEQ ID NO:1的氨基酸位置677、687、674、685、或641处的取代,诸如但不限于在SEQ ID NO:1的氨基酸位置677处的甘氨酸(G)取代野生型残基丙氨酸(A)(A677G);在SEQ ID NO:1的氨基酸位置687处的缬氨酸(V)取代野生型残基丙氨酸(A)(A687V);在SEQ ID NO:1的氨基酸位置674处的甲硫氨酸(M)取代野生型残基缬氨酸(V)(V674M);在SEQ ID NO:1的氨基酸位置685处的组氨酸(H)取代野生型残基精氨酸(R)(R685H);在SEQ ID NO:1的氨基酸位置685处的半胱氨酸(C)取代野生型残基精氨酸(R)(R685C);在SEQ ID NO:1的氨基酸位置641处的苯基丙氨酸(F)取代野生型残基酪氨酸(Y)(Y641F);在SEQ ID NO:1的氨基酸位置641处的组氨酸(H)取代野生型残基酪氨酸(Y)(Y641H);在SEQ ID NO:1的氨基酸位置641处的天冬酰胺(N)取代野生型残基酪氨酸(Y)(Y641N);在SEQ ID NO:1的氨基酸位置641处的丝氨酸(S)取代野生型残基酪氨酸(Y)(Y641S);或在SEQ ID NO:1的氨基酸位置641处的半胱氨酸(C)取代野生型残基酪氨酸(Y)(Y641C)。
本披露的突变还可包括在SEQ ID NO:3的氨基酸位置322处的丝氨酸(S)取代野生型残基天冬酰胺(N)(N322S)、在SEQ ID NO:3的氨基酸位置288处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R288Q)、在SEQ ID NO:3的氨基酸位置573处的异亮氨酸(I)取代野生型残基苏氨酸(T)(T573I)、在SEQ ID NO:3的氨基酸位置664处的谷氨酸(E)取代野生型残基天冬氨酸(D)(D664E)、在SEQ ID NO:5的氨基酸位置458处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R458Q)、在SEQ ID NO:3的氨基酸位置249处的赖氨酸(K)取代野生型残基谷氨酸(E)(E249K)、在SEQ ID NO:3的氨基酸位置684处的半胱氨酸(C)取代野生型残基精氨酸(R)(R684C)、在SEQ ID NO:21的氨基酸位置628处的组氨酸(H)取代野生型残基精氨酸(R)(R628H)、在SEQ ID NO:5的氨基酸位置501处的组氨酸(H)取代野生型残基谷氨酰胺(Q)(Q501H)、在SEQ ID NO:3的氨基酸位置192处的天冬酰胺(N)取代野生型残基天冬氨酸(D)(D192N)、在SEQ ID NO:3的氨基酸位置664处的缬氨酸(V)取代野生型残基天冬氨酸(D)(D664V)、在SEQ ID NO:3的氨基酸位置704处的亮氨酸(L)取代野生型残基缬氨酸(V)(V704L)、在SEQ ID NO:3的氨基酸位置132处的丝氨酸(S)取代野生型残基脯氨酸(P)(P132S)、在SEQ ID NO:21的氨基酸位置669处的赖氨酸(K)取代野生型残基谷氨酸(E)(E669K)、在SEQ ID NO:3的氨基酸位置255处的苏氨酸(T)取代野生型残基丙氨酸(A)(A255T)、在SEQ ID NO:3的氨基酸位置726处的缬氨酸(V)取代野生型残基谷氨酸(E)(E726V)、在SEQ ID NO:3的氨基酸位置571处的酪氨酸(Y)取代野生型残基半胱氨酸(C)(C571Y)、在SEQ ID NO:3的氨基酸位置145处的半胱氨酸(C)取代野生型残基苯丙氨酸(F)(F145C)、在SEQ ID NO:3的氨基酸位置693处的苏氨酸(T)取代野生型残基天冬酰胺(N)(N693T)、在SEQ ID NO:3的氨基酸位置145处的丝氨酸(S)取代野生型残基苯丙氨酸(F)(F145S)、在SEQ ID NO:21的氨基酸位置109处的组氨酸(H)取代野生型残基谷氨酰胺(Q)(Q109H)、在SEQ ID NO:21的氨基酸位置622处的半胱氨酸(C)取代野生型残基苯丙氨酸(F)(F622C)、在SEQ ID NO:3的氨基酸位置135处的精氨酸(R)取代野生型残基甘氨酸(G)(G135R)、在SEQ ID NO:5的氨基酸位置168处的谷氨酰胺(Q)取代野生型残基精氨酸(R)(R168Q)、在SEQ ID NO:3的氨基酸位置159处的精氨酸(R)取代野生型残基甘氨酸(G)(G159R)、在SEQ ID NO:5的氨基酸位置310处的半胱氨酸(C)取代野生型残基精氨酸(R)(R310C)、在SEQ ID NO:3的氨基酸位置561处的组氨酸(H)取代野生型残基精氨酸(R)(R561H)、在SEQ ID NO:21的氨基酸位置634处的组氨酸(H)取代野生型残基精氨酸(R)(R634H)、在SEQ ID NO:3的氨基酸位置660处的精氨酸(R)取代野生型残基甘氨酸(G)(G660R)、在SEQ ID NO:3的氨基酸位置181处的半胱氨酸(C)取代野生型残基酪氨酸(Y)(Y181C)、在SEQ ID NO:3的氨基酸位置297处的精氨酸(R)取代野生型残基组氨酸(H)(H297R)、在SEQ ID NO:21的氨基酸位置612处的丝氨酸(S)取代野生型残基半胱氨酸(C)(C612S)、在SEQ ID NO:3的氨基酸位置694处的酪氨酸(Y)取代野生型残基组氨酸(H)(H694Y)、在SEQ ID NO:3的氨基酸位置664处的丙氨酸(A)取代野生型残基天冬氨酸(D)(D664A)、在SEQ ID NO:3的氨基酸位置150处的苏氨酸(T)取代野生型残基异亮氨酸(I)(I150T)、在SEQ ID NO:3的氨基酸位置264处的精氨酸(R)取代野生型残基异亮氨酸(I)(I264R)、在SEQ ID NO:3的氨基酸位置636处的亮氨酸(L)取代野生型残基脯氨酸(P)(P636L)、在SEQ ID NO:3的氨基酸位置713处的苏氨酸(T)取代野生型残基异亮氨酸(I)(I713T)、在SEQ ID NO:5的氨基酸位置501处的脯氨酸(P)取代野生型残基谷氨酰胺(Q)(Q501P)、在SEQ ID NO:3的氨基酸位置243处的谷氨酰胺(Q)取代野生型残基赖氨酸(K)(K243Q)、在SEQ ID NO:5的氨基酸位置130处的天冬氨酸(D)取代野生型残基谷氨酸(E)(E130D)、在SEQ ID NO:3的氨基酸位置509处的甘氨酸(G)取代野生型残基精氨酸(R)(R509G)、在SEQ ID NO:3的氨基酸位置566处的组氨酸(H)取代野生型残基精氨酸(R)(R566H)、在SEQ ID NO:3的氨基酸位置677处的组氨酸(H)取代野生型残基天冬氨酸(D)(D677H)、在SEQ ID NO:5的氨基酸位置466处的天冬酰胺(N)取代野生型残基赖氨酸(K)(K466N)、在SEQ ID NO:3的氨基酸位置78处的组氨酸(H)取代野生型残基精氨酸(R)(R78H)、在SEQ ID NO:6的氨基酸位置1处的蛋氨酸(M)取代野生型残基赖氨酸(K)(K6M)、在SEQ ID NO:3的氨基酸位置538处的亮氨酸(L)取代野生型残基丝氨酸(S)(S538L)、在SEQ ID NO:3的氨基酸位置149处的谷氨酰胺(Q)取代野生型残基亮氨酸(L)(L149Q)、在SEQ ID NO:3的氨基酸位置252处的缬氨酸(V)取代野生型残基亮氨酸(L)(L252V)、在SEQ ID NO:3的氨基酸位置674处的缬氨酸(V)取代野生型残基亮氨酸(L)(L674V)、在SEQ ID NO:3的氨基酸位置656处的缬氨酸(V)取代野生型残基丙氨酸(A)(A656V)、在SEQ ID NO:3的氨基酸位置731处的天冬氨酸(D)取代野生型残基丙氨酸(A)(Y731D)、在SEQ ID NO:3的氨基酸位置345处的苏氨酸(T)取代野生型残基丙氨酸(A)(A345T)、在SEQ ID NO:3的氨基酸位置244处的天冬氨酸(D)取代野生型残基丙氨酸(A)(Y244D)、在SEQ ID NO:3的氨基酸位置576处的色氨酸(W)取代野生型残基半胱氨酸(C)(C576W)、在SEQ ID NO:3的氨基酸位置640处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N640K)、在SEQ ID NO:3的氨基酸位置675处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N675K)、在SEQ ID NO:21的氨基酸位置579处的酪氨酸(Y)取代野生型残基天冬氨酸(D)((D579Y)、在SEQ ID NO:3的氨基酸位置693处的异亮氨酸(I)取代野生型残基天冬酰胺(N)(N693I)、以及在SEQ ID NO:3的氨基酸位置693处的赖氨酸(K)取代野生型残基天冬酰胺(N)(N693K)。
本披露的突变可以是在SEQ ID NO:3、5或21的氨基酸位置730、391、461、441、235、254、564、662、715、405、685、64、73、656、718、374、592、505、730、或363或者编码SEQ ID NO:3、5、或21的核酸序列的相应核苷酸位置处的移码。EZH2的突变还可以是在SEQ ID NO:3、5或21的氨基酸位置148与149之间插入谷氨酸(E)。EZH2突变的另一个实例是在SEQ ID NO:3、5或21的氨基酸位置148和149处缺失谷氨酸(E)和亮氨酸(L)。突变体EZH2可进一步包括在SEQ ID NO:3、5或21的氨基酸位置733、25、317、62、553、328、58、207、123、63、137、或60处的无义突变。
对EZH2杂合的细胞预期会显示一个恶性表型,这是因为通过WT酶有效的形成H3-K27me1,并且随后通过突变体酶的形式有效的将这个源种类转换成H3-K27me2,以及特别是,H3-K27me3。
先前的结果指向对执行H3-K27单甲基化的酶之间的酶偶合以及对EZH2导致滤泡性淋巴瘤和弥漫性大B细胞淋巴瘤中发病机理的某些突变体形式的依赖性。例如,表达Y641突变体EZH2的细胞对小分子EZH2抑制剂可能比表达WT EZH2的细胞更敏感。具体而言,表达Y641突变体EZH2的细胞在EZH2抑制剂治疗后显示减少的生长、分裂或增生,甚至发生细胞凋亡或坏死。相比之下,表达WT EZH2的细胞不响应于EZH2抑制剂的抗增生作用(美国专利申请号13/230,703(现为美国专利8,895,245);通过引用以其全文结合在此)。
本披露的一个方面是一种用于通过向表达在如SEQ ID NO:6所定义的底物口袋结构域中包括突变的突变体EZH2的受试者给予治疗有效量如本文所述的EZH2抑制剂(例如,与适合同时地、顺序地、或交替地一起给予的另一种药剂组合的具有化学式(I)-(VIa)的化合物)来治疗或缓解受试者的癌症或癌前病状的症状的方法。
本披露的另一个方面是一种用于在受试者中抑制H3-K27转换成三甲基化的H3-K27的方法。该抑制可能涉及在受试者中抑制非甲基化的H3-K27转换成单甲基化H3-K27、单甲基化H3-K27转换成二甲基化H3-K27、二甲基化H3-K27转换成三甲基化H3-K27、或其任何组合,包括,例如单甲基化H3-K27转换成二甲基化H3-K27和二甲基化H3-K27转换成三甲基化H3-K27。如本文所用,非甲基化H3-K27是指组蛋白H3没有甲基基团共价连接到赖氨酸27的氨基基团。如本文所用,单甲基化H3-K27是指组蛋白H3有一个甲基基团共价连接到赖氨酸27的氨基基团。单甲基化H3-K27此处也被称为H3-K27mel。如本文所用,二甲基化H3-K27是指组蛋白H3有两个甲基基团共价连接到赖氨酸27的氨基基团。二甲基化H3-K27此处也被称为H3-K27me2。如本文所用,三甲基化H3-K27是指组蛋白H3有三个甲基基团共价连接到赖氨酸27的氨基基团。三甲基化H3-K27此处也被称为H3-K27me3。
组蛋白H3是136个氨基酸长的蛋白质,其序列是已知的。参见例如,GenBank登录号CAB02546,其内容通过引用结合在此。如本文进一步披露的,除全长组蛋白H3外,包含对应于全长组蛋白H3的K27的赖氨酸残基的组蛋白H3的肽片段可以用作EZH2的底物(并且同样用于EZH2的突变体形式)以评估H3-K27ml至H3-K27m2的转换,以及H3-K27m2至H3-K27m3的转换。在一个实施例中,这种肽片段对应于组蛋白H3的氨基酸残基21-44。这种肽片段具有氨基酸序列LATKAARKSAPATGGVKKPHRYRP(SEQ ID NO:19)。
本披露的组合物包含具有化学式(I)-(VIa)的化合物和一种或多种其他治疗剂或其药学上可接受的盐。具有化学式(I)-(VIa)的化合物适合于与一种或多种其他治疗剂或治疗方法作为组合疗法的一部分给予,适合于一起、顺序或交替给予。适用于本披露方法的具有化学式(I)-(VIa)的其他化合物描述于美国公开案20120264734中,将其内容通过引用以其全文而特此结合。
可以用于本文所述任何方法中的化合物(即EZH2抑制剂)可具有以下化学式I:
或其药学上可接受的盐;其中
R701是H、F、OR707、NHR707、-(C≡C)-(CH2)n7-R708、苯基、5元或6元杂芳基、C3-8环烷基或含有1-3个杂原子的4-7元杂环烷基,其中苯基、5元或6元杂芳基、C3-8环烷基或4-7元杂环烷基各自独立地任选地经一个或多个选自以下的基团取代:卤素、C1-3烷基、OH、O-C1-6烷基、NH-C1-6烷基和经C3-8环烷基或含有1-3个杂原子的4-7元杂环烷基取代的C1-3烷基,其中O-C1-6烷基和NH-C1-6烷基各自任选地经羟基、O-C1-3烷基或NH-C1-3烷基取代,O-C1-3烷基和NH-C1-3烷基各自任选地经O-C1-3烷基或NH-C1-3烷基进一步取代;
R702和R703各自独立地是H、卤素、C1-4烷基、C1-6烷氧基或C6-C10芳氧基,这些基团各自任选地经一个或多个卤素取代;
R704和R705各自独立地是C1-4烷基;
R706是经N(C1-4烷基)2取代的环己基,其中一个或两个C1-4烷基经C1-6烷氧基取代;或R706是四氢吡喃基;
R707是任选地经一个或多个选自以下的基团取代的C1-4烷基:羟基、C1-4烷氧基、氨基、单-或二-C1-4烷基氨基、C3-8环烷基和含有1-3个杂原子的4-7元杂环烷基,其中C3-8环烷基或4-7元杂环烷基各自独立地进一步任选地经C1-3烷基取代;
R708是任选地经一个或多个选自以下的基团取代的C1-4烷基:OH、卤素和C1-4烷氧基、含有1-3个杂原子的4-7元杂环烷基或O-C1-6烷基,其中4-7元杂环烷基可任选地进一步经OH或C1-6烷基取代;并且
n7是0、1或2。
例如,R706是由N(C1-4烷基)2取代的环己基,其中一个C1-4烷基未经取代且另一个经甲氧基取代。
例如,R706是
例如,化合物具有以下化学式II:
例如,R702是甲基或异丙基并且R703是甲基或甲氧基。
例如,R704是甲基。
例如,R701是OR707并且R707是任选地经OCH3或吗啉取代的C1-3烷基。
例如,R701是H或F。
例如,R701是四氢吡喃基、苯基、吡啶基、嘧啶基、吡嗪基、咪唑基或吡唑基,所述基团各自任选地经以下基团取代:甲基、甲氧基、经吗啉取代的乙基或-OCH2CH2OCH3。
例如,R708是吗啉、哌啶、哌嗪、吡咯烷、二氮杂环庚烷或氮杂环丁烷,所述基团各自任选地经以下基团取代:OH或C1-6烷基。
例如,R708是吗啉
例如,R708是经C1-6烷基取代的哌嗪。
例如,R708是甲基、叔丁基或C(CH3)2OH。
可用于本文所述任何方法中的化合物(即EZH2抑制剂)可具有以下化学式III:
或其药学上可接受的盐。
在此化学式中:
R801是C1-6烷基、C2-6烯基、C2-6炔基、C3-8环烷基、含有1-3个杂原子的4-7元杂环烷基、苯基或5元或6元杂芳基,所述基团各自经O-C1-6烷基-Rx或NH-C1-6烷基-Rx取代,其中Rx是羟基、O-C1-3烷基或NH-C1-3烷基,并且除了Rx是羟基时,Rx任选地进一步经O-C1-3烷基或NH-C1-3烷基取代;或R801是经-Q2-T2取代的苯基,其中Q2是键或任选地经卤素、氰基、羟基或C1-C6烷氧基取代的C1-C3烷基接头,并且T2是任选经取代的4元至12元杂环烷基;并且R801任选地经进一步取代;
R802和R803各自独立地是H、卤素、C1-4烷基、C1-6烷氧基或C6-C10芳氧基,这些基团各自任选地经一个或多个卤素取代;
R804和R805各自独立地是C1-4烷基;并且
R806是-Qx-Tx,其中Qx是键或C1-4烷基接头,Tx是H、任选经取代的C1-4烷基、任选经取代的C3-C8环烷基或任选经取代的4元至14元杂环烷基。
例如,Qx和Q2各自独立地是键或甲基接头,并且Tx和T2各自独立地是四氢吡喃基、经1、2或3个C1-4烷基取代的哌啶基或经N(C1-4烷基)2取代的环己基,其中一个或两个C1-4烷基任选地经C1-6烷氧基取代;
例如,R806是经N(C1-4烷基)2取代的环己基或R806是四氢吡喃基。
例如,R806是
例如,R801是经O-C1-6烷基-Rx取代的苯基或5元或6元杂芳基,或R801是经CH2-四氢吡喃基取代的苯基。
例如,本披露化合物具有以下化学式IVa或IVb:
其中Z’是CH或N,并且R807是C2-3烷基-Rx。
例如,R807是-CH2CH2OH、-CH2CH2OCH3或-CH2CH2OCH2CH2OCH3。
例如,R802是甲基或异丙基并且R803是甲基或甲氧基。
例如,R804是甲基。
本披露的化合物可具有以下化学式(V):
或其药学上可接受的盐或酯。
在此化学式中:
R2、R4和R12各自独立地是C1-6烷基;
R6是C6-C10芳基或5或6元杂芳基,这些基团各自任选地经一个或多个-Q2-T2取代,其中Q2是键或任选地经卤素、氰基、羟基或C1-C6烷氧基取代的C1-C3烷基接头,并且T2是H、卤素、氰基、-ORa、-NRaRb、-(NRaRbRc)+A-、-C(O)Ra、-C(O)ORa、-C(O)NRaRb、-NRbC(O)Ra、-NRbC(O)ORa、-S(O)2Ra、-S(O)2NRaRb、或Rs2,其中Ra、Rb和Rc各自独立地是H或RS3,A-是药学上可接受的阴离子,RS2和RS3各自独立地是C1-C6烷基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基或5元或6元杂芳基,或Ra和Rb与其所附接的N原子一起形成具有0或1个额外杂原子的4至12元杂环烷基环,并且RS2、RS3和由Ra和Rb形成的4至12元杂环烷基环任选地经一个或多个-Q3-T3取代,其中Q3是键或各自任选地经卤素、氰基、羟基或C1-C6烷氧基取代的C1-C3烷基接头,并且T3选自下组,该组由以下各项组成:卤素、氰基、C1-C6烷基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基、5元或6元杂芳基、ORd、COORd、-S(O)2Rd、-NRdRe和-C(O)NRdRe,Rd和Re各自独立地是H或C1-C6烷基或-Q3-T3是氧代基;或任两个相邻-Q2-T2与其所附接的原子一起形成任选地含有1-4个选自N、O和S的杂原子并且任选地经一个或多个选自由以下组成的组的取代基取代的5元或6元环:卤素、羟基、COOH、C(O)O-C1-C6烷基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基和5元或6元杂芳基;
R7是-Q4-T4,其中Q4是键、C1-C4烷基接头或C2-C4烯基接头,每一接头任选地经卤素、氰基、羟基或C1-C6烷氧基取代,并且T4是H、卤素、氰基、NRfRg、-ORf、-C(O)Rf、-C(O)ORf、-C(O)NRfRg、-C(O)NRfORg、-NRfC(O)Rg、-S(O)2Rf、或RS4,其中Rf和Rg各自独立地是H或RS5,RS4和RS5各自独立地是C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基或5元或6元杂芳基,并且RS4和RS5各自任选地经一个或多个-Q5-T5取代,其中Q5是键、C(O)、C(O)NRk、NRkC(O)、S(O)2或C1-C3烷基接头,Rk是H或C1-C6烷基,并且T5是H、卤素、C1-C6烷基、羟基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基、5元或6元杂芳基或S(O)qRq,其中q是0、1或2并且Rq是C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基或5元或6元杂芳基,并且除了T5是H、卤素、羟基或氰基时,T5任选地经一个或多个选自由以下组成的组的取代基取代:卤素、C1-C6烷基、羟基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基和5元或6元杂芳基;或-Q5-T5是氧代基;并且
R8是H、卤素、羟基、COOH、氰基、RS6、ORS6或COORS6,其中RS6是C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C8环烷基、4至12元杂环烷基、氨基、单-C1-C6烷基氨基或二-C1-C6烷基氨基,并且RS6任选地经一个或多个选自由以下组成的组的取代基取代:卤素、羟基、COOH、C(O)O-C1-C6烷基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基和二-C1-C6烷基氨基;或R7和R8与其所附接的N原子一起形成具有0至2个额外杂原子的4至11元杂环烷基环,并且由R7和R8形成的4至11元杂环烷基环任选地经一个或多个-Q6-T6取代,其中Q6是键、C(O)、C(O)NRm、NRmC(O)、S(O)2或C1-C3烷基接头,Rm是H或C1-C6烷基,并且T6是H、卤素、C1-C6烷基、羟基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基、5元或6元杂芳基或S(O)pRp,其中p是0、1或2并且Rp是C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基或5元或6元杂芳基,并且除了T6是H、卤素、羟基或氰基时,T6任选地经一个或多个选自由以下组成的组的取代基取代:卤素、C1-C6烷基、羟基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基、C3-C8环烷基、C6-C10芳基、4至12元杂环烷基和5元或6元杂芳基;或-Q6-T6是氧代基。
例如,R6是C6-C10芳基或5或6元杂芳基,这些基团各自任选地独立地经一个或多个-Q2-T2取代,其中Q2是键或C1-C3烷基接头,并且T2是H、卤素、氰基、-ORa、-NRaRb、-(NRaRbRc)+A-、-C(O)NRaRb、-NRbC(O)Ra、-S(O)2Ra或RS2,其中Ra和Rb各自独立地是H或RS3,RS2和RS3各自独立地是C1-C6烷基,或Ra和Rb与其所附接的N原子一起形成具有0或1个额外杂原子的4至7元杂环烷基环,并且RS2、RS3和由Ra和Rb形成的4至7元杂环烷基环各自任选地独立地经一个或多个-Q3-T3取代,其中Q3是键或C1-C3烷基接头并且T3选自下组,该组由以下各项组成:卤素、C1-C6烷基、4至7元杂环烷基、ORd、-S(O)2Rd和-NRdRe,Rd和Re各自独立地是H或C1-C6烷基或-Q3-T3是氧代基;或任两个相邻-Q2-T2与其所附接的原子一起形成任选地含有1-4个选自N、O和S的杂原子的5元或6元环。
例如,本披露化合物具有以下化学式(VI):
或其药学上可接受的盐,其中Q2是键或甲基接头,T2是H、卤素、-ORa、-NRaRb、-(NRaRbRc)+A-或-S(O)2NRaRb,R7是哌啶基、四氢吡喃、环戊基或环己基(这些基团各自任选地经一个-Q5-T5取代),并且R8是乙基。
本披露提供具有化学式(VIa)的化合物:
或其药学上可接受的盐或酯,其中R7、R8、Ra、以及Rb是在此所定义的。
具有化学式(VIa)的化合物可以包括一个或多个以下特征:
例如,Ra和Rb各自独立地是H或任选地经一个或多个-Q3-T3取代的C1-C6烷基。
例如,Ra和Rb中的一个是H。
例如,Ra和Rb与其所附接的N原子一起形成具有0或1个额外杂原子至N原子的4至7元杂环烷基环(例如,氮杂环丁烷基、吡咯烷基、咪唑烷基、吡唑烷基、恶唑烷基、异恶唑烷基、三唑烷基、哌啶基、1,2,3,6-四氢吡啶基、哌嗪基、吗啉基、1,4-二氮杂环庚烷基、1,4-氧氮杂环庚烷基、2-氧杂-5-氮杂双环[2.2.1]庚烷基、2,5-二氮杂双环[2.2.1]庚烷基等),并且该环任选地经一个或多个-Q3-T3取代。
例如,Ra和Rb与其所附接的N原子一起形成氮杂环丁烷基、吡咯烷基、咪唑烷基、吡唑烷基、恶唑烷基、异恶唑烷基、三唑烷基、四氢呋喃基、哌啶基、1,2,3,6-四氢吡啶基、哌嗪基或吗啉基,并且该环任选地经一个或多个-Q3-T3取代。
例如,一个或多个-Q3-T3是氧代基。
例如,Q3是键或未经取代或经取代的C1-C3烷基接头。
例如,T3是H、卤素、4至7元杂环烷基、C1-C3烷基、ORd、COORd、-S(O)2Rd、或-NRdRe。
例如,Rd和Re各自独立地是H或C1-C6烷基。
例如,R7是C3-C8环烷基或4至7元杂环烷基,这些基团各自任选地经一个或多个-Q5-T5取代。
例如,R7是哌啶基、四氢吡喃、四氢-2H-噻喃基、环戊基、环己基、吡咯烷基或环庚基,这些基团各自任选地经一个或多个-Q5-T5取代。
例如,R7是环戊基、环己基、或四氢-2H-噻喃基,这些基团各自任选地经一个或多个-Q5-T5取代。
例如,Q5是NHC(O),并且T5是C1-C6烷基或C1-C6烷氧基,
例如,一个或多个-Q5-T5是氧代基。
例如,R7是1-氧化物-四氢-2H-噻喃基或1,1-二氧化物-四氢-2H-噻喃基。
例如,Q5是键,并且T5是氨基、单-C1-C6烷基氨基、二-C1-C6烷基氨基。
例如,Q5是CO、S(O)2、或NHC(O);并且T5是C1-C6烷基、C1-C6烷氧基、C3-C8环烷基、或4至7元杂环烷基。
例如,R8是H或任选地经一个或多个选自由以下组成的组的取代基取代的C1-C6烷基:卤素、羟基、COOH、C(O)O-C1-C6烷基、氰基、C1-C6烷氧基、氨基、单-C1-C6烷基氨基和二-C1-C6烷基氨基。
例如,R8是H、甲基、或乙基。
在一个实施例中,本披露的化合物是化合物44
或其药学上可接受的盐。
在一些实施例中,可以用于本文所呈现的任何方法中的化合物是:
其立体异构体或其药学上可接受的盐和溶剂化物。
在一些实施例中,可以用于这里所呈现的任何方法中的化合物是GSK-126,其立体异构体或其药学上可接受的盐和溶剂化物。
在一个实施例中,本披露的化合物是化合物自身,即游离碱或“裸”分子。在另一个实施例中,该化合物是其盐,例如,裸分子的单-HCl盐或三-HCl盐、单-HBr盐或三-HBr盐。
本披露的代表性化合物包括列于表1的化合物。
在下表中,每次出现的应视为
表1
如本文所用,“烷基”、“C1、C2、C3、C4、C5或C6烷基”或“C1-C6烷基”旨在包括C1、C2、C3、C4、C5或C6直链(线性)饱和脂肪族烃基和C3、C4、C5或C6支链饱和脂肪族烃基。例如,C1-C6烷基旨在包括C1、C2、C3、C4、C5和C6烷基基团。烷基的实例包括具有一至六个碳原子的部分,诸如但不限于,甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、仲戊基或正己基。
在某些实施例中,直链或支链烷基具有六个或更少碳原子(例如,对于直链为C1-C6,对于支链为C3-C6),并且在另一实施例中,直链或支链烷基具有四个或更少碳原子。
如本文所用,术语“环烷基”是指具有3至30个碳原子(例如,C3-C10)的饱和或不饱和非芳香族烃单环或多环(例如,稠合环、桥接环或螺环)系统。环烷基的实例包括但不限于环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环戊烯基、环己烯基、环庚烯基和金刚烷基。除非另外指定,否则术语“杂环烷基”是指具有一个或多个杂原子(诸如O、N、S或Se)的饱和或不饱和非芳香族3-8元单环、7-12元二环(稠合环、桥接环或螺环)或11-14元三环系统(稠合环、桥接环或螺环)。杂环烷基基团的实例包括但不限于哌啶基、哌嗪基、吡咯烷基、二噁烷基、四氢呋喃基、异吲哚啉基、二氢吲哚基、咪唑烷基、吡唑烷基、噁唑烷基、异噁唑烷基、三唑烷基、四氢呋喃基、氧杂环丙烷基、氮杂环丁烷基、氧杂环丁基、硫杂环丁基、1,2,3,6-四氢吡啶基、四氢吡喃基、二氢吡喃基、吡喃基、吗啉基、1,4-二氮杂环庚烷基、1,4-氧杂氮杂环庚烷基、2-氧杂-5-氮杂二环[2.2.1]庚烷基、2,5-二氮杂二环[2.2.1]庚烷基、2-氧杂-6-氮杂螺[3.3]庚烷基、2,6-二氮杂螺[3.3]庚烷基、1,4-二氮杂-8-氮杂螺[4.5]癸烷基等。
术语“任选地经取代的烷基”是指未经取代的烷基或具有指定取代基替代烃主链的一个或多个碳上的一个或多个氢原子的烷基。所述取代基可包括例如烷基、烯基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。
“芳基烷基”或“芳烷基”部分是经芳基取代的烷基(例如,苯基甲基(苄基))。“烷基芳基”部分是经烷基取代的芳基(例如,甲基苯基)。
如本文所用,“烷基接头”旨在包括C1、C2、C3、C4、C5或C6直链(线性)饱和二价脂肪族烃基和C3、C4、C5或C6支链饱和脂肪族烃基。例如,C1-C6烷基接头旨在包括C1、C2、C3、C4、C5和C6烷基接头基团。烷基接头的实例包括具有一至六个碳原子的部分,诸如但不限于甲基(-CH2-)、乙基(-CH2CH2-)、正丙基(-CH2CH2CH2-)、异丙基(-CHCH3CH2-)、正丁基(-CH2CH2CH2CH2-)、仲丁基(-CHCH3CH2CH2-)、异丁基(-C(CH3)2CH2-)、正戊基(-CH2CH2CH2CH2CH2-)、仲戊基(-CHCH3CH2CH2CH2-)或正己基(-CH2CH2CH2CH2CH2CH2-)。
“烯基”包括在长度和可能的取代方面与上述烷基相似但含有至少一个双键的不饱和脂肪族基团。例如,术语“烯基”包括直链烯基(例如,乙烯基、丙烯基、丁烯基、戊烯基、己烯基、庚烯基、辛烯基、壬烯基、癸烯基)和支链烯基。在某些实施例中,直链或支链烯基在其主链中具有六个或更少碳原子(例如,对于直链为C2-C6,对于支链为C3-C6)。术语“C2-C6”包括含有两个至六个碳原子的烯基。术语“C3-C6”包括含有三个至六个碳原子的烯基。
术语“任选地经取代的烯基”是指未经取代的烯基或具有指定取代基替代一个或多个烃主链碳原子上的一个或多个氢原子的烯基。所述取代基可包括例如烷基、烯基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、杂环基、烷基芳基或芳香族或杂芳香族部分。
“炔基”包括在长度和可能的取代方面与上述烷基相似但含有至少一个三键的不饱和脂肪族基团。例如,“炔基”包括直链炔基基团(例如,乙炔基、丙炔基、丁炔基、戊炔基、己炔基、庚炔基、辛炔基、壬炔基、癸炔基)和支链炔基。在某些实施例中,直链或支链炔基基团在其主链中具有六个或更少碳原子(例如,对于直链为C2-C6,对于支链为C3-C6)。术语“C2-C6”包括含有二个至六个碳原子的炔基。术语“C3-C6”包括含有三个至六个碳原子的炔基。
术语“任选地经取代的炔基”是指未经取代的炔基或具有指定取代基替代一个或多个烃主链碳原子上的一个或多个氢原子的炔基。所述取代基可包括例如烷基、烯基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。
其他任选经取代的部分(诸如任选经取代的环烷基、杂环烷基、芳基或杂芳基)包括未经取代的部分和具有指定取代基中的一个或多个的部分二者。例如,经取代的杂环烷基包括经一个或多个烷基取代的杂环烷基,诸如2,2,6,6-四甲基-哌啶基和2,2,6,6-四甲基-1,2,3,6-四氢吡啶基。
“芳基”包括具有芳香性的基团,包括具有至少一个芳香族环的“共轭的”或多环系统并且在环结构中不含任何杂原子。实例包括苯基、苄基、1,2,3,4-四氢萘基等。
“杂芳基”是如上文所定义的芳基,但在环结构中具有一至四个杂原子,并且还可称为“芳基杂环”或“杂芳香族基团”。如本文所用,术语“杂芳基”旨在包括由碳原子以及一个或多个独立地选自由氮、氧和硫组成的组的杂原子(例如1或1-2或1-3或1-4或1-5或1-6个杂原子,或例如1、2、3、4、5或6个杂原子)组成的稳定的5元、6元或7元单环或7元、8元、9元、10元、11元或12元二环芳香族杂环。氮原子可经取代或未经取代(即,N或NR,其中R是H或如所定义的其他取代基)。氮和硫杂原子可任选地发生氧化(即,N→O和S(O)p,其中p=1或2)。应注意,芳香族杂环中S和O原子的总数不大于1。
杂芳基的实例包括吡咯、呋喃、噻吩、噻唑、异噻唑、咪唑、三唑、四唑、吡唑、噁唑、异噁唑、吡啶、吡嗪、哒嗪、嘧啶等。
另外,术语“芳基”和“杂芳基”包括多环芳基和杂芳基,例如三环、二环,例如萘、苯并噁唑、苯并二噁唑、苯并噻唑、苯并咪唑、苯并噻吩、亚甲基二氧苯基、喹啉、异喹啉、萘啶、吲哚、苯并呋喃、嘌呤、苯并呋喃、脱氮杂嘌呤、中氮茚。
在多环芳香族环的情况中,仅需要环中的一个是芳香族(例如,2,3-二氢吲哚),但所有环都可以是芳香族(例如,喹啉)。第二环也可以是稠合或桥接的。
环烷基、杂环烷基、芳基或杂芳基环可在一个或多个环位置经如上文所述的所述取代基取代(例如,成环碳或杂原子,诸如N),所述取代基例如烷基、烯基、炔基、卤素、羟基、烷氧基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、烷基氨基羰基、芳烷基氨基羰基、烯基氨基羰基、烷基羰基、芳基羰基、芳烷基羰基、烯基羰基、烷氧基羰基、氨基羰基、烷基硫代羰基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。芳基和杂芳基也可以与不是芳香族的脂环族环或杂环稠合或桥接,以形成多环系统(例如,四氢萘、亚甲基二氧苯基)。
如本文所用,“碳环”(“carbocycle”或“carbocyclic ring”)旨在包括任何具有指定碳数量的稳定单环、二环或三环,其中任一个可为饱和、不饱和或芳香族的。碳环包括环烷基和芳基。例如,C3-C14碳环旨在包括具有3、4、5、6、7、8、9、10、11、12、13或14个碳原子的单环、二环或三环。碳环的实例包括但不限于环丙基、环丁基、环丁烯基、环戊基、环戊烯基、环己基、环庚烯基、环庚基、环庚烯基、金刚烷基、环辛基、环辛烯基、环辛二烯基、芴基、苯基、萘基、茚满基、金刚烷基和四氢萘基。桥接环也包括在碳环的定义中,包括例如[3.3.0]二环辛烷、[4.3.0]二环壬烷、[4.4.0]二环癸烷和[2.2.2]二环辛烷。在一个或多个碳原子连接两个非相邻碳原子时出现桥接环。在一个实施例中,桥环具有一个或两个碳原子。应注意,桥总是将单环转化为三环。在环发生桥接时,针对该环所列举的取代基也可存在于桥上。还包括稠合(例如,萘基、四氢萘基)环和螺环。
如本文所用,“杂环”或“杂环基”包括含有至少一个环杂原子(例如,N、O或S)的任何环结构(饱和、不饱和或芳香族)。杂环包括杂环烷基和杂芳基。杂环的实例包括但不限于吗啉、吡咯烷、四氢噻吩、哌啶、哌嗪、氧杂环丁烷、吡喃、四氢吡喃、氮杂环丁烷和四氢呋喃。
杂环基团的实例包括但不限于吖啶基、吖辛因基、苯并咪唑基、苯并呋喃基、苯并硫代呋喃基、苯并苯硫基、苯并噁唑基、苯并噁唑啉基、苯并噻唑基、苯并三唑基、苯并四唑基、苯并异噁唑基、苯并异噻唑基、苯并咪唑啉基、咔唑基、4aH-咔唑基、咔啉基、色烷基、色烯基、噌啉基、十氢喹啉基、2H,6H-1,5,2-二噻嗪基、二氢氟[2,3-b]四氢呋喃、呋喃基、呋咱基、咪唑烷基、咪唑啉基、咪唑基、1H-吲唑基、吲哚烯基、二氢吲哚基、吲嗪基、吲哚基、3H-吲哚基、靛红酰基、异苯并呋喃基、异色烷基、异吲唑基、异吲哚啉基、异吲哚基、异喹啉基、异噻唑基、异噁唑基、亚甲基二氧苯基、吗啉基、萘啶基、八氢异喹啉基、噁二唑基、1,2,3-噁二唑基、1,2,4-噁二唑基、1,2,5-噁二唑基、1,3,4-噁二唑基、1,2,4-噁二唑5(4H)-酮、噁唑烷基、噁唑基、羟吲哚基、嘧啶基、菲啶基、菲咯啉基、吩嗪基、吩噻嗪基、吩噁噻基、吩噁噻嗯基(phenoxathinyl)、吩噁嗪基、酞嗪基、哌嗪基、哌啶基、哌啶酮基、4-哌啶酮基、胡椒基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑烷基、吡唑啉基、吡唑基、哒嗪基、吡啶并噁唑、吡啶并咪唑、吡啶并噻唑、吡啶基(pyridinyl)、吡啶基(pyridyl)、嘧啶基、吡咯烷基、吡咯啉基、2H-吡咯基、吡咯基、喹唑啉基、喹啉基、4H-喹嗪基、喹噁啉基、奎宁环基、四氢呋喃基、四氢异喹啉基、四氢喹啉基、四唑基、6H-1,2,5-噻二嗪基、1,2,3-噻二唑基、1,2,4-噻二唑基、1,2,5-噻二唑基、1,3,4-噻二唑基、噻蒽基、噻唑基、噻吩基、噻吩并噻唑基、噻吩并噁唑基、噻吩并咪唑基、苯硫基、三嗪基、1,2,3-三唑基、1,2,4-三唑基、1,2,5-三唑基、1,3,4-三唑基和呫吨基。
如本文所用,术语“经取代”意味着指定原子上的任何一个或多个氢经所选择的所指示基团替代,其条件是不超过指定原子的正常化合价,并且该取代得到稳定化合物。在取代基是氧代基或酮基(即,=O)时,则该原子上的2个氢原子被替代。酮基取代基不存在于芳香族部分上。如本文所用,环双键是在两个相邻环原子之间形成的双键(例如,C=C、C=N或N=N)。“稳定化合物”和“稳定结构”意欲指示足够稳健而可在从反应混合物分离至可用纯度、并且配制为有效治疗剂后幸存的化合物。
在与一取代基的键显示与连接环中两个原子的键交叉时,则这种取代基可键合到环中的任何原子上。在列示取代基但不指明这种取代基通过哪个原子键合到给定化学式的化合物的剩余部分时,则所述取代基可通过这种化学式中的任何原子键合。取代基和/或变量的组合是允许的,但仅在此类组合得到稳定化合物时允许。
在任何变量(例如,R1)在化合物的任何成分或化学式中出现多于一次时,其在每次出现时的定义与其在其他每次出现时的定义无关。因此,例如,如果显示一基团经0-2个R1部分取代,则该基团可任选地经多至两个R1部分取代,并且R1在每次出现时独立地选自R1的定义。另外,取代基和/或变量的组合是允许的,但仅在此类组合得到稳定化合物时允许。
术语“羟基”(“hydroxy”或“hydroxyl”)包括具有-OH或-O-的基团。
如本文所用,“卤代”或“卤素”是指氟、氯、溴和碘。术语“全卤化”通常是指其中所有氢原子都由卤素原子替代的部分。术语“卤代烷基”或“卤代烷氧基”是指经一个或多个卤素原子取代的烷基或烷氧基。
术语“羰基”包括含有以双键连接到氧原子的碳的化合物和部分。含有羰基的部分的实例包括但不限于醛、酮、羧酸、酰胺、酯、酸酐等。
术语“羧基”是指-COOH或其C1-C6烷基酯。
“酰基”包括含有酰基(R-C(O)-)或羰基的部分。“经取代的酰基”包括其中氢原子中的一个或多个由例如以下基团替代的酰基:烷基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。
“芳酰基”包括具有结合到羰基的芳基或杂芳香族部分的部分。芳酰基的实例包括苯基羧基、萘基羧基等。
“烷氧基烷基”、“烷基氨基烷基”和“硫代烷氧基烷基”包括如上文所述的烷基,其中氧、氮或硫原子替代一个或多个烃主链碳原子。
术语“烷氧基”(“alkoxy”或“alkoxyl”)包括共价连接到氧原子的经取代和未经取代的烷基、烯基和炔基。烷氧基或烷氧基团的实例包括但不限于甲氧基、乙氧基、异丙氧基、丙氧基、丁氧基和戊氧基。经取代烷氧基的实例包括卤化烷氧基。烷氧基可经诸如以下的基团取代:烯基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、磷酸根、膦酸基、次膦酸基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。卤素取代的烷氧基的实例包括但不限于氟甲氧基、二氟甲氧基、三氟甲氧基、氯甲氧基、二氯甲氧基和三氯甲氧基。
术语“醚”或“烷氧基”包括含有键合到两个碳原子或杂原子的氧的化合物或部分。例如,该术语包括“烷氧基烷基”,其是指共价键合到与烷基共价键合的氧原子的烷基、烯基或炔基。
术语“酯”包括含有结合到与羰基碳键合的氧原子的碳或杂原子的化合物或部分。术语“酯”包括烷氧基羧基,诸如甲氧基羰基、乙氧基羰基、丙氧基羰基、丁氧基羰基、戊氧基羰基等。
术语“硫代烷基”包括含有与硫原子连接的烷基的化合物或部分。硫代烷基可经诸如以下的基团取代:烷基、烯基、炔基、卤素、羟基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基、羧酸根、羧酸、烷基羰基、芳基羰基、烷氧基羰基、氨基羰基、烷基氨基羰基、二烷基氨基羰基、烷基硫代羰基、烷氧基、氨基(包括烷基氨基、二烷基氨基、芳基氨基、二芳基氨基和烷基芳基氨基)、酰基氨基(包括烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基)、脒基、亚氨基、巯基、烷硫基、芳硫基、硫代羧酸根、硫酸根、烷基亚磺酰基、磺酸基、氨磺酰基、磺酰胺基、硝基、三氟甲基、氰基、叠氮基、杂环基、烷基芳基或芳香族或杂芳香族部分。
术语“硫代羰基”或“硫代羧基”包括含有以双键连接到硫原子的碳的化合物和部分。
术语“硫醚”包括含有键合到两个碳原子或杂原子的硫原子的部分。硫醚的实例包括但不限于烷硫代烷基、烷硫代烯基和烷硫代炔基。术语“烷硫代烷基”包括具有键合到与烷基键合的硫原子的烷基、烯基或炔基的部分。类似地,术语“烷硫代烯基”是指其中烷基、烯基或炔基键合到与烯基共价键合的硫原子的部分;并且“烷硫代炔基”是指其中烷基、烯基或炔基键合到与炔基共价键合的硫原子的部分。
如本文所用,“胺”或“氨基”是指未经取代或经取代的-NH2。“烷基氨基”包括其中-NH2氮结合到至少一个烷基的化合物的基团。烷基氨基的实例包括苄基氨基、甲基氨基、乙基氨基、苯乙基氨基等。“二烷基氨基”包括其中-NH2的氮结合到至少两个其他烷基的基团。二烷基氨基的实例包括但不限于二甲基氨基和二乙基氨基。“芳基氨基”和“二芳基氨基”包括其中氮分别结合到至少一个或两个芳基的基团。“氨基芳基”和“氨基芳氧基”是指经氨基取代的芳基和芳氧基。“烷基芳基氨基”、“烷基氨基芳基”或“芳基氨基烷基”是指结合到至少一个烷基和至少一个芳基的氨基。“烷氨基烷基”是指结合到还与烷基结合的氮原子的烷基、烯基或炔基。“酰基氨基”包括其中氮结合到酰基的基团。酰基氨基的实例包括但不限于烷基羰基氨基、芳基羰基氨基、氨基甲酰基和脲基。
术语“酰胺”或“氨基羧基”包括含有结合到羰基或硫代羰基的碳上的氮原子的化合物或部分。该术语包括“烷氨基羧基”,其包括结合到与羰基或硫代羰基的碳结合的氨基上的烷基、烯基或炔基。其还包括“芳基氨基羧基”,其包括结合到与羰基或硫代羰基的碳结合的氨基上的芳基或杂芳基部分。术语“烷基氨基羧基”、“烯基氨基羧基”、“炔基氨基羧基”和“芳基氨基羧基”包括其中烷基、烯基、炔基和芳基部分分别结合到氮原子的部分,该氮原子又结合到羰基碳。酰胺可经诸如以下的取代基取代:直链烷基、支链烷基、环烷基、芳基、杂芳基或杂环。酰胺基团上的取代基可进一步经取代。
含有氮的本披露的化合物可以通过用氧化剂(例如,3-氯过氧苯甲酸(mCPBA)和/或过氧化氢)处理而转化成N-氧化物,以得到本披露的其他化合物。因此,当化合价和结构允许时,所有示出和要求保护的含氮化合物被认为包括如所示的化合物及其N-氧化物衍生物(可以被指定为N→O或N+-O-)。此外,在其他情况下,本披露的化合物中的氮可以转化成N-羟基或N-烷氧基化合物。例如,可以通过由诸如m-CPBA的氧化剂将母体胺氧化来制备N-羟基化合物。当化合价和结构允许时,所有示出和要求保护的含氮化合物还被认为覆盖如所示的化合物及其N-羟基(即,N-OH)和N-烷氧基(即,N-OR,其中R是经取代或经未取代的C1-C6烷基、C1-C6烯基、C1-C6炔基、3-14元碳环或3-14元杂环)衍生物。
“异构现象”意指具有相同分子式但其原子的键合顺序或其原子的空间排列不同的化合物。原子空间排列不同的异构体称为“立体异构体”。并非彼此的镜像的立体异构体称为“非对映异构体”,并且是彼此的不可重叠的镜像的立体异构体称为“对映异构体”或有时称为光学异构体。含有等量的具有相反手性的单独对映异构形式的混合物称为“外消旋混合物”。
键合到四个不相同取代基的碳原子称为“手性中心”。
“手性异构体”意指具有至少一个手性中心的化合物。具有多于一个手性中心的化合物可作为单独非对映异构体或作为非对映异构体的混合物存在,称为“非对映异构混合物”。在存在一个手性中心时,立体异构体可通过该手性中心的绝对构型(R或S)来表征。绝对构型是指附接到手性中心的取代基的空间排列。考虑中的附接至手性中心上的取代基按照Cahn、Ingold和Prelog的序列规则排序。(Cahn等人,Angew.Chem.Inter.Edit.[应用化学]1966,5,385;勘误表511;Cahn等人,Angew.Chem.[应用化学]1966,78,413;Cahn和Ingold,J.Chem.Soc.[化学学会会刊]1951(伦敦),612;Cahn等人,Experientia[实验]1956,12,81;Cahn,J.Chem.Educ.[化学教育杂志]1964,41,116)。
“几何异构体”意指因围绕双键或环烷基接头(例如,1,3-环丁基)的旋转受阻而存在的非对映异构体。这些构型的名称是通过前缀顺式和反式或Z和E来区分,这些前缀指示根据卡恩-英戈尔德-普雷洛格规则,基团位于分子中双键的同侧或对侧。
应理解,本披露的化合物可描述为不同手性异构体或几何异构体。还应当理解的是,在化合物具有手性异构体或几何异构体形式时,所有异构体形式均旨在被包括于本披露的范围内,并且化合物的命名不排除任何异构体形式。
此外,本披露中讨论的结构和其他化合物包括其所有阻转异构体。“阻转异构体”是其中两个异构体的原子空间排列不同的立体异构体类型。阻转异构体是因大基团围绕中心键的旋转受阻引起的旋转受限而存在。所述阻转异构体通常作为混合物存在,但由于色谱技术的最新发展,已可能在所选情形中分离两种阻转异构体的混合物。
“互变异构体”是平衡地存在并且易于从一种异构体形式转化成另一种形式的两种或更多种结构异构体之一。这种转化导致氢原子的形式迁移,伴随着相邻共轭双键的转换。互变异构体在溶液中作为互变异构体组的混合物存在。在可能进行互变异构化的溶液中,互变异构体将达到化学平衡。互变异构体的确切比率取决于若干因素,包括温度、溶剂和pH。可通过互变异构作用相互转化的互变异构体的概念被称为互变异构现象。
在可能的多种类型的互变异构现象中,通常观察到两种。在酮-烯醇互变异构现象中,电子和氢原子发生同时转移。环-链互变异构现象由于糖链分子中的醛基(-CHO)与同一分子中的一个羟基(-OH)反应使其形成如葡萄糖所展现的环状(环形状)形式而发生。
常见互变异构对为:杂环中(例如,在核碱基中,诸如鸟嘌呤、胸腺嘧啶和胞嘧啶)的酮-烯醇、酰胺-腈、内酰胺-内酰亚胺、酰胺-亚胺酸互变异构现象、亚胺-烯胺和烯胺-烯胺。如下文所示的,酮-烯醇平衡的实例在吡啶-2(1H)-酮与相应的吡啶-2-醇之间。
应该理解的是,本披露的这些化合物可以被描绘为不同互变异构体。还应当理解的是,在化合物具有互变异构形式时,所有互变异构形式都旨在被包括于本披露的范围内,并且化合物的命名不排除任何互变异构体形式。
本文所披露的具有化学式(I)-(VIa)的化合物包括化合物自身,以及其盐和其溶剂化物(若适用)。例如,盐可在阴离子与芳基-或杂芳基-取代的苯化合物上的带正电基团(例如,氨基)之间形成。适宜阴离子包括氯离子、溴离子、碘离子、硫酸根、硫酸氢根、氨基磺酸根、硝酸根、磷酸根、柠檬酸根、甲磺酸根、三氟乙酸根、谷氨酸根、葡糖醛酸根、戊二酸根、苹果酸根、马来酸根、琥珀酸根、延胡索酸根、酒石酸根、甲苯磺酸根、水杨酸根、乳酸根、萘磺酸根和乙酸根(例如,三氟乙酸根)。术语“药学上可接受的阴离子”是指适于形成药学上可接受的盐的阴离子。同样,盐也可在阳离子与芳基-或杂芳基-取代的苯化合物上的带负电基团(例如,羧酸根)之间形成。适合的阳离子包括钠离子、钾离子、镁离子、钙离子、以及铵阳离子(诸如四甲基铵离子)。芳基-或杂芳基-取代的苯化合物还包括那些含有季氮原子的盐。在盐形式中,应理解的是,化合物与盐的阳离子或阴离子的比率可以为1∶1,或不同于1∶1的任何比率,例如3∶1、2∶1、1∶2、或1∶3。
另外,本披露的化合物(例如,化合物的盐)可以水合或非水合(无水)形式或作为与其他溶剂分子的溶剂化物而存在。水合物的非限制性实例包括一水合物、二水合物等。溶剂化物的非限制性实例包括乙醇溶剂化物、丙酮溶剂化物等。
“溶剂化物”意指含有化学计量量或非化学计量量的溶剂的溶剂加成形式。一些化合物倾向于以结晶固态捕获固定摩尔比率的溶剂分子,从而形成溶剂化物。如果溶剂是水,那么所形成的溶剂化物是水合物;并且如果溶剂是醇,那么所形成的溶剂化物是醇合物。水合物是通过一个或多个水分子与该物质的一个分子组合形成,其中水保留其作为H2O的分子状态。
如本文所用,术语“类似物”是指在结构上与另一化合物类似、但在组成上稍微不同的化合物(如由不同元素的原子置换一个原子或在特定官能团存在下、或由另一官能团置换一个官能团)。因此,类似物是与参考化合物在功能和外观上,但不在结构或来源上类似或相当的化合物。
如本文所定义,术语“衍生物”是指具有共同的核心结构,并且被如本文所述的不同基团取代的化合物。例如,由化学式(I)表示的所有化合物均是芳基或杂芳基取代的苯化合物,并且具有化学式(I)作为共同核心。
术语“生物电子等排体”是指通过原子或原子团由大概相似的另一原子或原子团交换所得的化合物。生物电子等排置换的目标是产生具有与母体化合物相似的生物学特性的新化合物。生物电子等排置换可基于物理化学或拓扑学。羧酸生物电子等排体的实例包括但不限于酰基磺酰亚胺、四唑、磺酸酯和膦酸酯。参见例如,Patani和LaVoie,Chem.Rev.[化学评论]96,3147-3176,1996。
本披露旨在包括本披露化合物中存在的原子的所有同位素。同位素包括那些原子数相同但质量数不同的原子。借助一般实例且不受限制的,氢同位素包括氚和氘,并且碳同位素包括C-13和C-14。
如本文所述,本披露的具有化学式(I)-(VIa)的任何化合物可以是EZH2抑制剂。
在本披露的某些方面,当EZH2的抑制剂抑制突变体EZH2的组蛋白甲基转移酶活性比它抑制野生型EZH2的组蛋白甲基转移酶活性更有效时,EZH2的抑制剂“选择性地抑制”突变体EZH2的组蛋白甲基转移酶活性。例如,在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低40%。在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低50%。在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低60%。在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低70%。在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低80%。在一个实施例中,选择性的抑制剂具有一个IC50,该IC50对于突变体EZH2比对于野生型EZH2的IC50至少低90%。
在一个实施例中,突变体EZH2的选择性抑制剂对野生型EZH2基本上没有抑制作用。
在本披露的某些方面,该抑制剂抑制从H3-K27me2至H3-K27me3的转换。在一个实施例中,该抑制剂据说是抑制H3-K27的三甲基化。因为从H3-K27me1至H3-K27me2的转换先于从H3-K27me2至H3-K27me3的转换,从H3-K27me1至H3-K27me2的转换天然地也抑制从H3-K27me2至H3-K27me3的转换,即,它抑制H3-K27的三甲基化。也有可能抑制从H3-K27me2至H3-K27me3的转换而不会抑制从H3-K27me1至H3-K27me2的转换。这种类型的抑制尽管不会抑制H3-K27的二甲基化,但也将导致H3-K27的三甲基化的抑制。
在一个实施例中,该抑制剂抑制从H3-K27me1至H3-K27me2的转换和从H3-K27me2至H3-K27me3的转换。这样的抑制剂可以直接单独抑制从H3-K27me1至H3-K27me2的转换。可替换地,这样的抑制剂可以直接抑制从H3-K27me1至H3-K27me2的转换和从H3-K27me2至H3-K27me3的转换。
在本披露的某些方面,该抑制剂化合物抑制组蛋白甲基转移酶活性。组蛋白甲基转移酶活性的抑制可以使用任何适合的方法来检测。该抑制可以被测量,例如,依据组蛋白甲基转移酶的活性比率或者依照组蛋白甲基转移酶活性的产物来测量。
相比合适的对照该抑制是一种可测量的抑制。在一个实施例中,相比合适的对照,抑制是至少10%的抑制。即,使用抑制剂时酶活性的比率或产物的量是少于或等于不使用抑制剂时产生的相应的比率和的量的90%。在各种其他实施例中,相比合适的对照,抑制是至少20%、25%、30%、40%、50%、60%、70%、75%、80%、90%或95%抑制。在一个实施例中,相比合适的对照,抑制是至少99%抑制。即,使用抑制剂时酶活性的比率或产物的量是少于或等于不使用抑制剂时产生的相应的比率和量的1%。
本披露的组合物包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种其他治疗剂或其药学上可接受的盐。本披露提供了用于具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种治疗剂或其药学上可接受的盐(作为共同配制品或单独的配制品)的给药,其中配制品的给予是同时的、顺序的或交替的。在某些实施例中,其他治疗剂可以是被本技术领域认为是对治疗正在经本披露的组合物治疗的疾病或病状有用的药剂。在某些实施例中,其他治疗药剂可以是未被本技术领域认为是对治疗正在经本披露的组合物治疗的疾病或病状有用的药剂。在一个方面,其他治疗剂可以是赋予本披露的组合物有益属性的药剂(例如,影响组合物粘性的药剂)。对本披露的组合物有益的属性包括但不限于从具有化学式(I)-(VIa)的化合物与一种或多种其他治疗剂的组合产生的药剂代谢动力学或药效学共同作用。例如,该一种或多种其他治疗剂可以是抗癌剂或化学治疗剂。例如,该一种或多种其他治疗剂可以是糖皮质激素。例如,该一种或多种其他治疗剂可以是选自强的松、泼尼松龙、环磷酰胺、长春新碱、阿霉素、马磷酰胺、顺铂、阿糖胞苷、依维莫司、地西他滨、地塞米松、或其功能类似物、衍生物、前药和代谢物。在另一方面,其他治疗药剂可以是强的松或它的活性代谢物、泼尼松龙。
以下阐述的治疗剂是为了说明的目的,而不是意在限制。本披露包括从以下列表中选出的至少一种其他治疗药剂。本披露可以包括多于一种其他治疗剂,例如,两种、三种、四种、或五种其他治疗剂以至于本披露的组合物可以执行其预期功能。
在一个实施例中,该其他治疗剂是抗癌剂。在一个实施例中,该抗癌剂是影响组蛋白修饰的化合物,例如HDAC抑制剂。在某些实施例中,抗癌剂选自下组,该组由以下各项组成:化学治疗剂(诸如2CdA、5-FU、6-巯基嘌呤、6-TG、AbraxaneTM、放线菌素D、全反式维甲酸、氨甲蝶呤、阿糖胞苷、Azacitadine、BCNU、氯法拉滨、ClolarTM、盐酸柔红霉素、DIC、磷酸依托泊苷、六甲嘧胺、伊沙匹隆、L-天冬酰胺酶、脂质体阿糖胞苷、L-PAM、Lysodren、光辉霉素(mithracin)、丝裂霉素C、尼洛替尼、氮芥子气、具有卡莫司汀植入物的prolifeprospan 20、TESPA、VidazaTM、硫酸长春新碱、VM 26、以及);生物制剂(诸如α干扰素、卡介苗、埃罗替尼、白介素-2、来那度胺、TarcevaTM、以及ZevalinTM);皮质类固醇(诸如地塞米松磷酸钠、和);激素疗法(诸如PlenaxisTM以及);以及放射性药剂(诸如以及Samarium SM-153)。
在另一个实施例中,该其他治疗剂是选自包括以下各项的组的化学治疗剂(也称为抗肿瘤剂或抗增生剂):烷基化剂;抗生素;抗代谢药;解毒剂;干扰素;多克隆或单克隆抗体;EGFR抑制剂;HER2抑制剂;组蛋白脱乙酰酶抑制剂;激素;有丝分裂抑制剂;MTOR抑制剂;多激酶抑制剂;丝氨酸/苏氨酸激酶抑制剂;酪氨酸激酶抑制剂;VEGF/VEGFR抑制剂;紫杉烷或紫杉烷衍生物、芳香酶抑制剂、蒽环霉素、微管靶向药物、拓扑异构酶药物、分子靶标或酶的抑制剂(例如,激酶或蛋白质甲基转移酶)、胞苷类似物药物或列于www.cancer.org/docroot/cdg/cdg_0.asp的任何化学治疗剂、抗肿瘤剂或抗增生剂。
示例性烷基化剂包括但不限于环磷酰胺(Cytoxan;Neosar);苯丁酸氮芥(Leukeran);美法仑(Alkeran);卡莫司汀(BiCNU);白消安(Busulfex);洛莫司汀(CeeNU);达卡巴嗪(DTIC-Dome);奥沙利铂(Eloxatin);卡莫司汀(Gliadel);异环磷酰胺(Ifex);氮芥(Mustargen);白消安(Myleran);卡铂(Paraplatin);顺铂(CDDP;Platinol);替莫唑胺(Temodar);噻替哌(Thioplex);苯达莫司汀(Treanda);或链脲霉素(Zanosar)。
示例性抗生素包括但不限于阿霉素(Adriamycin);阿霉素脂质体(Doxil);米托蒽醌(Novantrone);博莱霉素(Blenoxane);柔红霉素(Cerubidine);柔红霉素脂质体(DaunoXome);更生霉素(Cosmegen);表柔比星(Ellence);伊达比星(Idamycin);普卡霉素(Mithracin);丝裂霉素(Mutamycin);喷司他丁(Nipent);或戊柔比星(Valstar)。
示例性抗代谢药包括但不限于氟尿嘧啶(Adrucil);卡培他滨(Xeloda);羟基脲(Hydrea);巯基嘌呤(Purinethol);培美曲塞(Alimta);氟达拉滨(Fludara);奈拉滨(Arranon);克拉屈滨(Cladribine Novaplus);氯法拉滨(Clolar);阿糖胞苷(Cytosar-U);地西他滨(Dacogen);阿糖胞苷脂质体(DepoCyt);羟基脲(Droxia);普拉曲沙(Folotyn);氟尿苷(FUDR);吉西他滨(Gemzar);克拉屈滨(Leustatin);氟达拉滨(Oforta);甲氨蝶呤(MTX;Rheumatrex);甲氨蝶呤(Trexall);硫鸟嘌呤(Tabloid);Ts-1或阿糖胞苷(Tarabine PFS)。
示例性解毒剂包括但不限于氨磷汀(Ethyol)或美司钠(Mesnex)。
示例性干扰素包括但不限于干扰素α-2b(Intron A)或干扰素α-2a(Roferon-A)。
示例性多克隆或单克隆抗体类包括但不限于曲妥珠单抗(Herceptin);奥法木单抗(Arzerra);贝伐单抗(Avastin);利妥昔单抗(Rituxan);西妥昔单抗(Erbitux);帕尼单抗(Vectibix);托西莫单抗/碘131托西莫单抗(Bexxar);阿仑单抗(Campath);替伊莫单抗(Zevalin;In-111;Y-90 Zevalin);吉妥珠单抗(Mylotarg);依库珠单抗(Soliris)或狄诺塞麦。
示例性EGFR抑制剂包括但不限于吉非替尼(Iressa);拉帕替尼(Tykerb);西妥昔单抗(Erbitux);埃罗替尼(Tarceva);帕尼单抗(Vectibix);PKI-166;卡纳替尼(CI-1033);马妥珠单抗(Emd7200)或EKB-569。
示例性HER2抑制剂包括但不限于曲妥珠单抗(Herceptin);拉帕替尼(Tykerb)或AC-480。
组蛋白脱乙酰酶抑制剂包括但不限于伏立诺他(Zolinza)。
示例性激素包括但不限于它莫西芬(Soltamox;Nolvadex);雷洛昔芬(Evista);甲地孕酮(Megace);亮丙瑞林(Lupron;Lupron Depot;Eligard;Viadur);氟维司群(Faslodex);来曲唑(Femara);曲普瑞林(Trelstar LA;Trelstar Depot);依西美坦(Aromasin);戈舍瑞林(Zoladex);比卡鲁胺(Casodex);阿那曲唑(Arimidex);氟甲睾酮(Androxy;Halotestin);甲羟孕酮(Provera;Depo-Provera);雌氮芥(Emcyt);氟他胺(Eulexin);托瑞米芬(Fareston);地加瑞克(Firmagon);尼鲁米特(Nilandron);阿巴瑞克(Plenaxis);或睾内酯(Teslac)。
示例性有丝分裂抑制剂包括但不限于紫杉醇(Taxol;Onxol;Abraxane);多西他赛(Taxotere);长春新碱(Oncovin;Vincasar PFS);长春碱(Velban);依托泊苷(Toposar;Etopophos;VePesid);替尼泊苷(Vumon);伊沙匹隆(Ixempra);诺考达唑;埃博霉素;长春瑞滨(Navelbine);喜树碱(CPT);伊立替康(Camptosar);托泊替康(Hycamtin);安吖啶或片螺素D(LAM-D)。
示例性MTOR抑制剂包括但不限于依维莫司(Afinitor)或坦罗莫司(Torisel);雷帕呜、地磷莫司;或AP23573。
示例性VEGF/VEGFR抑制剂包括但不限于贝伐单抗(Avastin);索拉非尼(Nexavar);舒尼替尼(Sutent);兰尼单抗;哌加他尼;或凡德他尼。
示例性微管靶向药物包括但不限于紫杉醇、多西他赛、长春新碱、长春碱、诺考达唑、埃博霉素以及诺维本。
示例性拓扑异构酶药物包括但不限于替尼泊苷、依托泊苷、阿霉素、喜树碱、柔红霉素、更生霉素、米托蒽醌、安吖啶、表柔比星以及伊达比星。
示例性紫杉烷或紫杉烷衍生物包括但不限于紫杉醇和多西他赛。
示例性一般化学治疗剂、抗肿瘤剂、抗增生剂包括但不限于六甲蜜胺(Hexalen);异维甲酸(Accutane;Amnesteem;Claravis;Sotret);维甲酸(Vesanoid);阿扎胞苷(Vidaza);硼替佐米(Velcade);天冬酰胺酶(Elspar);左旋咪唑(Ergamisol);米托坦(Lysodren);丙卡巴肼(Matulane);培门冬酶(Oncaspar);地尼白介素-毒素连接物(Ontak);卟吩姆(Photofrin);阿地白介素(Proleukin);来那度胺(Revlimid);贝沙罗汀(Targretin);沙利度胺(Thalomid);坦罗莫司(Torisel);三氧化二砷(Trisenox);维替泊芬(Visudyne);含羞草氨酸(Leucenol);(1M替加氟-0.4M 5-氯-2,4-二羟基嘧啶-1M氧嗪酸钾)、或洛伐他汀。
在另一方面,该其他治疗剂是化学治疗剂或细胞因子,诸如G-CSF(粒细胞集落刺激因子)。
在另一方面,该其他治疗剂可以是标准化学疗法组合,诸如但不限于CMF(环磷酰胺、甲氨蝶呤和5-氟尿嘧啶)、CAF(环磷酰胺、阿霉素和5-氟尿嘧啶)、AC(阿霉素和环磷酰胺)、FEC(5-氟尿嘧啶、表柔比星和环磷酰胺)、ACT或ATC(阿霉素、环磷酰胺和紫杉醇)、利妥昔单抗、Xeloda(卡培他滨)、顺铂(CDDP)、卡铂、TS-1(摩尔比为1∶0.4∶1的替加氟、吉美司特和奥斯他特钾)、喜树碱-11(CPT-11、伊立替康或CamptosarTM)、CHOP(环磷酰胺、羟基柔红霉素、长春新碱(oncovin)、和强的松或泼尼松龙)、R-CHOP(利妥昔单抗、环磷酰胺,羟基柔红霉素、长春新碱、强的松或泼尼松龙)、或CMFP(环磷酰胺、甲氨蝶呤、5-氟尿嘧啶和强的松)。
在另一方面,该其他治疗剂可以是酶(诸如受体或非受体激酶)的抑制剂。受体或非受体激酶是例如,酪氨酸激酶或丝氨酸/苏氨酸激酶。本文所述的激酶抑制剂是小分子、多核酸、多肽、或抗体。
示例性激酶抑制剂包括但不限于贝伐单抗(靶向VEGF)、BIBW 2992(靶向EGFR和Erb2)、西妥昔单抗/Erbitux(靶向Erb1)、伊马替尼/Gleevic(靶向Bcr-Ab1)、曲妥珠单抗(靶向Erb2)、吉非替尼/Iressa(靶向EGFR)、兰尼单抗(靶向VEGF)、哌加他尼(靶向VEGF)、埃罗替尼/Tarceva(靶向Erb1)、尼洛替尼(靶向Bcr-Ab1)、拉帕替尼(靶向Erb1和Erb2/Her2)、GW-572016/二甲苯磺酸拉帕替尼(靶向HER2/Erb2)、帕尼单抗/Vectibix(靶向EGFR)、凡德他尼(靶向RET/VEGFR)、E7080(包括RET和VEGFR多个靶向)、Herceptin(靶向HER2/Erb2)、PKI-166(靶向EGFR)、卡纳替尼/CI-1033(靶向EGFR)、舒尼替尼/SU-11464/Sutent(靶向EGFR和FLT3)、马妥珠单抗/Emd7200(靶向EGFR)、EKB-569(靶向EGFR)、Zd6474(靶向EGFR和VEGFR)、PKC-412(靶向VEGR和FLT3)、伐他拉尼/Ptk787/ZK222584(靶向VEGR)、CEP-701(靶向FLT3)、SU5614(靶向FLT3)、MLN518(靶向FLT3)、XL999(靶向FLT3)、VX-322(靶向FLT3)、Azd0530(靶向SRC)、BMS-354825(靶向SRC)、SKI-606(靶向SRC)、CP-690(靶向JAK)、AG-490(靶向JAK)、WHI-P154(靶向JAK)、WHI-P131(靶向JAK)、索拉非尼/Nexavar(靶向RAF激酶、VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-β、KIT、FLT-3、以及RET)、达沙替尼/Sprycel(BCR/ABL和Src)、AC-220(靶向Flt3)、AC-480(靶向所有HER蛋白质,“panHER”)、二磷酸莫替沙尼(靶向VEGF1-3、PDGFR、和c-kit)、狄诺塞(靶向RANKL,抑制SRC)、AMG888(靶向HER3)、以及AP24534(包括Flt3多个靶向)。
示例性丝氨酸/苏氨酸激酶抑制剂包括但不限于雷帕呜(靶向mTOR/FRAP1)、地磷莫司(靶向mTOR)、Certican/依维莫司(靶向mTOR/FRAP1)、AP23573(靶向mTOR/FRAP1)、Eril/法舒地尔盐酸化物(靶向RHO)、黄酮吡多(靶向CDK)、塞利西利/CYC202/Roscovitrine(靶向CDK)、SNS-032/BMS-387032(靶向CDK)、芦波妥林(靶向PKC)、Pkc412(靶向PKC)、苔藓抑素(靶向PKC)、KAI-9803(靶向PKC)、SF1126(靶向PI3K)、VX-680(靶向极光激酶)、Azd1152(靶向极光激酶)、Arry-142886/AZD-6244(靶向MAP/MEK)、SCIO-469(靶向MAP/MEK)、GW681323(靶向MAP/MEK)、CC-401(靶向JNK)、CEP-1347(靶向JNK)、以及PD 332991(靶向CDK)。
示例性酪氨酸激酶抑制剂包括但不限于埃罗替尼(Tarceva);吉非替尼(Iressa);伊马替尼(Gleevec);索拉非尼(Nexavar);舒尼替尼(Sutent);曲妥珠单抗(Herceptin);贝伐单抗(Avastin);利妥昔单抗(Rituxan);拉帕替尼(Tykerb);西妥昔单抗(Erbitux);帕尼单抗(Vectibix);依维莫司(Afinitor);阿仑单抗(Campath);吉妥珠单抗(Mylotarg);坦罗莫司(Torisel);帕唑帕尼(Votrient);达沙替尼(Sprycel);尼洛替尼(Tasigna);伐他拉尼(Ptk787;ZK222584);CEP-701;SU5614;MLN518;XL999;VX-322;Azd0530;BMS-354825;SKI-606CP-690;AG-490;WHI-P154;WHI-P131;AC-220;或AMG888。适合与具有化学式(I)-(VIa)的化合物或其药学上可接受的盐组合使用的其他治疗剂的更多实例在2014年5月13日提交的共同未决的美国申请号61/992,881、2014年12月8日提交的国际申请号PCT/US2014/069167中公开,将其各自的内容通过引用以其全文结合在此。
本披露提供用于组合疗法的方法,其中将包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种其他治疗剂的组合物给予需要治疗疾病或癌症的受试者。组合疗法还可以是被给予癌症细胞以抑制增生或诱导细胞死亡。在一个方面,在给予了包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种其他治疗剂的本披露的组合物之后,给予具有化学式(I)-(VIa)的化合物或其药学上可接受的盐。在一个方面,在给予了包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种其他治疗剂的本披露的组合物之前,给予具有化学式(I)-(VIa)的化合物或其药学上可接受的盐。在一个方面,在给予了一种或多种其他治疗剂之后给予具有化学式(I)-(VIa)的化合物或其药学上可接受的盐,以至于这些其他治疗剂是在单一的组合物中或在两种或更多种组合物中被给予,例如,同时地、顺序地、或交替地被给予。在一个方面,在给予了一种或多种其他治疗剂之前给予具有化学式(I)-(VIa)的化合物或其药学上可接受的盐,以至于这些其他治疗剂是在单一的组合物中或在两种或更多种组合物中被给予,例如,同时地、顺序地、或交替地被给予。
在一个实施例中,本披露的组合物包括具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种抗癌剂,例如,CHOP(环磷酰胺、羟基柔红霉素、长春新碱、以及强的松或泼尼松龙)或R-CHOP(利妥昔单抗从、环磷酰胺、羟基柔红霉素、长春新碱、强的松或泼尼松龙)。在一个实施例中,本披露的组合物包括具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和强的松或泼尼松龙。本披露的方法包括给予具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和抗癌剂(其中抗癌剂是CHOP、R-CHOP、强的松或泼尼松龙)的组合疗法。
在某些实施例中,“组合疗法”旨在包括以顺序方式给予这些治疗剂,其中在不同时间给予每种治疗剂,以及同时或以基本上同时的方式给予这些治疗剂或这些治疗剂中的至少两种。可以例如通过向受试者给予具有固定比率的每种治疗剂的单一胶囊或以针对这些治疗剂中的每种的多个单一胶囊来完成基本上同时给予。依次或基本上同时给予每一治疗剂,可通过任何适当的途径来实现,包括但不限于口服途径、静脉途径、肌内途径、并通过粘膜组织直接吸收。可以通过相同途径或通过不同途径给予这些治疗剂。例如,可以通过静脉注射给予选定组合的第一治疗剂,同时可以口服地给予该组合的其他治疗剂。可替代地,例如,可以口服地给予所有治疗剂或可以通过静脉注射给予所有治疗剂。也可以交替地给予治疗剂。
在本披露的某些方面,本披露所展示的组合疗法可以在疾病或癌症治疗中产生协同效应。“协同效应”被定义为治疗剂的组合的功效比任何给定的药剂单独的效果的总和要大。协同效应还可以是任何化合物或其他治疗剂作为单一药剂给予时所不能达到的效果。协同效应可以包括但不限于通过减小肿瘤大小、抑制肿瘤生长或增加受试者存活来治疗癌症的效果。协同效应还可以包括降低癌症细胞活力、诱发癌症细胞死亡、和抑制或延迟癌症细胞生长。
在本披露的某些方面,“组合疗法”还包括进一步组合其他生物活性成分和非药物治疗(例如,手术或放射治疗)来给予如上文描述的治疗剂。其中组合疗法进一步包括非药物治疗,该非药物治疗可以在任何合适的时间进行,只要能达到来自所述治疗剂与非药物治疗共同作用的有益效果。例如,在适当的情况下,当非药物治疗暂时从治疗剂的给药中移除时(也许是几天甚至几个星期),仍然能获得有益的效果。
在另一方面,本披露的组合物或其药学上可接受的盐、溶剂化物、类似物或衍生物可与放射疗法组合给予。放射治疗也能与本披露的组合物和本文所述的作为多药剂治疗的一部分的其他化学治疗药剂组合给予。
组合疗法可以通过给予两种或更多种药剂来达到,这些药剂是例如具有化学式(I)-(VIa)的化合物和一种或多种其他治疗剂,其中每一种单独配制和给予或通过单一配制品给予两种或更多种药剂。组合疗法也包括其他的组合。例如,两种药剂可以一起配制和与包含第三药剂的单独配制品一起给予。然而组合疗法中的两种或更多种药剂可以同时给予,它们不需要如此。例如,给予第一药剂(或药剂的组合)可以先于给予第二药剂(或药剂的组合)几分钟、几小时、几天、或几个星期。因此,两种或更多种药剂可以在间隔几分钟内给予,或间隔1、2、3、6、9、12、15、18、或24小时内给予,或间隔1、2、3、4、5、6、7、8、9、10、12、14天给予,或间隔2、3、4、5、6、7、8、9、或10星期给予。在一些情况下可能是甚至更长的间隔。然而在许多情况下,最好是在组合疗法中使用的两种或更多种药剂可以同时存在于病人的体内,这不必如此。
本披露还提供了药物组合物,这些药物组合物包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐以及本文披露的一种或多种其他治疗剂,并且混合药学上合适剂量的载体或赋形剂来治疗或预防的如本文所述的疾病或病状。在一个方面,本披露还提供了药物组合物,这些药物组合物包含表I的任何化合物或其药学上可接受的盐和一种或多种治疗剂,并混合药学上合适剂量的载体或赋形剂来治疗或预防的如本文所述的疾病或病状。在另一方面,本披露还提供了药物组合物,这些药物组合物包含化合物44
或其药学上可接受的盐和一种或多种其他治疗剂,并且混合药学上合适剂量的载体或赋形剂来治疗或预防的如本文所述的疾病或病状。本披露的药物组合物也可以与其他治疗剂或治疗方法同时地、顺序地、或交替地组合给予。
本披露的组合物混合物也可以作为简单的混合物或合适的配制的药物组合物给予患者。例如,本披露的一个方面涉及一种药物组合物,该药物组合物包含治疗有效剂量的EZH2抑制剂或具有化学式(I)-(VIa)的化合物或其药学上可接受的盐、水合物、对映体或立体异构体;一种或多种其他治疗剂,和药学上可接受的稀释剂或载体。
“药物组合物”是呈适合给予受试者的形式的含有本披露化合物的配制品。本文所述的具有化学式(I)-(VIa)的化合物和一种或多种其他治疗剂每一个可以单独配制或配制成由任何活性成分组合的药物组合物。相应地,可以基于每种药物组合物的剂型选择合适的一种或多种给药途径。可替代地,本文所述的具有化学式(I)-(VIa)的化合物和一种或多种其他治疗剂可以配制成一种药物组合物。
在一个实施例中,药物组合物呈整块或呈单位剂型。单位剂型是多种形式中的任一种,包括例如胶囊、IV袋、片剂、气溶胶吸入器上的单泵或小瓶。组合物单位剂量中活性成分(如披露的化合物、盐、水化物、溶剂化物或其同分异构物的配制品)的量是有效的量且根据涉及的特定的治疗有所不同。本领域的技术人员应当了解有时候有必要根据患者的年龄和病状对剂量进行常规变化。剂量也取决于给药的途径。考虑了多种途径,包括经口、经肺、经直肠、肠胃外、经皮、皮下、静脉内、肌内、腹膜内、吸入、经颊、舌下、胸膜内、鞘内、鼻内等。用于局部或经皮给予本披露的化合物的剂型包括散剂、喷雾剂、软膏剂、糊剂、乳膏剂、洗剂、凝胶剂、溶液、贴剂以及吸入剂。在一个实施例中,在无菌条件下将活性化合物与药学上可接受的载体以及需要的任何防腐剂、缓冲剂、或推进剂混合。
如本文所用,短语“药学上可接受的”是指在合理医学判断范围内的那些化合物、阴离子、阳离子、材料、组合物、载体、和/或剂型,这些是适合用于与人类和动物组织接触使用而没有过多的毒性、刺激、过敏反应或其他问题或并发症,与合理的益处/风险比相称。
“药学上可接受的赋形剂”意指用于制备药物组合物的赋形剂,它们通常是安全的、无毒性的和没有生物学上或其他方面不期望的,并且包括兽医使用或人类药物使用可接受的赋形剂。如本说明书和权利要求书中使用的,“药学上可接受的赋形剂”包括一种和多于一种此类赋形剂。
本披露的药物组合物被配制成与其预期的给予途径相容。给药途径的实例包括包括(例如)肠胃外、静脉内、皮内、皮下、口腔(例如,吸入)、经皮(局部)、和经粘膜给药。用于肠胃外、真皮内或皮下施用的溶液或悬浮液可包括以下组分:无菌稀释剂,诸如注射用水、盐水溶液、不挥发性油、聚乙二醇、甘油、丙二醇或其他合成溶剂;抗细菌剂,诸如苄基醇或对羟苯甲酸甲酯;抗氧化剂,诸如抗坏血酸或亚硫酸氢钠;螯合剂,诸如乙二胺四乙酸;缓冲剂,诸如乙酸盐、柠檬酸盐或磷酸盐,及用于调节张力的试剂,诸如氯化钠或右旋糖。可以用酸或碱诸如盐酸或氢氧化钠调节pH。肠胃外制剂可以被封装在由玻璃或塑料制成的安瓿、一次性注射器或多剂量小瓶中。
本披露的组合物可以许多目前用于化学治疗的熟知的方法给予受试者。例如,对于治疗癌症,本披露的化合物可以直接被注射进肿瘤,注射进血流或体腔或口服或使用贴剂应用于皮肤。选择的剂量应该足以构成有效治疗,但不会高到引起不可接受的副作用。疾病病状状况(例如,癌症、癌前病变等)和患者的健康状况在治疗期间和治疗后的合理期限内应优选地被密切监测。
如本文所用,“治疗有效量”是指对治疗、改善或预防目标疾病或病状所需的治疗剂的量或表现出可检测的治疗或抑制作用所需的治疗剂的量。该作用可以通过本技术领域已知的任何测定方法来检测。用于受试者的精确有效量取决于受试者的体重、身材和健康状况;病状的性质和程度;以及选择用于给予的治疗剂或治疗剂的组合。给定情况的治疗有效量可以通过临床医师的技术和判断范围内的常规实验来确定。在一个优选方面中,待治疗的疾病或病状是癌症。在另一方面,待治疗的疾病或病状是细胞增生性病症。
在某些实施例中,相比单独使用各个药剂的单药治疗,当组合使用各个药剂时各个药剂的治疗有效量会更低。这种更低的治疗有效量能负担的治疗方案的毒性更低。
对于任何化合物,最初治疗有效量可以在细胞培养分析(如肿瘤细胞的细胞培养分析)中或者在动物模型(通常大鼠、小鼠、兔、狗、或猪)中估算。动物模型还可以用于确定给药的适当浓度范围和途径。然后这类信息可以用于确定对于在人中给药的适用剂量和途径。治疗/预防功效和毒性可通过标准药剂程序在细胞培养物或实验性动物中来确定,例如,ED50(在50%群体中治疗有效的剂量)和LD50(对50%的群体具杀伤性的剂量)。毒性和治疗效果之间的剂量比率是治疗指数,并且可以表达为比率LD50/ED50。呈现大的治疗指数的药物组合物是优选的。该剂量可以根据所采用的剂型、患者的敏感性、和给药途径在此范围内变化。
调整剂量和给药以提供一种或多种活性药剂的足够水平或维持期望的效果。可考虑的因素包括疾病状况的严重性、受试者的一般健康状况、受试者的年龄、体重以及性别、饮食、给药时间和频率、一种或多种药剂组合、响应灵敏度、以及对疗法的耐受/响应。长效药物组合物根据特定配制品的半衰期和清除率可以每3至4天、每周、或每两周一次给药。
包括本披露的活性化合物的药物组合物可以通过本领域公知的方法制备,例如通过常规混合、溶解、制粒、制糖衣、磨粉、乳化、胶囊化、包埋、或冻干工艺的方式。药物组合物可以常规方式使用一种或多种促进将活性化合物加工成可在药学上使用的制剂的药学上可接受的载体(包括赋形剂和/或助剂)来配制。当然,适当的配制品取决于所选的给药途径。
适合于可注射使用的药物组合物包括无菌水溶液(在水溶性的情况下)或分散体以及用于临时制备无菌可注射溶液或分散液液的无菌粉末。对于静脉内给药,适合的载体包括生理盐水、抑菌水、克列莫佛(Cremophor)ELTM(BASF,新泽西州派西派尼市(Parsippany,NJ))或磷酸盐缓冲盐水(PBS)。在所有情况下,该组合物必须是无菌的并且必须具有达到容易注射的程度的流动性。它在制备和存储的条件下必须是稳定的并且必须抗微生物(诸如细菌和真菌)的污染作用而保存。该载体可以是包含以下物质的溶剂或分散介质:例如,水、乙醇、多元醇(例如,甘油、丙二醇和液体聚乙二醇等),以及其适合的混合物。恰当的流动性可例如通过使用诸如卵磷脂的包衣来维持,在分散液的情况下通过维持所需粒径来维持,以及通过使用表面活性剂来维持。防止微生物的作用可以通过不同的抗细菌以及抗真菌剂,例如对羟苯甲酸酯、三氯叔丁醇、苯酚、抗坏血酸、硫柳汞等来实现。在许多情况下,优选在组合物中包括等渗剂(例如糖)、多元醇(诸如甘露醇和山梨糖醇)、和氯化钠。可通过在组合物中包括延迟吸收的试剂(例如单硬脂酸铝和明胶)来实现可注射组合物的延长吸收。
按照需要,可以通过将活性化合物以需要的量加入具有以上列举的组分中的一种或多种组合的适当溶剂中,随后进行无菌过滤来制备无菌注射液。通常,通过将活性化合物加入无菌载体中来制备分散液,该无菌载体包含基础分散介质以及来自以上列举的其他所需成分。在用于制备无菌可注射溶液的无菌粉末的情况下,制备方法为真空干燥和冷冻干燥,其产生活性成分的粉末以及来自其先前经无菌过滤溶液的任何其他所需成分。
口服组合物通常包括惰性稀释剂或可食用的药学上可接受的载体。它们可以被封装在明胶胶囊中或被压成片剂。出于口服治疗性给药的目的,可以将活性化合物随赋形剂一起加入并且以片剂、锭剂或胶囊剂的形式使用。可以使用用以口腔清洗的流体载体来制备口服组合物,其中流体载体中的化合物经口服、漱、咳出或咽下。可以包括药学上相容的粘合剂和/或佐剂材料作为组合物的一部分。片剂、丸剂、胶囊、锭剂等可含有以下成分中的任一者或具有类似性质的化合物:粘合剂,诸如微晶纤维素、黄蓍胶或明胶;赋形剂,诸如淀粉或乳糖;崩解剂,例如海藻酸、普拉莫胶(Primogel)或玉米淀粉;润滑剂,诸如硬脂酸镁或氢化植物油;助流剂,诸如胶体二氧化硅;甜味剂,诸如蔗糖或糖精;或调味剂,诸如薄荷、水杨酸甲酯或橙味剂。
对于通过吸入给药,化合物从加压容器或分配器(其中包含合适的推进剂,例如气体(诸如二氧化碳))、或喷雾器以气溶胶喷雾形式递送。
系统给药也可以通过经粘膜或经皮肤的方式。对于经粘膜或经皮肤给药,在该配制品中使用适合有待渗透的障碍的渗透剂。这样的渗透剂通常是本技术领域所熟知的,并且对于经粘膜给药包括,例如洗涤剂、胆汁盐和夫西地酸衍生物。经粘膜给药可以通过使用鼻喷雾剂或栓剂来实现。对于经皮给药,如本领域中公知的,将活性化合物配制成软膏剂、油膏剂、凝胶剂、或乳膏。
将这些活性化合物与保护这些化合物免于从体内快速消除的药学上可接受的载体一起制备,诸如控制释放配制品,包括植入物和微囊化的递送系统。可以使用可生物降解、生物相容的聚合物,诸如乙烯乙酸乙烯酯、聚酐类、聚乙醇酸、胶原、聚原酸酯类、以及聚乳酸。用于制备此类配制品的方法对本领域的普通技术人员而言应是清楚的。材料也可以从Alza公司和Nova制药公司购买得到,脂质体悬浮液(包括靶向受感染细胞的具有针对病毒抗原的单克隆抗体的脂质体)也可以用作药学上可接受的载体。这些可以根据本领域的普通技术人员已知的方法制备,如例如描述在美国专利号4,522,811中的。
特别有利的是以剂量单位形式配制口服或肠胃外组合物以便给药和剂量统一。如本文所用的单位剂型是指适合作为用于待治疗受试者的单一剂量的物理离散单位;每一单位含有经计算以产生所需治疗效应的预定量的活性化合物与所需药物载体的结合。对于本披露的单位剂型的规格被指示为并且直接取决于活性化合物的独特特征和有待实现的特定治疗效果。
在治疗应用中,本文所述的EZH2抑制剂化合物、本文所述的其他治疗剂、包含具有化学式(I)-(VIa)的化合物和一种或多种其他治疗剂的组合物、或在按照本披露使用的药物组合物的剂量变化,在影响所选剂量的其他因素中,取决于药剂、接受的患者的年龄、体重和临床状况,以及给予治疗的临床医师或执业医师的经验和判断。通常,剂量应足以导致肿瘤生长的减慢,并且优选是消退,并且还优选是使肿瘤完全消退。剂量的范围可以是从每天大约0.01mg/kg至每天大约5000mg/kg。在优选方面中,剂量的范围可以是从每天大约1mg/kg至每天大约1000mg/kg。在一个方面中,剂量范围将为约0.1mg/天至约50g/天;约0.1mg/天至约25g/天;约0.1mg/天至约10g/天;约0.1mg至约3g/天;或约0.1mg至约1g/天,呈单一、分开或连续剂量(该剂量可针对患者的体重(kg)、体表面积(m2)和年龄(岁)来调整)。药剂的有效量是它提供了由临床医生或其他合格的观察者注意到的一个客观可识别的改进。例如,患者肿瘤的消退可以参考肿瘤的直径来测量。肿瘤直径减小指示消退。治疗停止后肿瘤复发失败也表示消退。如本文所用,术语“剂量有效的方式”是指活性化合物在受试者或细胞中产生期望的生物效应的量。
药物组合物可以与给药的说明书一起包括在容器、包装、或分配器中。
本披露的组合物能够进一步形成盐。本披露的组合物每个分子能够形成多于一个(例如单-、二-、三-)盐。所有这些形式也被考虑在要求保护的披露的范围内。
如本文所用,“药学上可接受的盐”是指本披露的化合物的衍生物,其中母体化合物通过制备其酸或碱的盐来修饰。药学上可接受的盐的实例包括但不限于碱性残基的矿物盐或有机酸盐,诸如胺,酸性残基(如羧酸等)的碱盐或有机盐。药学上可接受的盐包括常规的无毒盐或例如由无毒的无机酸或有机酸形成的母体化合物的季铵盐。例如,这样的常规无毒盐包括但不限于来源于无机酸和有机酸的那些,这些无机酸和有机酸选自2-乙酰氧基苯甲酸、2-羟基乙磺酸、乙酸、抗坏血酸、苯磺酸、苯甲酸、重碳酸、碳酸、柠檬酸、依地酸、乙二磺酸、1,2-乙磺酸、富马酸、葡庚糖酸、葡糖酸、谷氨酸、乙醇酸、乙二醇阿散酸、己基间苯二酚酸(hexylresorcinic)、水巴米克酸(hydrabamic)、氢溴酸、盐酸、氢碘酸、羟基马来酸、羟萘甲酸、羟乙磺酸、乳酸、乳糖酸、月桂基磺酸、马来酸、苹果酸、扁桃酸、甲磺酸、萘磺酸、硝酸、草酸、扑酸、泛酸、苯乙酸、磷酸、聚半乳糖醛酸、丙酸、水杨酸、硬脂酸、亚乙酸(subacetic)、琥珀酸、氨基磺酸、对氨基苯磺酸、硫酸、鞣酸、酒石酸、甲苯磺酸,以及常见的氨基酸,例如甘氨酸、丙氨酸、苯丙氨酸、精氨酸等。
药学上可接受的盐的其他实例包括己酸、环戊烷丙酸、丙酮酸、丙二酸、3-(4-羟基苯甲酰基)苯甲酸、肉桂酸、4-氯苯磺酸、2-萘磺酸、4-甲苯磺酸、樟脑磺酸、4-甲基二环-[2.2.2]-辛-2-烯-1-甲酸、3-苯基丙酸、三甲基乙酸、叔丁基乙酸、粘糠酸等。本披露还涵盖在以下发生时形成的盐:母体化合物中存在的酸性质子被金属离子(例如,碱金属离子、碱土金属离子或铵离子)替代;或与有机碱(诸如乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、N-甲基葡糖胺等)配位。
应当理解的是,所有引用的药学上可接受的盐包括相同盐的溶剂加成形式(溶剂化物)。
本披露的组合物还可被制备为酯,例如,药学上可接受的酯。例如,化合物中的羧酸官能团可以转化成其相应的酯,例如甲酯、乙酯或其他酯。另外,化合物中的醇基可以转化成其相应的酯,例如乙酸酯、丙酸酯或其他酯。
组合物或其药学上可接受的盐或溶剂化物的给药方式有口服、经鼻、经皮、经肺、吸入、经颊、舌下、腹腔内、皮下、肌内、静脉内、直肠、胸膜内、鞘内和肠胃外。在一个实施例中,化合物是口服给予的。本领域的技术人员应认识到某些给药途径的优点。
利用这些化合物的剂量方案是根据多种因素来选择的,包括患者的类型、物种、年龄、体重、性别和医学病状;待治疗病状的严重性;给药途径;患者的肾功能和肝功能;以及所采用的具体化合物或其盐。普通技术的医师或兽医可以容易地确定并开出所需药物的有效量以预防、对抗病状、或阻止病状的进展。
本披露所披露的化合物的配制和给药技术可见于雷明顿:药学科学与实践(Remington:the Science and Practice of Pharmacy),第19版,麦克出版公司,伊斯顿,宾夕法尼亚州(1995)。在一个实施例中,本文所述的化合物及其药学上可接受的盐在药物配制中与药学上可接受的载体或稀释剂组合使用。适合的药学上可接受的载体包括惰性固体填充剂或稀释剂和无菌水或有机溶液。化合物将以足以提供在本文所述范围内的期望剂量的量存在于此类药物组合物中。
除非另行说明,本文所用的所有百分比和比率均以重量计。本发明的其他特征和优势将从不同实例变得清楚。所提供的实例说明在实施本发明中有用的不同组分和方法。这些实例并不限制要求保护的发明。基于本披露,技术人员可以识别并利用可用于实施本发明的其他组分和方法。
本披露提供了用于治疗病状和疾病的组合物和方法,其治疗的过程可以通过调节组蛋白或其他蛋白质的甲基化状态被影响,其中所述甲基化状态是至少部分地由EZH2的活性介导的。组蛋白的甲基化状态的调节可以反过来影响由甲基化激活的靶基因和/或由甲基化抑制的靶基因的表达水平。该方法包括给予需要这种治疗的受试者治疗有效量的本披露的组合物或其药学上可接受的盐或溶剂化物。
至少基于异常组蛋白甲基化已被发现与某些癌症和癌前状态有关联的事实,在受试者中使用突变体EZH2用于治疗癌症或癌前病状的方法包括给予需要的受试者治疗有效量的抑制甲基化的化合物。在一个实施例中,用于在受试者中治疗癌症或癌前病状的方法包括向对其有需要的受试者给予抑制非甲基化H3-K27向单甲基化H3-K27(H3-K27mel)转换的治疗有效量的化合物。在一个实施例中,用于在受试者中治疗癌症或癌前病状的方法包括向对其有需要的受试者给予抑制单甲基化H3-K27向二甲基化H3-K27(H3-K27me2)转换的治疗有效量的化合物。在一个实施例中,用于在受试者中治疗癌症或癌前病状的方法包括向对其有需要的受试者给予抑制H3-K27me2向三甲基化H3-K27(H3-K27me3)转换的治疗有效量的化合物。在一个实施例中,用于在受试者中治疗癌症或癌前病状的方法包括向对其有需要的受试者给予抑制H3-K27me1向H3-K27me2转换和H3-K27me2向H3-K27me3转换的治疗有效量的化合物。值得注意的是,在没有组蛋白或蛋白质甲基化细胞水平的整体增加时,在关键基因位点的染色质上可以发生疾病特异的甲基化的增加。例如,针对整体组蛋白或蛋白质低甲基化的背景,在关键疾病相关基因上可能发生异常的高甲基化。
通常,甲基化的调节剂可用于调节细胞增生。例如,在一些情况下,使用减少甲基化的药剂可以减少过度增生,反之使用增加甲基化的药剂可以刺激不足的增生。相应地,待治疗的疾病包括过度增生性疾病,诸如良性细胞生长和恶性细胞生长(癌症)。
EZH2介导的蛋白质甲基化起作用的病症可以是癌症、细胞增生性病症、或癌前病状。本披露进一步提供本披露组合物、或其药学上可接受的盐或溶剂化物对需要这样的治疗的受试者的用途,制备用于治疗癌症的药剂的用途。待治疗的示例性的癌症包括淋巴瘤,包括非霍奇金淋巴瘤、滤泡性淋巴瘤(FL)和弥漫性大B细胞淋巴瘤(DLBCL);黑色素瘤;以及白血病,包括CML。示例性癌前病状包括骨髓增生异常综合征(MDS;以前称为白血病前期)。
通常,作为甲基化调节剂的化合物可以用于调节细胞增生。例如,在一些情况下,使用减少甲基化的药剂可以减少过度增生,反之使用增加甲基化的药剂可以刺激不足的增生。相应地,可以通过本披露的化合物治疗的疾病包括过度增生性疾病,诸如良性细胞生长和恶性细胞生长。
如本文所用,“对其有需要的受试者”是具有EZH2介导的蛋白质甲基化起作用的疾病的受试者,或者相对于大的群体具有增加的发展这样的疾病风险的受试者。对其有需要的受试者可以患有癌前病状。优选地,对其有需要的受试者具有癌症。“受试者”包括哺乳动物。该哺乳动物可以是(例如)任何哺乳动物,例如人类、灵长类、鸟类、小鼠、大鼠、家禽、狗、猫、牛、马、山羊、骆驼、绵羊或猪。优选地,该哺乳动物是人类。
本披露的受试者包括已经被诊断为癌症或癌前病状、具有癌症或癌前病状的症状、或具有发展癌症或癌前病状的风险的任何人类受试者。本披露的受试者包括任何表达突变体EZH2的人类受试者。例如,突变体EZH2包括一个或多个突变,其中该突变是取代、点突变、无义突变、错义突变、缺失、或插入或任何其他本文所述的EZH2突变。
对其有需要的受试者可以具有难治性或抗性的癌症。“难治性或抗性的癌症”意味着癌症对治疗不响应。该癌症可以从治疗开始就有抗性,或在治疗中变得有抗性。在一些实施例中,对其有需要的受试者在最近治疗缓解后有癌症复发。在一些实施例中,对其有需要的受试者接受了用于治疗癌症的所有已知的有效治疗并都失败了。在一些实施例中,对其有需要的受试者接受了至少一次前治疗。在某些实施例中,前治疗是单药治疗。在某些实施例中,前治疗是组合疗法。
在一些实施例中,对其有需要的受试者由于以前的治疗可以具有第二癌症。“第二癌症”是指由于或因为以前的致癌治疗(诸如化学疗法)发生的癌症。
受试者也可以对EZH2组蛋白甲基转移酶抑制剂或任何其他治疗药剂显示出抗性。
本披露的特征还在于为具有癌症的受试者选择组合疗法的方法。该方法包括以下步骤:从受试者的样品中检测本文所述的一个或多个EZH2突变;并且基于该一个或多个EZH2突变的存在,选择用于治疗癌症的组合疗法。在一个实施例中,该治疗包括给予受试者本披露的组合物。在一个实施例中,该方法进一步包括给予受试者治疗有效量的本披露的组合物。可以使用任何本技术领域已知的合适方法来检测EZH2突变。更多方法描述在美国专利公开US 20130040906,其通过引用以其全文结合在此。
本文所述的方法和用途可包括在向受试者给予本披露的组合物(例如,包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种治疗剂的组合物)之前和/或之后,从对其有需要的受试者的样品中检测本文所述的一个或多个EZH2突变的步骤。在测试样品中存在本文所述的一个或多个EZH2突变表明所述受试者响应于本披露的组合疗法。
通过在受试者中遗传筛查本文所述的一个或多个EZH2突变,本披露提供了个性化的药物、治疗和/或癌症管理给受试者。例如,本披露通过确定受试者对组合疗法的响应性且当受试者响应于所述组合疗法时给予受试者本披露的组合物而提供了用于治疗或减轻有需要的受试者的癌症或癌前病状的症状的方法。通过从受试者获得样品,并检测本文所述的一个或多个EZH2突变来确定响应性,并且本文所述的这样的一个或多个EZH2突变的存在表明所述受试者响应于本披露的组合物。一旦受试者的响应被确定,可以给予治疗有效量的组合物,例如,包含具有化学式(I)-(VIa)的化合物或其药学上可接受的盐和一种或多种治疗剂的组合物。组合物的治疗有效量可以由本领域的普通技术人员来确定。
如本文所用,术语“响应性”是与术语“响应”、“敏感”、和“敏感性”可互换,并且它是指当给予本披露的组合物时受试者表现出治疗响应,例如,受试者的肿瘤细胞或肿瘤组织经受细胞凋亡和/或坏死,和/或显示降低的生长、分裂或增生。该术语也指当给予本披露的组合物时,受试者相对于大的群体会或具有更高的概率表现治疗响应,例如,受试者的肿瘤细胞或肿瘤组织经受细胞凋亡和/或坏死和/或显示降低的生长、分裂或增生。
所谓“样品”意思是源自受试者的任何生物样品,包括但不限于细胞、组织样品、体液(包括但不限于粘液、血液、血浆、血清、尿液、唾液、和精液)、肿瘤细胞、以及肿瘤组织。优选地,样品选自骨髓、外周血细胞、血液、血浆以及血清。样品可以由正在治疗或测试的受试者提供。可替代地,样品可以根据本领域的常规实践由医师获得。
如本文所用,术语“细胞增生性病症”是指一种病状,在其中细胞的非常规或异常的生长或两者可导致有害的病状或病症,其可以是或可以不是癌性的发展。本披露的示例性细胞增生性病症包括各种病状,其细胞分裂是失调的。示例性细胞增生性病症包括但不限于肿瘤、良性肿瘤、恶性肿瘤、癌前期病状、原位肿瘤、包封的肿瘤、转移瘤、液体肿瘤、实体肿瘤、免疫肿瘤、血液瘤、癌症、癌、白血病、淋巴瘤、肉瘤和快速分裂细胞。如本文所用,术语“快速分裂的细胞”被定义为相同的组织内超过或高于相邻或并列的细胞所预期的或观察到的分裂速率的任何细胞。细胞增生性病症包括癌前期或癌前病状。细胞增生性病症包括癌症。优选地,本文所提供的方法用于治疗或减轻癌症的症状。“癌症”一词包括实体瘤以及血液肿瘤和/或恶性肿瘤。“癌前期细胞”或“癌前细胞”是体现细胞增生性病症是癌前期或癌前病状的细胞。“癌细胞”或“癌性细胞”是体现细胞增生性病症是癌症的细胞。任何可重复测量方式可以被用来识别癌细胞和癌前细胞。癌细胞或癌前细胞可通过组织样品(例如,活组织检查样品)的组织学分型或分级来鉴定。癌细胞和癌前期细胞可以通过使用适当的分子标记来鉴定。
示例性非癌性病状或病症包括但不限于类风湿性关节炎;炎症;自身免疫性疾病;淋巴细胞增生病状;肢端肥大症;类风湿性脊椎炎;骨关节炎;痛风、其他关节炎病状;败血症;感染性休克;内毒素休克;革兰氏阴性败血症;中毒性休克综合征;哮喘;成人呼吸窘迫综合征;慢性阻塞性肺病;慢性肺部炎症;炎症性肠病;克罗恩病;银屑病;湿疹;溃疡性结肠炎;胰纤维化;肝纤维化;急性和慢性肾脏疾病;肠道易激综合症;发热;再狭窄;脑型疟;中风和缺血性损伤;神经创伤;阿尔茨海默病;亨廷顿氏病;帕金森氏病;急性和慢性疼痛;过敏性鼻炎;过敏性结膜炎;慢性心力衰竭;急性冠脉综合征;恶病质;疟疾;麻风病;利什曼病;莱姆病;莱特尔综合征;急性滑膜炎;肌肉退化、滑囊炎;肌腱炎;腱鞘炎;突出、破裂、或脱垂的椎间盘综合征;骨硬化病;血栓形成;再狭窄;矽肺;肺肉瘤病;骨质吸收疾病,诸如骨质疏松症;移植物抗宿主反应;多发性硬化症;狼疮;纤维肌痛;AIDS以及其他病毒性疾病,诸如带状疱疹、I或II型单纯疱疹、流感病毒和巨细胞病毒;以及糖尿病。
示例性癌症包括但不限于肾上腺皮质癌、AIDS相关癌症、AIDS相关性淋巴瘤、肛门癌、肛门直肠癌、肛管癌、阑尾癌、儿童小脑星形细胞瘤、儿童脑星形细胞瘤、基底细胞癌、皮肤癌(非黑色素瘤)、胆道癌、肝外胆管癌、肝内胆管癌、膀胱癌(bladder cancer)、膀胱癌(urinary bladder cancer)、骨和关节癌、骨肉瘤和恶性纤维组织细胞瘤、脑癌、脑肿瘤、脑干神经胶质瘤、小脑星形细胞瘤、脑星形细胞瘤/恶性胶质瘤、室管膜瘤、成神经管细胞瘤、脑幕上原始神经外胚层肿瘤、视神经通路和下丘脑胶质瘤、乳腺癌、支气管腺瘤/类癌、类癌肿瘤、胃肠、神经系统癌症、神经系统淋巴瘤、中枢神经系统癌症、中枢神经系统淋巴瘤、子宫颈癌、儿童期癌症、慢性淋巴细胞性白血病、慢性骨髓性白血病、慢性骨髓增生性病症、结肠癌、结肠直肠癌、皮肤T淋巴细胞瘤、淋巴肿瘤、蕈样霉菌病、塞泽里综合征(Seziary Syndrome)、子宫内膜癌、食管癌、颅外生殖细胞瘤、性腺外生殖细胞瘤、肝外胆道癌、眼癌、眼内黑素瘤、视网膜母细胞瘤、胆囊癌、胃部(胃)癌、胃肠类癌肿瘤、胃肠道间质瘤(GIST)、生殖细胞瘤、卵巢生殖细胞瘤、妊娠滋养层肿瘤胶质瘤、头颈癌、肝细胞(肝)癌、霍奇金淋巴瘤、下咽癌、眼内黑素瘤、眼癌、胰岛细胞瘤(内分泌胰腺)、卡波西肉瘤、肾癌(kidney cancer)、肾癌(renal cancer)、肾癌、喉癌、急性成淋巴细胞白血病、急性髓细胞性白血病、慢性淋巴细胞性白血病、慢性骨髓性白血病、毛细胞白血病、唇和口腔癌、肝癌、肺癌、非小细胞肺癌、小细胞肺癌、AIDS相关性淋巴瘤、非霍奇金淋巴瘤、原发性中枢神经系统淋巴瘤、华氏巨球蛋白血症(Waldenstram macroglobulinemia)、成神经管细胞瘤、黑素瘤、眼内(眼)黑素瘤、梅克尔细胞癌、恶性间皮瘤、间皮瘤、转移性鳞状颈癌、口腔癌、舌癌、多发性内分泌肿瘤综合征、蕈样霉菌病、骨髓增生异常综合征、骨髓增生异常/骨髓增生性疾病、慢性骨髓性白血病、急性髓细胞性白血病、多发性骨髓瘤、慢性骨髓增生性病症、鼻咽癌、神经母细胞瘤、口腔癌(oral cancer)、口腔癌、口咽癌、卵巢癌、卵巢上皮癌、卵巢低度恶性潜能的肿瘤、胰腺癌、胰岛细胞胰腺癌、鼻旁窦和鼻腔癌、甲状旁腺癌、阴茎癌、咽癌、嗜铬细胞瘤、成松果体细胞瘤和脑幕上原始神经外胚层肿瘤、垂体瘤、浆细胞肿瘤/多发性骨髓瘤、胸膜肺胚细胞瘤、前列腺癌、直肠癌、肾盂和输尿管、移行细胞癌、视网膜母细胞瘤、横纹肌肉瘤、唾液腺癌、卡波西肉瘤、软组织肉瘤、子宫癌、子宫肉瘤、皮肤癌(非黑色素瘤)、皮肤癌(黑色素瘤)、梅克尔细胞皮肤癌、小肠癌、软组织肉瘤、鳞状细胞癌、胃(胃部)癌、脑幕上原始神经外胚层肿瘤、睾丸癌、喉癌、胸腺瘤、胸腺瘤和胸腺癌、甲状腺癌、肾盂、输尿管和其他泌尿器官的移行细胞癌、妊娠滋养层肿瘤、尿道癌、子宫内膜子宫癌、子宫肉瘤、子宫体癌、阴道癌、外阴癌、以及威尔姆氏肿瘤。
“血液系统的细胞增生性病症“是涉及血液系统细胞的细胞增生性病症。血液系统的细胞增生性病症可以包括淋巴瘤、白血病、髓样肿瘤、肥大细胞肿瘤、脊髓发育不良、良性单克隆丙种球蛋白病、淋巴瘤样肉芽肿、淋巴瘤样丘疹病、真性红细胞增多症、慢性髓细胞白血病、特发性髓样化生、以及特发性血小板增多症。血液系统的细胞增生性病症可以包括血液系统细胞的增生、发育异常、和化生。优选地,本披露的组合物可用于治疗选自本披露的血液学癌症或本披露的血液细胞增生性病症组成的组的癌症。本披露的血液学癌症可以包括多发性骨髓瘤、淋巴瘤(包括霍奇金淋巴瘤、非霍奇金淋巴瘤、儿童期淋巴瘤、以及淋巴细胞和皮肤起源的淋巴瘤)、白血病(包括儿童期白血病、毛细胞白血病、急性淋巴细胞性白血病、急性髓细胞白血病、慢性淋巴细胞性白血病、慢性髓细胞白血病、慢性骨髓性白血病、以及肥大细胞白血病)、髓样肿瘤以及肥大细胞肿瘤。
“肺的细胞增生性病症”是涉及肺细胞的细胞增生性病症。肺的细胞增生性病症可以包括影响肺细胞的所有形式的细胞增生性病症。肺的细胞增生性病症可以包括肺癌、肺的癌前期或癌前病状、肺的良性生长或病灶、和肺的恶性生长或病灶,以及身体除肺外的组织和器官中的转移病灶。优选地,本披露的组合物可用于治疗肺癌或肺的细胞增生性病症。肺癌可以包括所有形式的肺的癌症。肺癌可以包括恶性肺肿瘤、原位癌、典型类癌肿瘤,以及非典型类癌肿瘤。肺癌可以包括小细胞肺癌(“SCLC”)、非小细胞肺癌(“NSCLC”)、鳞状细胞癌、腺癌、小细胞癌、大细胞癌、腺鳞状细胞癌、以及间皮瘤。肺癌可以包括“瘢痕癌”、细支气管肺泡癌、巨细胞癌、梭形细胞癌,以及大细胞神经内分泌癌。肺癌可以包括具有组织学和超微结构异质性的肺肿瘤(例如,混合细胞类型)。
肺的细胞增生性病症可以包括影响肺细胞的所有形式的细胞增生性病症。肺的细胞增生性病症可以包括肺癌、肺的癌前病状。肺的细胞增生性病症可以包括肺的增生、化生、和发育异常。肺的细胞增生性病症可以包括石棉诱导的增生、鳞状化生、和良性反应性间皮化生。肺的细胞增生性病症可以包括柱状上皮被复层鳞状上皮置换和粘膜发育异常。暴露于吸入性有害环境因素(诸如香烟烟雾和石棉)的个体可能会增发展肺的细胞增生性病症的风险。可使个体易于发展肺的细胞增生性病症的先前的肺部疾病可以包括慢性间质性肺病、坏死性肺病、硬皮病、类风湿病、结节病、间质性肺炎、肺结核、反复性肺炎、特发性肺纤维化、肉芽肿、石棉肺、纤维性肺泡炎、以及霍奇金病。
“结肠的细胞增生性病症”是涉及结肠细胞的细胞增生性病症。优选地,结肠的细胞增生性病症是结肠癌。优选地,本披露的组合物可用于治疗结肠癌或结肠的细胞增生性病症。结肠癌可以包括所有形式的结肠的癌症。结肠癌可以包括散发性和遗传性结肠癌。结肠癌可以包括恶性结肠肿瘤、原位癌、典型类癌肿瘤,以及非典型类癌肿瘤。结肠癌可以包括腺癌、鳞状细胞癌、和腺鳞状细胞癌。结肠癌可以与选自下组的遗传性综合征相关,该组由以下各项组成:遗传性非息肉性结肠直肠癌、家族性多发性腺癌、加德纳氏综合征、波伊茨-耶格综合征、Turcot综合征以及青少年息肉病。结肠癌可能起因于选自下组的遗传性综合征,该组由以下各项组成:遗传性非息肉性结肠直肠癌、家族性多发性腺癌、加德纳氏综合征、波伊茨-耶格综合征、Turcot综合征以及青少年息肉病。
结肠的细胞增生性病症可以包括影响结肠细胞的所有形式的细胞增生性病症。结肠的细胞增生性病症可以包括结肠癌、结肠的癌前病状、结肠的腺瘤性息肉以及结肠的异时性病灶。结肠的细胞增生性病症可以包括腺瘤。结肠的细胞增生性病症的特征可能在于结肠的增生、化生、和发育异常。可使个体易于发展结肠的细胞增生性病症的先前的结肠病可以包括括先前的结肠癌。可使个体易于发展结肠的细胞增生性病症的当前疾病可以克罗恩病和溃疡性结肠炎。结肠的细胞增生性病症可能与选自p53、ras、FAP以及DCC组成的组的基因中的突变相关。由于在选自p53、ras、FAP以及DCC组成的组的基因中存在突变,因此个体可能具有升高的发展结肠的细胞增生性病症的风险。
“胰腺的细胞增生性病症”是涉及胰腺细胞的细胞增生性病症。胰腺的细胞增生性病症可以包括影响胰腺细胞的所有形式的细胞增生性病症。胰腺的细胞增生性病症可以包括胰腺癌、胰腺的癌前期或癌前病状、胰腺增生、和胰腺的发育异常、胰腺的良性生长或病灶、和胰腺的恶性生长或病灶,以及身体除胰腺外的组织和器官中的转移病灶。胰腺癌包括所有形式的胰腺的癌症。胰腺癌可以包括导管腺癌、腺鳞癌、多形性巨细胞癌、粘液腺癌、破骨细胞样巨细胞癌、粘液性囊腺癌、腺泡癌、未分类大细胞癌、小细胞癌、胰母细胞瘤、乳头状肿瘤、粘液性囊腺瘤、乳头状囊性肿瘤、以及浆液性囊腺瘤。胰腺癌还可以包括具有组织学和超微结构异质性的胰腺肿瘤(例如,混合细胞类型)。
“前列腺的细胞增生性病症”是涉及前列腺细胞的细胞增生性病症。前列腺的细胞增生性病症可以包括影响前列腺细胞的所有形式的细胞增生性病症。前列腺的细胞增生性病症可以包括前列腺癌、前列腺的癌前期或癌前病状、前列腺的良性生长或病灶、和前列腺的恶性生长或病灶,以及身体除前列腺外的组织和器官中的转移病灶。前列腺的细胞增生性病症可以包括前列腺的增生、化生、和发育异常。
“皮肤的细胞增生性病症”是涉及皮肤细胞的细胞增生性病症。皮肤的细胞增生性病症可以包括影响皮肤细胞的所有形式的细胞增生性病症。皮肤的细胞增生性病症可以包括皮肤的癌前期或癌前病状、皮肤的良性生长或病灶、黑色素瘤、恶性黑色素瘤和皮肤的其他恶性生长或病灶,以及身体除皮肤外的组织和器官中的转移病灶。皮肤的细胞增生性病症可以包括皮肤的增生、化生、和发育异常。
“卵巢的细胞增生性病症”是涉及卵巢细胞的细胞增生性病症。卵巢的细胞增生性病症可以包括影响卵巢细胞的所有形式的细胞增生性病症。卵巢的细胞增生性病症可以包括卵巢的癌前期或癌前病状、卵巢的良性生长或病灶、卵巢癌、卵巢的恶性生长或病灶,以及身体除卵巢外的组织和器官中的转移病灶。皮肤的细胞增生性病症可以包括卵巢细胞的增生、化生、和发育异常。
“乳腺的细胞增生性病症”是涉及乳腺细胞的细胞增生性病症。乳腺的细胞增生性病症可以包括影响乳腺细胞的所有形式的细胞增生性病症。乳腺的细胞增生性病症可以包括乳腺癌、乳腺的癌前期或癌前病状、乳腺的良性生长或病灶、和乳腺的恶性生长或病灶,以及身体除乳腺外的组织和器官中的转移病灶。乳腺的细胞增生性病症可以包括乳腺的增生、化生、和发育异常。
乳腺的细胞增生性病症可以是乳腺的癌前病状。本披露的组合物可用于治疗乳腺的癌前病状。乳腺的癌前病状可以包括乳腺的不典型增生、原位导管癌(DCIS)、导管内癌、原位小叶癌(LCIS)、小叶瘤形成、以及乳腺的0阶段或0级生长或病灶(例如,0阶段或0级乳腺癌、或原位癌)。乳腺的癌前病状可以根据美国癌症联合委员会(AJCC)所接受的TNM分类方案来分阶段,其中原发性肿瘤(T)已被指定为T0或Tis阶段;并且其中局部淋巴结(N)已被指定为N0阶段;并且远程转移(M)已被指定为M0阶段。
乳腺的细胞增生性病症可以是乳腺癌。优选地,本披露的组合物可用于治疗乳腺癌。乳腺癌包括所有形式的乳腺的癌症。乳腺癌可以包括原发性上皮乳腺癌。乳腺癌可以包括其中乳腺与其他肿瘤(诸如淋巴瘤、肉瘤或黑色素瘤)相关的癌症。乳腺癌可以包括乳癌、乳腺导管癌、乳腺小叶癌、乳腺未分化癌、乳腺叶状囊肉瘤、乳腺血管肉瘤、以及乳腺原发性淋巴瘤。乳腺癌可以包括I、II、IIIA、IIIB、IIIC和IV阶段乳腺癌。乳腺导管癌可以包括浸润性癌、具有主要导管内成分的原位浸润性癌、炎性乳腺癌、以及具有选自下组的组织类型的乳腺导管癌,该组由以下各项组成:粉刺、粘液(胶体)、髓质、具有淋巴细胞浸润、乳头状、硬癌和管状的髓质。乳腺的小叶癌可以包括具有主要原位成分的浸润性小叶癌、浸润性小叶癌和浸透性小叶癌。乳腺癌可以包括佩吉特病、佩吉特病伴导管内癌、和佩吉特病伴浸润性导管癌。乳腺癌可以包括具有组织学和超微结构异质性的乳腺肿瘤(例如,混合细胞类型)。
优选地,本披露的化合物或其药学上可接受的盐或溶剂化物,可用于治疗乳腺癌。待治疗的乳腺癌可以包括家族性乳腺癌。待治疗的乳腺癌可以包括散发性乳腺癌。待治疗的乳腺癌可以在男性受试者中出现。待治疗的乳腺癌可以在女性受试者中出现。待治疗的乳腺癌可以在绝经前的女性受试者或绝经后的女性受试者中出现。待治疗的乳腺癌可以在等于或大于30岁的受试者或小于30岁的受试者中出现。待治疗的乳腺癌已经在等于或大于50岁的受试者或小于50岁的受试者中出现。待治疗的乳腺癌可以在等于或大于70岁的受试者或小于70岁的受试者中出现。
待治疗的乳腺癌要可以被分型以识别BRCA1、BRCA2、或p53中的家族性或自发性突变。待治疗的乳腺癌可以被分型为具有HER2/neu基因扩增,如过度表达HER2/neu,或具有低、中等或高水平的HER2/neu表达。待治疗的乳腺癌可以针对选自下组的标记物被分型,该组由以下各项组成:雌激素受体(ER)、黄体酮受体(PR)、人类表皮生长因子受体-2、Ki-67、CA15-3、CA 27-29、以及c-Met。待治疗的乳腺癌可以被分型为ER-未知、ER-富含或ER-贫乏。待治疗的乳腺癌可以被分型为ER-阴性或ER-阳性。可通过任何可再现的手段进行乳腺癌的ER分型。可按照Onkologie 27:175-179(2004)中所列出进行乳腺癌的ER分型。待治疗的乳腺癌可以被分型为PR-未知、PR-富含或PR-贫乏。待治疗的乳腺癌可以被分型为PR-阴性或PR-阳性。待治疗的乳腺癌可以被分型为受体阳性或受体阴性。待治疗的乳腺癌可以被分型为与CA 15-3或CA 27-29或两者升高的血液水平相关。
待治疗的乳腺癌可以包括乳腺的局部肿瘤。待治疗的乳腺癌可以包括与阴性前哨淋巴结(SLN)活组织检查相关的乳腺肿瘤。待治疗的乳腺癌可以包括与阳性前哨淋巴结(SLN)活组织检查相关的乳腺肿瘤。待治疗的乳腺癌可以包括与一个或多个阳性腋淋巴结相关的乳腺肿瘤,其中这些腋淋巴结已经通过任何适用的方法被分期。待治疗的乳腺癌可以包括已经被分型为具有淋巴结阴性状态(例如,淋巴结阴性)或淋巴结阳性状态(例如,淋巴结阳性)的乳腺肿瘤。待治疗的乳腺癌可以包括已经转移到体内其他位置的乳腺肿瘤。待治疗的乳腺癌可以被分类为已经转移到选自骨、肺、肝、或脑组成的组的位置。待治疗的乳腺癌可以根据选自下组的特征分类,该组由以下各项组成:转移性、局部性、区域性、局部区域性、局部晚期、远程、多中心、双侧、同侧、对侧、新诊断、复发、以及不能手术。
本披露的化合物或其药学上可接受的盐或溶剂化物可用于在相对于大的群体具有增加的发展乳腺癌风险的受试者中治疗或预防乳腺的细胞增生性病症,或者用于治疗或预防乳腺癌。相对于大的群体具有增加的发展乳腺癌风险的受试者是具有家族史或乳腺癌个人病史的女性受试者。相对于大的群体具有增加的发展乳腺癌风险的受试者是在BRCA1或BRCA2或两者中均具有种系或自发突变的女性受试者。相对于大的群体具有增加的发展乳腺癌风险的受试者是具有乳腺癌家族史以及在BRCA1或BRCA2或两者中的种系或自发突变的女性受试者。相对于大的群体具有增加的发展乳腺癌风险的受试者是大于30岁、大于40岁、大于50岁、大于60岁、大于70岁、大于80岁、或大于90岁的女性。相对于大的群体具有增加的发展乳腺癌风险的受试者是具有乳腺的不典型增生、原位导管癌(DCIS)、导管内癌、原位小叶癌(LCIS)、小叶瘤形成、或乳腺的0阶段生长或病灶(例如,0阶段或0级乳腺癌、或原位癌)。
待治疗的乳腺癌可以根据Scarff-Bloom-Richardson系统进行组织学分级,其中乳腺肿瘤已被指定为具有1、2、或3的有丝分裂计数得分;1、2、或3的核多态性得分;1、2、或3的小管形成得分;以及3与9之间的总Scarff-Bloom-Richardson得分。待治疗的乳腺癌可以根据关于乳腺癌治疗的国际共识小组被指定为选自1级、1-2级、2级、2-3级、或3级组成的组的肿瘤等级。
待治疗的癌症可以根据美国癌症联合委员会(AJCC)TNM分类系统来分阶段,其中肿瘤(T)已被指定为TX、T1、T1mic、T1a、T1b、T1c、T2、T3、T4、T4a、T4b、T4c、或T4d阶段;并且其中局部淋巴结(N)已被指定为NX、N0、N1、N2、N2a、N2b、N3、N3a、N3b、或N3c阶段;并且其中远程转移(M)可以被指定为MX、M0、或M1阶段。待治疗的癌症可以根据美国癌症联合委员会(AJCC)分类来分阶段为I阶段、IIA阶段、IIB阶段、IIIA阶段、IIIB阶段、IIIC阶段、或IV阶段。待治疗的癌症可以根据AJCC分类被指定为GX级(例如,不能评估等级)、1级、2级、3级或4级。待治疗的癌症可以根据AJCC病理分类(pN)来分阶段为pNX、pN0、PN0(I-)、PN0(I+)、PN0(mol-)、PN0(mol+)、PN1、PN1(mi)、PN1a、PN1b、PN1c、pN2、pN2a、pN2b、pN3、pN3a、pN3b、或pN3c。
待治疗的癌症可以包括已被确定为直径小于或等于约2厘米的肿瘤。待治疗的癌症可以包括已被确定为直径是从约2厘米至约5厘米的肿瘤。待治疗的癌症可以包括已被确定为直径大于或等于约3厘米的肿瘤。待治疗的癌症可以包括已被确定为直径大于5厘米的肿瘤。待治疗的癌症可以通过微观外观分类为高度分化、中度分化,低分化、或未分化。待治疗的癌症可以关于有丝分裂计数(例如,细胞分裂量)或核多态性(例如,细胞变化)通过微观外观分类。待治疗的癌症可以通过微观外观被分类为与坏死区域(例如,死亡或退化细胞的区域)相关。待治疗的癌症可以被分类为具有异常核型、染色体异常数目、或具有外观异常的一个或多个染色体。待治疗的癌症可以被分类为非整倍体、三倍体、四倍体、或具有改变的倍数性。待治疗的癌症可以被分类为具有染色体易位、或整个染色体的缺失或复制、或染色体的一部分的缺失、复制或扩增区域。
待治疗的癌症可以通过DNA细胞计量术、流式细胞术或图像细胞仪来评估。待治疗的癌症可分型为具有10%、20%、30%、40%、50%、60%、70%、80%、或90%的在细胞分裂的合成阶段(如在细胞分裂的S期)的细胞。待治疗的癌症可以分型为具有低S期分数或高S期分数。
如本文所用,“正常细胞”是不能被分类为“细胞增生性病症”的一部分的细胞。一个正常细胞缺乏可导致不希望的病状或疾病发展的非常规生长或异常生长或两者。优选地,正常细胞具有正常功能的细胞周期检验点控制机制。
如本文所用,“接触细胞”是指一种状态,其中化合物或其他物质组合物直接接触细胞,或足够接近以在细胞内诱导期望的生物效果。
如本文所用,“候选化合物”是指已经或将要在一个或多个体外或体内生物测试中测定,以确定该化合物是否可能在细胞、组织、系统、动物或人类中引起正在被研究人员或临床医师所寻求的期望的生物学或医学响应的本披露的化合物或其药学上可接受的盐或溶剂化物。候选化合物是本披露的化合物、或其药学上可接受的盐或溶剂化物。生物学或医学响应可以是癌症的治疗。生物学或医学响应可以是细胞增生性病症的治疗或预防。体外或体内生物测定可以包括但不限于酶活性测定、电泳迁移率变动分析、报告基因测定、体外细胞活力测定、以及本文所述的测定。
如本文所用,“治疗”(“treating”或“treat”)描述了出于防治疾病、病状或病症的目的对患者的管理和护理,并包括给予本披露的化合物或其药学上可接受的盐或溶剂化物,以减轻疾病、病状或病症的症状或并发症,或消除疾病、病状或病症。
本披露的组合物或其药学上可接受的盐或溶剂化物,还可以用于预防疾病、病状或病症。如本文所用,“预防”(“preventing”或“prevent”)描述了减少或消除疾病、病状或病症的症状或并发症的发作。
如本文所用,术语“减轻”是指用来描述一个过程,其中疾病的体征或症状的严重性降低。重要的是,体征或症状可以减轻而不被消除。在一个优选的实施例中,给予本披露的药物组合物导致体征或症状的消除,然而,不需要消除。有效剂量预期降低体征或症状的严重性。例如,可能发生在多个位置的病症诸如癌症的体征或症状得以减轻的条件是该癌症的严重性在多个位置中的至少一个内下降了。
如本文所用,术语“严重性”是指描述癌症从癌前期或良性的状态转变成恶性状态的潜力。可替代地或另外地,严重性是指例如根据TNM分期系统(由国际抗癌联盟(UICC)和美国癌症组合委员会(AJCC)接受)或者由其他技术领域公认的方法描述的癌症阶段。癌症阶段指的是基于因素诸如原发肿瘤的位置、肿瘤大小、肿瘤数和受累的淋巴结(癌症扩散到淋巴结)癌症的程度或严重性。可替代地或另外地,严重性是指由本领域公认的方法(参见,美国国家癌症研究所,www.cancer.gov)描述的肿瘤分级。肿瘤等级是按照癌细胞在显微镜下的外观和肿瘤生长和扩散的可能速度,用于癌细胞分类的系统。当判断肿瘤等级的时候,包括细胞的结构和生长方式的很多因素被考虑。用于确定肿瘤等级的具体因素因每个类型癌症的不同而不同。严重性还描述了一种组织学分级,也称为分化,其是指有多少肿瘤细胞类似于同一组织类型的正常细胞(参见,美国国家癌症研究所,www.cancer.gov)。此外,严重性描述了一种核级,其是指肿瘤细胞中的核的大小和形状以及正在分裂的肿瘤细胞的百分比(参见,美国国家癌症研究所,www.cancer.gov)。
本披露的另一个方面,严重性描述了肿瘤已分泌生长因子、降低细胞外基质、成为血管、失去粘附到并列组织或转移的程度。此外,严重性描述了原发肿瘤已经转移的位置的数量。最后,严重性包括治疗不同类型和位置的肿瘤的难度。例如,不能手术的肿瘤、那些更多的接近多个身体系统(血液学和免疫学肿瘤)的那些癌症、和最抵抗传统治疗的那些被认为是最严重的。在这些情况下,延长受试者的预期寿命和/或减轻疼痛,减少癌细胞的比例或者限制细胞于一个系统,以及改善癌症阶段/肿瘤分级/组织学分级/核级被认为是减轻癌症的体征或症状。
如本文所用,术语“症状”被定义为疾病、病、损伤或某些在体内不正常的指示。症状被经历该症状的个人感觉到或注意到,但可能不容易被其他人注意。其他人被定义为非医护人员。
如本文所用,术语“体征”也被定义为某些在体内不正常的指示。但体征被定义为可以由医生、护士和其他卫生保健专业人士可以看出的事物。
癌症是一组可以导致几乎所有的体征或症状的疾病。症状和体征将取决于癌症在哪里、癌症的尺寸、它对附近器官或结构有多大的影响。如果癌症传播(发生转移),那么症状可出现在身体的不同部位。
其中EZH2介导的蛋白质甲基化起作用的病症可以是神经系统疾病。因此,本披露的化合物还可以用于治疗神经疾病,诸如癫痫、精神分裂症、双相性精神障碍或其他心理和/或精神障碍、神经病、骨骼肌萎缩、以及神经变性疾病(例如,神经变性疾病)。示例性神经变性疾病包括:阿尔茨海默病、肌萎缩性侧索硬化(ALS)、和帕金森氏病。另一类神经变性疾病包括至少部分地由聚谷氨酰胺的聚集引起的疾病。此类疾病包括:亨廷顿氏病、脊髓延髓性肌肉萎缩(SBMA或肯尼迪氏病)、齿状核红核苍白球路易体萎缩(DRPLA)、脊髓小脑性共济失调1(SCA1)、脊髓小脑性共济失调2(SCA2)、马-约病(MJD;SCA3)、脊髓小脑性共济失调6(SCA6)、脊髓小脑性共济失调7(SCA7)、以及脊髓小脑性共济失调12(SCA12)。
其中由EZH2介导的表观遗传甲基化发挥作用的任何其他疾病可以使用本文所述的组合物和方法来治疗或预防。
治疗癌症可导致肿瘤尺寸的减小。肿瘤尺寸的减小也可以被称为“肿瘤消退”。优选地,在治疗后,肿瘤大小相对于治疗前的肿瘤大小减小5%或更多;更优选地,肿瘤大小减小10%或更多;更优选地,减小20%或更多;更优选地,减小30%或更多;更优选地,减小40%或更多;甚至更优选地,减小50%或更多;并且最优选地,减小多于75%或更多。肿瘤尺寸可通过任何可重复测量方式来测量。肿瘤的大小可以测量为肿瘤的直径。
治疗癌症可导致肿瘤体积的减小。优选地,在治疗后,肿瘤体积相对于治疗前的肿瘤尺寸减小5%或更多;更优选地,肿瘤体积减小10%或更多;更优选地,减小20%或更多;更优选地,减小30%或更多;更优选地,减小40%或更多;甚至更优选地,减小50%或更多;并且最优选地,减小多于75%或更多。肿瘤体积可通过任何可重复测量方式来测量。
治疗癌症可导致肿瘤数量的减少。优选地,在治疗后,肿瘤数相对于治疗前的数量减小5%或更多;更优选地,肿瘤数减少10%或更多;更优选地,减小20%或更多;更优选地,减小30%或更多;更优选地,减小40%或更多;甚至更优选地,减小50%或更多;并且最优选地,减少多于75%。肿瘤数可通过任何可重复测量方式来测量。肿瘤数可以通过计数肉眼或在指定的放大率下可见的肿瘤来测量。优选地,指定的放大率是2x、3x、4x、5x、10x、或50x。
治疗癌症可以导致原生肿瘤位点遥远的其他组织或器官的转移病灶数量的减少。优选地,在治疗后,转移病灶数量相对于治疗前的数量减小5%或更多;更优选地,转移病灶数量减少10%或更多;更优选地,减小20%或更多;更优选地,减小30%或更多;更优选地,减小40%或更多;甚至更优选地,减小50%或更多;并且最优选地,减少多于75%。转移病灶数量可通过任何可重复测量方式来测量。转移病灶数量可以通过计数肉眼或在指定的放大率下可见的肿瘤来测量。优选地,指定的放大率是2x、3x、4x、5x、10x、或50x。
相对于仅接受载体的群体,治疗癌症可以导致接受治疗的受试者群体的平均存活时间增加。优选地,平均存活时间延长超过30天;更优选地,延长超过60天;更优选地,延长超过90天;并且最优选地,延长超过120天。群体的平均存活时间的延长可以通过任何可重复的方式来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗开始后计算群体的平均存活长度来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗第一轮完成后计算群体的平均存活长度来测量。
相对于未治疗的群体,治疗癌症可以导致接受治疗的受试者群体的平均存活时间增加。优选地,平均存活时间延长超过30天;更优选地,延长超过60天;更优选地,延长超过90天;并且最优选地,延长超过120天。群体的平均存活时间的延长可以通过任何可重复的方式来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗开始后计算群体的平均存活长度来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗第一轮完成后计算群体的平均存活长度来测量。
相比于接受单药治疗(使用的药物不是本披露的化合物或其药学上可接受的盐、溶剂化物、类似物或衍生物)的群体,治疗癌症可以导致增加接受治疗的受试者群体的平均存活时间。优选地,平均存活时间延长超过30天;更优选地,延长超过60天;更优选地,延长超过90天;并且最优选地,延长超过120天。群体的平均存活时间的延长可以通过任何可重复的方式来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗开始后计算群体的平均存活长度来测量。群体的平均存活时间的延长可以例如通过使用活性化合物治疗第一轮完成后计算群体的平均存活长度来测量。
相对于仅接受载体的群体,治疗癌症可以导致接受治疗的受试者群体的死亡率降低。相对于未接受治疗的群体,治疗癌症可以导致接受治疗的受试者群体的死亡率降低。相对于接受单药治疗(使用的药物不是本披露的化合物或其药学上可接受的盐、溶剂化物、类似物或衍生物)的群体,治疗癌症可以导致接受治疗的受试者群体的死亡率降低。优选地,死亡率降低超过2%;更优选地,降低超过5%;更优选地,降低超过10%;并且最优选地,降低超过25%。接受治疗的受试者群体的死亡率的降低可以通过任何可重复的方式来测量。群体的死亡率的降低可以例如通过使用活性化合物治疗开始后计算每个单位时间内群体的疾病相关死亡的平均数量来测量。群体的死亡率的降低可以例如通过使用活性化合物治疗第一轮完成后计算每个单位时间内群体的疾病相关死亡的平均数量来测量。
治疗癌症可导致肿瘤生长率降低。优选地,在治疗后,肿瘤生长速率相对于治疗之前的数值降低至少5%;更优选地,肿瘤生长速率降低至少10%;更优选地,降低至少20%;更优选地,降低至少30%;更优选地,降低至少40%;更优选地,降低至少50%;甚至更优选地,降低至少50%;并且最优选地,降低至少75%。肿瘤生长率可通过任何可重复测量方式来测量。肿瘤生长率可以根据每个单位时间内肿瘤直径的变化来测量。
治疗癌症可导致肿瘤再生长的降低。优选地,在治疗后,肿瘤再生长低于5%;更优选地,肿瘤再生长低于10%;更优选地,低于20%;更优选地,低于30%;更优选地,低于40%;更优选地,低于50%;甚至更优选地,低于50%;并且最优选地,低于75%。肿瘤再生长可通过任何可重复测量方式来测量。肿瘤再生长例如是通过测量治疗后的居前肿瘤收缩后肿瘤直径的增加来测量。治疗停止后肿瘤复发的失败表示肿瘤再生长的降低。
治疗或预防细胞增生性病症可以导致细胞增生率的降低。优选地,在治疗后,细胞增生率降低至少5%;更优选地,至少10%;更优选地,至少20%;更优选地,至少30%;更优选地,至少40%;更优选地,至少50%;甚至更优选地,至少50%;并且最优选地,至少75%。细胞增生率可通过任何可重复测量方式来测量。细胞增生率的测量(例如)是通过测量每个单位时间内组织样品中分裂细胞的数量。
治疗或预防细胞增生性病症可以导致增生细胞的比例的下降。优选地,在治疗后,增生细胞的比例降低至少5%;更优选地,至少10%;更优选地,至少20%;更优选地,至少30%;更优选地,至少40%;更优选地,至少50%;甚至更优选地,至少50%;并且最优选地,至少75%。增生细胞的比例可通过任何可重复测量方式来测量。优选地,增生细胞的比例例如是通过定量相对于非分裂细胞的数量而言组织样品中分裂细胞的数量来测量。增生细胞的比例可以是等同于细胞有丝分裂指数。
治疗或预防细胞增生性病症可以导致细胞增生面积或区域大小的降低。优选地,在治疗后,细胞增生的面积或区域的大小相对于其治疗前的大小减少至少5%;更优选地,减少至少10%;更优选地,减少至少20%;更优选地,减少至少30%;更优选地,降低至少40%;更优选地,降低至少50%;甚至更优选地,降低至少50%;并且最优选地,降低至少75%。细胞增生面积或区域大小可通过任何可重复测量方式来测量。细胞增生面积或区域大小可以通过细胞增生面积或区域的直径或宽度来测量。
治疗或预防细胞增生性病症可以导致具有异常外观或形态的细胞的数量或比例的下降。优选地,在治疗后,具有异常形态的细胞数相对于其在治疗之前的数量减少至少5%;更优选地,减少至少10%;更优选地,减少至少20%;更优选地,减少至少30%;更优选地,减少至少40%;更优选地,减少至少50%;甚至更优选地,减少至少50%;并且最优选地,减少至少75%。异常细胞的外观或形态可通过任何可重复测量方式来测量。异常细胞形态可通过显微镜检查,例如使用倒置的组织培养显微镜来测量。异常细胞形态可以采取核多形性的形式。
如本文所用,术语“选择性地”是指倾向于在一个群体中比另一个群体中以更高的频率发生。比较的群体可以细胞群体。优选地,本披露的化合物或其药学上可接受的盐或溶剂化物选择性地对癌症或癌前细胞起作用,但不作用于正常细胞。优选地,本披露的化合物或其药学上可接受的盐或溶剂化物选择性地调节一种分子靶(例如,靶蛋白甲基转移酶),但不显著调节另一种分子靶(例如,非靶蛋白甲基转移酶)。本披露还提供了一种选择性地抑制酶(如蛋白质甲基转移酶)的活性的方法。优选地,如果事件在群体A中的发生频率大于在群体B中发生频率的两倍,那么相对于群体B,该事件选择性地发生于群体A中。如果事件在群体A中的发生频率大于5倍,那么该事件选择性地发生。如果事件在群体A中的发生频率大于群体B的10倍;更优选地,大于50倍;甚至更优选地,大于100倍;并且最优选地,在群体A中的频率大于1000倍,那么该事件选择性地发生。例如,如果细胞死亡在癌细胞中的发生频率大于正常细胞的两倍,那么将认为细胞死亡在癌细胞中选择性地发生。
本披露组合物(例如,包含具有化学式(I)-(VIa)的化合物44或其药学上可接受的盐和一种或多种其他治疗剂(诸如强的松)的组合物)可以调节分子靶标(例如,靶蛋白甲基转移酶)的活性。调节是指刺激或抑制分子靶标的活性。优选地,如果相对于分子靶标在相同的条件下仅缺乏本披露的化合物的存在时的活性,分子靶标的活性被刺激或抑制了至少2倍,那么本披露的化合物或其药学上可接受的盐或溶剂化物调节分子靶标的活性。更优选地,如果相对于分子靶标在相同的条件下仅缺乏本披露的化合物的存在时的活性,分子靶标的活性被刺激或抑制了至少5倍、至少10倍、至少20倍、至少50倍、至少100倍,那么本披露的化合物或其药学上可接受的盐或溶剂化物调节分子靶标的活性。分子靶标的活性可以用任何可重复的方法测量。分子靶标的活性可以在体外或体内测量。例如,分子靶标的活性在体外可以通过酶活性试验或DNA结合试验测量,或分子靶标的活性在体内可以通过报告基因的表达试验来测量。
优选地,如果相对于分子靶标在相同的条件下但仅缺乏本披露的化合物的存在时的活性,所述化合物的加入不能刺激或抑制分子靶标的活性超过10%,那么本披露的化合物不能显著地调节分子靶标的活性。
如本文所用,术语“同功酶选择性”是指相比酶的第二同种型,优先抑制或刺激酶的第一同种型(例如相比蛋白质甲基转移酶的同工酶β,优先抑制或刺激蛋白质甲基转移酶同工酶α)。优选地,本披露的化合物或其药学上可接受的盐或溶剂化物,在实现生物效应所需的剂量中显示出最小4倍差异,优选是10倍差异,更优选是50倍差异。优选地,本披露的化合物或其药学上可接受的盐或溶剂化物,显示出这种横跨抑制范围的差别,并且该差别的示例为IC50,即,对于感兴趣的分子靶标50%的抑制。
向对其有需要的细胞或受试者给予本披露的组合物可导致感兴趣的蛋白甲基转移酶活性的调节(即刺激或抑制)。
向对其有需要的细胞或受试者给予本披露的化合物(例如,包含具有化学式(I)-(VIa)的任何化合物或其药学上可接受的盐和一种或多种其他治疗剂(诸如强的松)的组合物)导致细胞内靶标(诸如底物)活性的调节(即刺激或抑制)。几个细胞内靶标可以使用本披露的化合物、包括但不限于蛋白质甲基转移酶来调节。
激活是指放置物质组合物(例如蛋白质或核酸)进入适合执行期望的生物学功能的状态。能够被激活的物质组合物也具有非激活的状态。激活的物质组合物可以具有抑制或刺激的生物学功能或两者。
提高是指物质组合物(例如,蛋白质或核酸)的期望的生物学活性的增加。提高可以通过物质组合物的浓度增加而发生。
如本文所用,“细胞周期关卡通路”是指涉及细胞周期关卡调节的生物化学通路。细胞周期关卡通路可以在包括细胞周期关卡的一个或多个功能中具有刺激或抑制效应或两者都是。细胞周期关卡通路是由至少两种物质组合物(优选是蛋白质)构成,其中两种都对细胞周期关卡的调节有贡献。细胞周期关卡通路可以通过细胞周期关卡通路中一个或多个成员的激活来激活。优选地,细胞周期关卡通路是生物化学信号通路。
如本文所用,“细胞周期关卡调节子”是指可以至少部分在细胞周期关卡调节中起作用的物质组合物。细胞周期关卡调节子在包括细胞周期关卡的一个或多个功能上可以具有刺激或抑制效应或两者都是。细胞周期关卡调节子可以是蛋白质或不是蛋白质。
治疗癌症或细胞增生性病症可导致细胞死亡,并且优选地细胞死亡导致群体中细胞数量减少至少10%。更优选地,细胞死亡意指减少至少20%;更优选地,减少至少30%;更优选地,减少至少40%;更优选地,减少至少50%;最优选地,减少至少75%。群体中细胞数可以通过任何可重复的方式来测量。群体中细胞数可以通过荧光激活细胞分选(FACS)、免疫荧光显微镜和光学显微镜来测量。测量细胞死亡的方法示于Li等人,Proc Natl Acad Sci U S A.[美国国家科学院院刊]100(5):2674-8,2003中。在一个方面,细胞死亡通过细胞凋亡发生。
优选地,本披露的组合物或其药学上可接受的盐或溶剂化物的有效量是对正常细胞没有显著的细胞毒性。如果给予治疗有效量的化合物不会引起正常细胞大于10%的细胞死亡,那么治疗有效量的化合物对正常细胞没有显著的细胞毒性。如果给予治疗有效量的化合物不会引起正常细胞大于10%的细胞死亡,那么治疗有效量的化合物不会显著地影响正常细胞的活力。在一个方面,细胞死亡通过细胞凋亡发生。
使细胞接触本披露的组合物或其药学上可接受的盐或溶剂化物可选择性地诱发或激活癌细胞的细胞死亡。向对其有需要的受试者给予本披露的化合物或其药学上可接受的盐或溶剂化物可选择性地诱发或激活癌细胞的细胞死亡。使细胞接触本披露的组合物或其药学上可接受的盐或溶剂化物可选择性地诱发受细胞增生性病症影响的一种或多种细胞的细胞死亡。优选地,向对其有需要的受试者给予本披露的组合物或其药学上可接受的盐或溶剂化物选择性地诱发受细胞增生性病症影响的一种或多种细胞的细胞死亡。
本披露涉及通过向对其有需要的受试者给予本披露的组合物或其药学上可接受的盐或溶剂化物来治疗或预防癌症的方法,其中,给予本披露的组合物或其药学上可接受的盐或溶剂化物导致以下项中的一个或多个:通过在细胞周期的一个或多个阶段(例如,G1期、G1/S期、G2/M期)中的细胞累积、或诱导细胞的衰老、或促进肿瘤细胞分化来阻止癌细胞的增生;通过细胞毒性、坏死或凋亡促进癌细胞的细胞死亡,而不引起大量正常细胞的细胞死亡,并且在动物中抗肿瘤活性具有至少2的治疗指数。如本文所用,“治疗指数”是由有效剂量除最大耐受剂量。
本领域的技术人员可以参考一般参考文献中在此讨论的已知技术或等效技术的详细说明。这些文献包括Ausubel等人,Current Protocols in Molecular Biology[当前分子生物学方案],John Wiley and Sons,Inc.[约翰成利父子公司](2005);Sambrook等人,Molecular Cl0ning,A Laboratory Manual[分子克隆,实验室手册](第3版),Cold Spring Harbor Press[冷泉港出版社],冷泉港,纽约(2000);Coligan等人,Current Protocols in Immunology[当前免疫学方案],约翰成利父子公司,纽约;Enna等人,Current Protocols in Pharmacology[当前药理学方案],约翰成利父子公司,纽约;Fingl等人,The Pharmacological Basis of Therapeutics[治疗的药理学基础](1975),Remington′s Pharmaceutical Sciences[雷明顿制药科学],Mack Publishing Co.[麦克出版公司],伊斯顿,宾夕法尼亚州,第18版(1990)。当然,在制备或使用本披露的方面中可参考这些文献。
实例1
临床前数据已经表明,用于组蛋白甲基转移酶EZH2的小分子抑制剂代表了用于表达EZH2功能突变变化的非霍奇金淋巴瘤(NHL)的潜在的新的治疗方式。先前已经报道,EZH2的选择性抑制导致在体外和体内具有EZH2突变体的淋巴瘤细胞的特异性杀伤,对非突变淋巴瘤细胞具有最小的影响[Knutson等人Nature Chemical Biology[自然化学生物学]20121;Keilhack等人Blood[血液](ASH年度会议摘要)2012,120,摘要37122]。由于表观遗传改变已被认为涉及癌细胞对许多抗癌剂的抗性,因此研究了化合物44与用于NHL药物的护理标准、二线疗法或在此适应症中正在探索的靶向疗法的组合。通过连续暴露于化合物44,两种不同EZH2突变体细胞系的基于细胞的测定显示了与CHOP化学疗法方案、二线疗法,以及几种靶向疗法(例如其他表观遗传药物、PI3K途径或其他抑制剂)的所有组分的组合益处。在活化的B细胞类型的EZH2野生型淋巴瘤细胞系中未观察到这些效果。在两种不同的EZH2突变体异种移植模型中还观察到与CHOP的强的组合益处。例如,在SUDHL6 Y646N异种移植模型中,单独的化合物44和CHOP化学疗法均不会诱导显著的抗肿瘤效果,即使在停止给药后,它们的组合仍然产生持久的肿瘤消退。重要的是,在使用另一种EZH2突变体异种移植模型的第三项研究中,当从CHOP化学疗法方案中省略阿霉素时此效果被保留。随后本文所呈现的数据显示,糖皮质激素受体激动作用可能是与CHOP所观察到的组合益处的关键机制,因为在几种EZH2突变体淋巴瘤细胞系(体外)中,化合物44的抗增生作用被单独的泼尼松龙或地塞米松增强。综合这些数据表明,化合物44在EZH2突变体NHL中的单一药剂活性可通过合理的组合策略被进一步增强和扩大。
本文所呈现的数据表明:
化合物44和糖皮质激素受体激动剂配合以显著地增强EZH2突变体和野生型GCB淋巴瘤系(包括EZH2i不敏感突变系,而不是体外活化的B细胞类型)中的抗增生活性。
在突变体EZH2GCB淋巴瘤细胞中,也观察到与CHOP化学疗法方案、第二线和其他靶向疗法的所有单一组分的组合益处,包括与BCL2抑制剂、纳威托克斯和mTOR抑制剂依维莫司的强协同作用。
在两种不同的EZH2突变体异种移植模型中观察到与CHOP的强的组合益处,并且此效果在第三种EZH2突变体异种移植模型(其中从化学疗法方案中省略阿霉素)的研究中被保留。
综合这些结果表明,糖皮质激素受体激动剂可在化合物44和CHOP的组合在EZH2突变体淋巴瘤异种移植物中所观察到的扩增的抗肿瘤活性中以及观察到的强的体外协同作用中起关键作用,其中当前在NHL中所评估的几种新型疗法有必要进一步调查合理的组合方法。
实例2:EZH2抑制剂和糖皮质激素受体激动剂在生发中心非霍奇金淋巴瘤中的协同抗肿瘤活性
结果
当化合物44仅与CHOP的糖皮质激素受体激动剂(GRag)泼尼松龙或与其他GRag(诸如地塞米松)组合时观察到显著的协同作用。当与CHOP组合时化合物44的抗增生效应被大大地增加了,并且这种协同作用的大部分可以归因于CHOP的GRag成分泼尼松龙(强的松的活性代谢物)。引人注目地,化合物44和泼尼松龙的组合延伸了对EZH2抑制敏感的细胞的范围,从仅携带突变的GCB NHL细胞延伸至所有GCB NHL细胞。
两个EZH2突变体细胞系(WSU-DLCL2和SU-DHL10)使用化合物44预处理4天,然后使用化合物44和单独CHOP组分共处理另外的3天(4+3模型)。马磷酰胺(环磷酰胺的类似物)、阿霉素和长春新碱都自身显示出在突变体细胞系中浓度依赖性的生长抑制。因此,在与化合物44组合时获得它们药物的组合指数(使用Calcusyn软件计算的CI)。然而,这些细胞系它们自身没有显示对泼尼松龙(强的松的活性代谢物)的敏感性。因此,在这种情况下不能确定CI,反而基于使用泼尼松龙的浓度响应曲线观察的化合物44的IC50的转变计算出效力的增加。
化合物44+马磷酰胺的组合导致在两个EZH2突变体细胞系中的总体加和组合益处(图3C、F)。在WSU-DLCL2细胞中,化合物44+阿霉素在4+3模型中协同作用(图3A),而这种组合在SU-DHL10细胞中是加和的(图3D)。化合物44+长春新碱的组合在两个EZH2突变体细胞系中显示加和性(图3B、E)。当使用泼尼松龙+化合物44处理WSU-DLCL2细胞时,观察到比化合物44大9倍的效力转变。使用不同的GRag(地塞米松)处理导致甚至更大的转变,是化合物44的IC50的17倍(图4A、B)。在SU-DHL10细胞中观察到化合物44效力转变的类似趋势(图4C、D)。
然后调查了化合物44+CHOP的组合效应是否使得WT EZH2淋巴瘤细胞系(GCB和ABC两种亚型)对化合物44敏感。因为化合物44单独治疗不会诱发EZH2 WT淋巴瘤系的生长抑制,基于CHOP组分各自的浓度响应曲线来计算效力的转变。在测试的四种CHOP组分中,仅GRag+化合物44的组合导致了WT GCB淋巴瘤细胞系的效力转变(图5A、B和表3)。相比之下,在具有4个CHOP组分中的任一个的WT ABC淋巴瘤系中没有观察到效力转变(图5C、D和表3),这表明GRag+EZH2i组合益处对淋巴瘤GCB亚型的生物学是特异性的。
鉴于相对于单个药剂,GRag+EZH2i组合在EZH2 WT和突变体GCB淋巴瘤细胞系中诱发了显著地增加的抗增生效应,确定了化合物治疗的持续时间和/或添加的顺序是否影响敏感性。细胞系组也被延伸至包括两个EZH2 WT、两个EZH2突变体、化合物44敏感型、以及两个EZH2突变体、化合物44不敏感型细胞系(先前被McCabe等人报道,和内部未发表数据)。在先前的4+3模型中,效力转变是基于化合物44(在EZH2 Y646敏感型细胞系中)或泼尼松龙(在EZH2 WT细胞系中)的暴露。对于这组实验,在泼尼松龙的固定浓度下化合物44 IC50的转变被用于确定接受4+3模型、4天或7天共处理、或4天泼尼松龙预处理加3天共处理的处理的细胞系中的组合益处。当EZH2突变体、化合物44敏感型细胞系被共处理4天时,观察到化合物44 IC50降低30-60倍,显示了与4+3处理方案相似的趋势(表2)。使用7天共处理和4+3模型观察到了相似的结果(表2)。在EZH2 WT GCB细胞系中,尽管4天后没有产生可测量的化合物44IC50,但是在与泼尼松龙共处理4天后两个细胞系都显示出降低的增生和可测量的化合物44IC50(表2)。EZH2 WT GCB细胞也响应4+3模型和/或7天共处理方案(表2)。引人注目地,EZH2突变体、化合物44不敏感型细胞系,处理4天后也不显示可测量的化合物44 IC50,而共处理4天后显示降低的增生,使用4+3处理方案以及7天共处理对该组合显示出更大的响应(表2)。当使用泼尼松龙预处理细胞,然后使用化合物44+泼尼松龙共处理,这6个细胞系中仅1个细胞系显示组合益处,表明药物添加的顺序对协同效应是重要的(表2)。
表2.化合物44/GRag组合在EZH2 Y646细胞系中增加EZH2i敏感性并且在抵抗EZH2i的细胞系中克服EZH2i的不敏感性
为了研究化合物44+GRAG的这种组合益处在这些细胞系中起作用的作用机制,分析了组蛋白H3赖氨酸27(H3K27)残基的全局甲基化和乙酰化。用化合物44、泼尼松龙或其组合处理WSU-DLCL2、OCI-LY19和RL细胞4天,并且通过ELISA或蛋白质印迹评估H3K27修饰。单独的泼尼松龙对WSU-DCL2或RL细胞中的H3K27三甲基化(H3K27me3)没有任何影响,但在OCI-LY19细胞中在较高剂量下确实导致H3K27me3的适度增加。化合物44+泼尼松龙的组合在任何细胞系中不转变对H3K27me3抑制的化合物44IC50(图11A)。类似地,H3K27乙酰化的整体水平不被单独的泼尼松龙或化合物44+泼尼松龙组合影响(图11B)。
已经发现H3K27乙酰化或三甲基化的整体水平不被影响,研究了GR信号通路的转录调节。使用化合物44、泼尼松龙、或其组合的单一浓度处理WSU-DLCL2、SU-DHL10、RL、SU-DHL4、OCI-LY19、和DOHH2细胞4天,并且使用糖皮质激素信号PCR试验分析基因表达(表4)。总体上在所有细胞系中使用泼尼松龙和组合治疗的很多基因被下调了,表明GR在基因表达中起到激活剂和抑制剂的作用。这里聚焦了GR的激活功能且描述了在细胞系组中使用组合治疗具有协同上调的3个基因。抑制mTOR信号(ref)的候选肿瘤抑制基因瑟斯崔因在4个EZH2突变体细胞系中(而不是在EZH2WT细胞系中)使用组合治疗以协同作用的方式被识别为通常上调的基因(图10A)。TNF表达仅在EZH2突变体、化合物44不敏感型细胞系(图10B)、和TSC22D3/GILZ中协同上调,虽然在所有细胞系中被泼尼松龙上调,但仅在EZH2突变体、化合物44敏感型细胞系中通过组合治疗协同地增强(图10C)。
表3.与化合物44组合的汇总
最后,在3个不同的EZH2突变体淋巴瘤异种移植模型中评估了肿瘤生长抑制。使用化合物44和化学疗法(CHOP或COP(没有阿霉素的CHOP))共同处理SCID或携带皮下淋巴瘤异种移植物的裸鼠,并且与单一药剂处理进行了比较。在携带WSU-DLCL2异种移植物的小鼠中,所有化合物44剂量和应用的方案实现了肿瘤生长抑制,并且比单独的CHOP化学疗法更好(图9A)。同时,化合物44与CHOP组合疗法诱发了强的抗肿瘤应答且比单独的任何单一药剂(对于CHOP和化合物44分别是45%和71%)显著地(p<0.001)更好的肿瘤生长抑制(93%)。所有单一治疗都被耐受;在第一个周期后化合物44/CHOP组合物组的有小的体重损失(11.3%),其后小鼠在下一个周期治疗前恢复了。
在SU-DHL6异种移植模型中,与由Beguelin等人先前公布的结果相反,使用EZH2抑制剂GSK503在单独的CHOP或化合物44(图9B,顶部图)没有观察到显著的肿瘤生长抑制。引人注目地,化合物44/CHOP的组合导致肿瘤消退。当在第28天停止给药且小鼠的肿瘤生长延迟被观察到第60天时,这个组合导致58%的小鼠无肿瘤地存活(图9B,底部图)。
CHOP的阿霉素组分具有它的心脏毒性具有<550mg/m2的终生累积剂量限制。因此,调查了排除这个组分的化合物44/化学疗法方案的组合益处。在第三个研究中,使用增加剂量的化合物44(BID)、无阿霉素的化学疗法方案(COP)、或COP和化合物44的组合处理携带SU-DHL10异种移植物的小鼠28天。在所有化合物44剂量和包含COP的组中观察到肿瘤生长抑制(图9C,顶部图)。如通过重复测验ANOVA和邓尼特事后检验评估的,266mg/kg、532mg/kg以及COP/化合物44组合治疗导致与运载体显著不同的消退(p>0.001),其中化合物44/COP组合物组显示出最好的总体响应。给药28天后,具有最小肿瘤负荷的小鼠亚组(每组8只小鼠)保持存活,没有针对肿瘤生长延迟终点的进一步给药。使用化合物44处理的小鼠具有明显的剂量依赖性肿瘤生长延迟益处,而COP治疗的肿瘤比化合物44处理的那些进展要更快(图9C,中间图)。给予小鼠最大耐受剂量的化合物44或化合物44/COP的组合物显示在第60天100%存活,而组合物组显示最小的终点肿瘤重量,与所有其他治疗组(包括化合物44的最大耐受剂量组)存在统计学上的差异(p>0.05)(图9C,底部图)。
B细胞NHL的标准治疗是组合化学疗法方案,该方案包括环磷酰胺、阿霉素、长春新碱和泼尼松龙。完全响应率可以达到40-50%,很大比例的患者旧病复发,而3年总体存活率仅为约30%。复发的淋巴瘤可以对很大范围的抗癌药物显示抗性,其给临床上管理这些激进的恶性肿瘤提出了严峻的挑战。在淋巴瘤中获得抗药性的部分原因是由于肿瘤细胞的遗传异质性和不稳定性。从而,化学抗性NHL的成功治疗将需要药物的合理组合,这些药物靶向对B细胞NHL不同亚型特异的多种途径。例如,在活化的B细胞型淋巴瘤中,NFkB途径的组成型活化已经涉及治疗抗性,并且几种新型靶向疗法已经在这种亚型显示出潜力。
表观遗传效应物,诸如多梳蛋白,也被牵连于癌细胞的化学抗性。多梳蛋白抑制复合物2(PRC2)的催化亚基EZH2是生发中心起源的B细胞淋巴瘤中的关键的致癌因子。这些更原始的B细胞恶性肿瘤,尤其是表达EZH2突变体具有改变的催化活性的变体需要EZH2来增生和存活。从临床前研究获得的结果预测EZH2抑制剂治疗这种遗传定义的癌症具有很大的潜力,并且EZH2抑制剂也可缓和化学疗法抗性。本文所呈现的数据显示临床阶段EZH2抑制剂化合物44与CHOP的组分一起显示出从加和效应到协同效应的不同程度的组合益处。那些组合益处被特异地在生发中心起源的淋巴瘤中发现,并且在环磷酰胺、阿霉素和长春新碱的情况下,被限制在携带EZH2突变体的细胞中。当化合物44与CHOP在体内共同给药时,在杀死淋巴瘤细胞方面也发现显著的协同作用。在SU-DHL6异种移植模型中尤其是这样,其中单一药剂都没有显示显著的抗肿瘤活性,但是该组合在>50%的小鼠中诱发了持续的消退。这重申了过于活跃的EZH2在EZH2突变体淋巴瘤的化学抗性中潜在的重要性。在CHOP组分中,化合物44与强的松的组合诱发了最强的抗增生活性,并且无论EZH2突变的状态如何,该组合也可以使不敏感的GCB淋巴瘤细胞系对EZH2抑制敏感。另外,当化合物44和泼尼松龙一起或以序列特异的方式给药时,这种组合益处更明显;因此,用EZH2抑制剂引发细胞,接着用GR激动剂治疗证明是特别有效的。这个意外的发现对于在临床上应用EZH2抑制剂具有潜在的重要意义。首先,广泛使用的GRag通常与抗癌药物共同给药以防止药物引发的过敏反应并减轻疼痛、恶心和呕吐,而且它们在治疗造血系统的恶性肿瘤中很关键,因为它们在这些癌症中具有诱发细胞凋亡的能力。相比其他的CHOP组分,GRag诱发了最少的严重副作用。同时,如在SU-DHL10异种移植模型中的数据显示,在保留与化合物44的组合益处的同时,从CHOP方案中排除阿霉素的机会可以让患者免受阿霉素的剂量限制的心脏毒素副作用。最后,临床前研究显示单一药剂EZH2抑制剂仅在携带EZH2突变体的淋巴瘤(其代表一部分(20%)具有高的未满足的临床需求的GCB淋巴瘤患者)中诱发显著的细胞死亡。在此的这些结果显示GRag/EZH2抑制剂组合可能在所有生发中心起源的B细胞淋巴瘤中具有临床实用性。
结合糖皮质激素的GR分子移动至细胞核并可以作为转录激活子或抑制子起作用,这取决于细胞环境。已经表明,GR不断地采集核小体以产生相互作用,并且染色体修饰酶的目的是给DNA提供GR的受调节的接近、它的辅因子和基本的转录机制。其他研究显示GR经常结合至原有的开放染色质区域,并识别在给定的细胞类型中的染色质结构使得GR可以以组织特异性方式起作用。GR结合位点的可及性可以通过ATP依赖性的染色质重塑进一步提高,并且SWI/SNF复合物在这个活动中起到关键的作用。不想被特定的理论或具体的机制约束,可想象的是,由EZH2介导的H3K27超三甲基化诱发的异常染色质抑制可以阻止一些其他可及的GR结合位点,干扰正常GR介导的基因诱导或抑制。事实上,所有EZH2突变体淋巴瘤细胞系对GRag治疗不敏感,而在EZH2 WT细胞中观察到浓度依赖性的细胞死亡。使用泼尼松龙预处理,接着使用化合物44处理在几乎所有测试的细胞系中不会诱发协同作用,该观察指出EZH2抑制剂诱导的染色质重塑的可能是GR增加活性的速率限制性步骤。另外,PRC2已知与SWI/SNF功能拮抗,并且SWI/SNF复合物的核心亚基(SMARCA4、ARID1A与INI1)的下调与急性淋巴母细胞性T细胞白血病中对泼尼松龙的抗性有关联。由于INI1损失和EZH2过度激活的关系已经在横纹肌样瘤中建立,调查了是否整体INI1蛋白质水平将在暴露于化合物44和泼尼松龙的各种淋巴瘤细胞中增加,这在SWI/SNF功能增加后潜在地允许GR更多地接近它的结合位点。
GR通路基因表达试验揭示使用化合物44、泼尼松龙和它们的组合处理几种GCB淋巴瘤细胞(EZH2 WT和突变体)后增加和减少的基因表达确认了GR的双向功能。在所有EZH2突变体淋巴瘤细胞中与该组合协同上调的唯一基因是SESN1,它是对DNA损伤和氧化胁迫具有细胞响应功能的TP53肿瘤抑制子。瑟斯崔因通过激活AMP活化的蛋白激酶、导致mTOR通路的抑制来抑制细胞生长。因此,SESN1介导的mTOR通路抑制可以是化合物44处理后在EZH2突变体淋巴瘤细胞中重新引入GRag敏感性的重要机制。
相反地,GRag/化合物44组合治疗也可以在被报道为对EZH2抑制剂治疗顽固的那些EZH2突变体淋巴瘤细胞系(RL、SU-DHL4)中诱导细胞死亡。使用组合治疗,SESN1也在那些细胞系中被诱导,但是尤其是在RL和SU-DHL4细胞中观察到另外的TNF(强的炎性细胞因子)协同上调。这一观察似乎令人惊讶,因为TNF和糖皮质激素通常拮抗作用。TNF通过它的受体TNFR-1可以诱导细胞凋亡,但是也具有转换生存信号的能力,主要是通过NFkB通路。因而,有可能在GR激动剂抑制NFkB介导的转录的背景下由化合物44/泼尼松龙组合诱导的增加的TNF表达可以转变TNF作用朝向细胞凋亡。然而,为什么在化合物44不敏感的EZH2突变体细胞中这种机制会导致协同的细胞死亡还不清楚。通过观察到的GILZ在使用该组合的6个细胞系的2个中被协同地上调,其进一步支持了在GRag抗性中NFkB通路的负调节因子的异常抑制的潜在重要性和介导它的EZH2的潜在作用。
方法
培养基通量试验
淋巴瘤细胞被接种进烧瓶(WSU-DLCL2和DOHH2为50,000细胞/mL,SU-DHL10为10,000细胞/mL,以及Toledo为100,000细胞/mL),并且使用7个剂量的化合物44预处理或使用DMSO预处理4天或对于Toledo试验预处理6天。然后细胞分为50,000细胞/mL(WSU-DLCL2和DOHH2)或30,000细胞/mL(SU-DHL-10),并且使用HP D300数字分配器(Tecan)用化合物44和感兴趣的化合物共处理。两种药物进行连续稀释2倍,并且沿板的对角线以恒定比率组合在基质中,其中最终DMSO含量为0.11%(体积/体积)。共处理3天(对于Toledo试验是5天)后,通过ATP含量使用(Promega公司)测量了细胞活力,并且使用SpectraMax M5酶标仪(分子器件公司(Molecular Devices))检测了发光。
使用Chou-Talalay方法对药物组合进行了协同作用定量(参考文献1)。组合指数(CI)方程式提供了加和作用(CI=1)、协同作用(CI<1)和拮抗作用(CI>1)的数量定义。此公式采用来自恒定比率的药物组合的分数效应(Fa)值来确定CI值。所得的曲线图(Fa-CI曲线图)示出了由95%置信区间括号内的所得CI值。这些Fa-CI曲线图是使用Windows软件Calcusyn产生的(参考文献2)。CI值<1且置信区间线也低于1表明统计学上显著的协同作用。
用于药物组合,其中只有一种药物显示出超过50%的抑制,对效力转变进行了测定。剂量响应使用图板棱柱(GraphPad Prism)作图,并且50%或60%的抑制浓度从剂量响应曲线内推。当剂量响应的置信区间不重叠时,效力转变被认为是显著的。
细胞系、化合物、和治疗概述
WSU-DLCL2、SU-DHL10、RL、SU-DHL4、OCI-Ly19和DOHH2在以前描述过(自然化学生物学(NatChemBio),2012)。对于组合物研究,使用在悬浮细胞中增生测定的修改版本,如先前所述(Daigle等人)。简要地说,在第0天,将细胞一式三份以初始密度接种于96孔板,以确保4天以上的线性对数生长期。使用化合物44的任一剂量曲线(在1μM的顶剂量开始)、泼尼松龙(目录号和制造商)的单剂量(比该药物的第4天IC50低10倍以上的浓度)或化合物44+泼尼松龙的组合处理细胞。在第4天,在番石榴easyCyte流式细胞仪中使用Viacount试剂计数细胞,并且活细胞数目用于以原始密度重新铺板细胞另外3天。用化合物44预处理的细胞单独接受连续的化合物44或化合物44+泼尼松龙(恒定剂量);用泼尼松龙预处理的细胞接受连续的泼尼松龙或泼尼松龙+化合物44;共处理4天的细胞继续接受7天的共处理。
异种移植研究
所有涉及到动物处理的过程,本研究中的护理和治疗方法是根据CRL皮埃蒙特和上海化学伙伴的机构动物护理和使用委员会(IACUC,Institutional Animal Care and Use Committee(IACUC)of CRL Piedmont and Shanghai ChemPartner)核准的准则以及实验动物管理评估和评审委员会(AAALAC,Association for Assessment and Accreditation of Laboratory Animal Care)的指导意见进行。WSU-DLCL2、SU-DHL6或SU-DHL10细胞在中期对数生长期间收获,并重新悬浮于含有50%基质胶TM(BD Biosciences公司)的PBS中,并注射到免疫缺陷小鼠中。每只小鼠在右胁皮下接受1×107个细胞(0.2mL细胞悬浮液),并且一旦肿瘤达到预定大小,小鼠在不同时间口服不同剂量的化合物44持续至28天和/或按以下方案服用CHOP/COP:环磷酰胺经腹腔(i.p.)给药,且阿霉素和长春新碱分别经推注尾静脉注射(i.v.)给药;在SU-DHL6研究中在第1天和第8天每种每天给药一次,以及在WSU-DLCL2和SU-DHL10研究中在第1天和第22天每种每天给药一次。强的松经口服给药进行两个5天剂量的周期,在SU-DHL6研究中起始于第1和8天((qd×5)×2,第1和8天),并且在WSU-DLCL2和SU-DHL10研究中在第1和22天((qd×5)×2,第1和22天)。每个剂量是以0.2mL/20g小鼠(10L/kg)的体积递送,并针对最后记录的单独动物的体重进行调整。每周两次收集肿瘤测量和体重,所有研究持续28天。为了在SU-DHL10和SU-DHL6研究中确定肿瘤生长延迟,当肿瘤达到2000立方毫米的终点体积或在研究的最后一天(第60天)(无论哪种情况居先)对每个测试动物实施安乐死。
定量PCR
使用DMSO、1uM化合物44(SU-DHL10使用100nM化合物44处理)、低于4天IC5010倍浓度的泼尼松龙、或药物的组合平行处理WSU-DLCL2、SU-DHL10、RL、SU-DHL4、OCI-LY19和DOHH2细胞4天。细胞被收集且总mRNA使用RNeasy Plus Mini Kit(Qiagen;74134)从细胞沉淀中提取。对于RT2糖皮质激素信号PCR试验(RT2 Glucocorticoid Signaling PCR array)(Qiagen;PAHS-154ZE-4),使用RT2第一链试剂盒(RT2 First Strand Kit)(Qiagen;330401)制备cDNA。使用RT2 SYBR Green ROX qPCR Mastermix(Qiagen;330521),在ViiA 7实时PCR系统[应用生物系统公司(Applied Biosystems)(AB)]上进行阵列RT-PCR分析。基因表达归一化至阵列的B2M且使用ΔΔCt方法计算相对于DMSO的倍数变化。为了验证阵列数据,使用TaqMan Fast Advanced Master Mix(AB;4444964)以及用于瑟斯崔因(AB;Hs00902787_m1)和TNF(AB;Hs01113624_m1)的TaqMan引物/探针组进行基于TaqMan探针的qPCR。如上述计算倍数变化,归一化至RPLPO(AB;4333761F)。
ELISA
如上所述,组蛋白从肿瘤样品中提取。以当量浓度在涂层缓冲液(PBS+0.05%BSA)中制备组蛋白,得到0.5ng/l的样品,并将100ul的样品或标准品一式两份加入到2个96孔ELISA板(热实验室系统公司(Thermo Labsystems),Immulon 4HBX#3885)。将平板密封并在4℃温育过夜。第二天,使用300 1/孔PBST(PBS+0.05%Tween 20;10X PBST,KPL#51-14-02)在Bio Tek洗板器上洗涤平板3次。用300 1/孔稀释液(PBS+2%BSA+0.05%Tween 20)封闭平板,在室温下温育2小时,并用PBST洗涤3次。所有抗体在稀释液中稀释。100ul/孔的抗H3K27me3(CST#9733,50%甘油储液1∶1,000)或抗总H3(Abcam ab1791,50%甘油1∶10,000)被添加到每个板。平板在室温温育90分钟并使用PBST洗涤3次。将100ul/孔的抗Rb-IgG-HRP(细胞信号技术公司(Cell Signaling Technology),7074)以1∶2,000加入到H3K27Me3板并以1∶6,000加入到H3板,并在室温温育90分钟。将平板在PBST中洗涤4次。对于检测,加入100ul/孔TMB底物(BioFx实验室公司(BioFx Laboratories),#TMBS)并且将板在室温暗处温育5分钟。使用100ul/孔1N H2SO4停止反应。
在SpectraMax M5酶标仪上读取450nm的吸光值。
实例3:EZH2抑制在透明细胞肾细胞癌模型中克服了对舒尼替尼的抗性
在基因启动区中包括组蛋白修饰和超甲基化的表观遗传机制的改变已经被认为是癌症中抗药性的机制。表观遗传调节物在诸如组蛋白甲基转移酶EZH2中的交替已经在多种癌症类型中报道,包括晚期肾细胞癌(RCC)。先前的研究表明,舒尼替尼可具有直接的抗肿瘤作用,并且获得的性舒尼替尼抗性可在肿瘤细胞中而不仅在内皮细胞中被诱导。在本研究中,在透明细胞肾细胞癌中的舒尼替尼抗性中研究了EZH2的作用。
方法:对人类RCC细胞系786-0进行处理并且暴露于增加浓度的舒尼替尼以开发抗性细胞系786-0R。用舒尼替尼、GSK126(EZH2抑制剂)或两者处理亲本和抗性细胞系。同时,EZH2在786-0细胞中被敲低,并且暴露于增加浓度的舒尼替尼。通过使用在570nm处的光谱仪的结晶紫染色细胞的吸光度来定量细胞活力。在第二组实验中,收集对照和处理的细胞用于蛋白质分析。携带人类ccRCC患者衍生异种移植物(PDX)的小鼠;将RP-R-01、RP-R-02和RP-R-02LM(由RP-R-02建立的转移性CCRCC模型)植入SCID小鼠。当肿瘤达到50mm3的平均体积时,将小鼠随机分为2个组;对照、舒尼替尼治疗(40mg/kg,5天/周)或EZH2i EPZ011989(500mg/kg,2次/天,5天/周)。每周一次评估肿瘤体积和体重。收集肿瘤组织和肺用于免疫组织化学分析。所有的评估和定量均盲法地进行。
结果:体外和体内数据显示EZH2对舒尼替尼抗性的表达增加。此外,体外和体内研究中EZH2的抑制与舒尼替尼在亲本和抗性细胞系中的抗肿瘤作用显著增加相关。
结论:总体而言,数据表明表观遗传改变的潜在作用,特别是EZH2过表达及其与对舒尼替尼抗性的关系。
本文引用的全部公开案和专利文献都通过引用并入本文,如同每一份这样的公开案或文献都被具体地且分别地指出已通过引用结合在此。公开和专利文件的引用不旨在为承认任何这些是相关的现有技术,也不构成对关于相同内容或日期的任何承认。本发明现在已经通过书面描述的方式进行了描述,本领域的技术人员将认识到本发明可以以各种实施例的方式来实施,并且在前面的描述和下面的实例是为了说明的目的,而不是限制后面的权利要求。
在不背离本发明的精神或本质特征的情况下,本发明可以体现为其他具体形式。因此前述实施例应当在所有方面被视为是说明性的而非限制本文所述的发明。因此本发明的范围是由所附权利要求书而非前述说明书指示的,并且属于权利要求书的含义和等效范围内的所有变化意图被包含在其中。
- 还没有人留言评论。精彩留言会获得点赞!