苦豆碱衍生物在制备治疗肿瘤的药物中的应用的制作方法
本发明属于医药用途领域,特别涉及苦豆碱衍生物在制备治疗肿瘤的药物中的应用。
背景技术:
肿瘤的发生与机体免疫功能存在密切的相关性。通常情况下,宿主的免疫系统会对肿瘤细胞产生一定的杀伤作用,这一作用机制主要是,含有特异性抗原的肿瘤细胞被树突细胞吞噬,经树突细胞呈递给T细胞,而后激活的T细胞识别肿瘤细胞并释放淋巴因子,或直接杀伤肿瘤细胞。而部分恶性肿瘤细胞可以通过不同的方式逃避免疫细胞的杀伤作用。其实现免疫逃逸的途径主要包括:(1)肿瘤自身修饰和代谢;(2)肿瘤微环境的改变。因此,研究肿瘤微环境是肿瘤治疗的一种新的探索方式。靶向肿瘤微环境的肿瘤免疫疗法正在逐步成为研究热点。
众所周知,作为免疫系统中重要的成员,T细胞激活需双重信号的协同作用,即抗原提呈信号(抗原肽-MHC/TCR)和B7/CD28提供的协同刺激信号。协同刺激分子能通过为淋巴细胞提供正性或负性调控信号参与机体免疫应答,程序性死亡分子1(PD-1),及其配体(PD-L1和PD-L2)同属于B7/CD28家族。研究表明,PD-L1在多种肿瘤中处于高表达状态,如黑色素瘤、乳腺癌、肺癌和肾小细胞癌等。激活的T细胞也会通过分泌IFN-γ等细胞因子来增强其杀伤作用,而在IFN-γ的刺激下,肿瘤细胞表面的程序性死亡因子1配体(PD-L1)的表达也会发生明显的上调。许多研究表明,PD-1/PD-L1作为一对免疫抑制性共刺激因子,其信号通路的激活可抑制T细胞介导的免疫应答,甚至诱导T细胞的凋亡,从而导致免疫抑制性肿瘤微环境形成,使肿瘤细胞逃避机体免疫监视和杀伤。而阻止这个信号通路可以增强内源性的抗肿瘤免疫效应。因此PD-1/PD-L1信号通路有望成为肿瘤免疫治疗的新靶点。
目前,抗PD-L1抗体及抗PD-1抗体在临床试验中对黑色素瘤,肺癌,肾癌,膀胱癌等肿瘤均具有良好的活性。这些单克隆抗体作为肿瘤治疗的药物有着较高的靶向性、安全性、副作用小、阻断作用强且不会产生耐药性等优点。但其也有相应的局限性,如半衰期短,生产成本较高等,对于一些实体瘤,由于单克隆抗体分子量较大不能进入到肿瘤细胞间隙中,因而发挥作用的范围仅限于肿瘤的边缘处。
苦豆碱是从苦豆子中分离出的生物碱,其结构式如下:
研究表明,苦豆碱具有包括抗心律失常、负性肌力、抗菌、抗病毒、抗炎、舒张平滑肌、免疫抑制、抗癌、清除自由基、毒副作用、杀虫作用等多种生理活性。
此外,申请人以苦豆碱为母核,设计合成了一系列苦豆碱衍生物,其结构通式如下:
其中,
X基团为-CH2-、-C(O)-或-S(O)a-,其中a为1或2;
Y基团为C1-10烷基、或由选自C、N、O和S的原子形成的3-15元环烷基、芳基或杂环基;任选地,其中所述C1-10烷基、3-15元环烷基、芳基或杂环基各自独立地被一个或多个选自卤素、氨基、氰基、硝基、羧基、C1-6烷基、C1-6烷氧基、卤代C1-6烷基、卤代C1-6烷氧基、C1-6烷氧羰基、C1-6烷基酰基、C1-6烷基胺酰基、C1-6烷基酰氨基、C1-6烷基磺酰基和8-15元苯并杂环基的取代基取代。
以上苦豆碱衍生物具有优异的抗病毒活性。但是现有技术还未报道过该类苦豆碱衍生物的其他用途。
技术实现要素:
本发明提供一种苦豆碱衍生物的新用途,即苦豆碱衍生物在制备治疗肿瘤的药物中的应用。
本发明具体技术方案如下:
本发明提供一种苦豆碱衍生物的新用途,即式Ⅰ所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐在制备用于治疗肿瘤的药物中的应用,
其中,R为被一个或多个选自卤素、三氟甲氧基、三氟甲基、氨基、硝基、氰基、羧基、C1-4烷基、C1-4烷氧基、C1-4烷氧羰基、C1-4烷基胺酰基、C1-6烷基酰氨基或C1-6烷基磺酰基取代的苯基、咪唑基、苄基、吡啶基、噻吩基、噻唑基、哌嗪基或苯并呋喃基。
进一步的改进,R为被一个或多个选自卤素、三氟甲氧基、三氟甲基、氨基、硝基、氰基、羧基、C1-4烷基、C1-4烷氧基取代的苯基或咪唑基。
进一步的改进,R为被一个选自三氟甲氧基、甲基取代的苯基或咪唑基。
进一步的改进,R为甲基取代的咪唑基。
进一步的改进,R为三氟甲氧基取代的苯基。
进一步的改进,式Ⅰ所示的苦豆碱衍生物为12-N-(1-甲基-1H-咪唑-4-磺酰基)苦豆碱(NKD-49),其结构式如下:
进一步的改进,式Ⅰ所示的苦豆碱衍生物为12-N-(4-三氟甲氧基苯磺酰基)苦豆碱(NKD-50),其结构式如下:
进一步的改进,式Ⅰ所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐在制备用于降低肿瘤细胞中PD-L1总蛋白的表达的药物中的应用。
进一步的改进,所述肿瘤选自黑色素瘤、乳腺癌、肺腺癌、肺癌、肾小细胞癌中的一种。
本发明中所用术语C1-4烷基包括甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基等,所述烷基可以是取代的或未取代的,当被取代时,包括三氟甲基、三氟乙基、羟甲基、羟基乙基、氨基甲基、氨基乙基等。
本文所用术语C1-4烷氧基包括甲氧基、乙氧基、丙氧基、异丙氧基和叔丁氧基等。
本发明所用术语C1-4烷氧羰基包括甲氧羰基、乙氧羰基、丙氧羰基、叔丁氧羰基等。
本发明所用术语C1-4烷基酰基包括乙酰基、丙酰基、丁酰基等。
本发明所用术语C1-4烷基胺酰基包括甲氨酰基、乙氨酰基、N,N-二甲氨酰基、N,N-二乙氨酰基等。
本发明所用术语C1-4烷基酰氨基包括甲酰氨基、乙酰氨基等。
本发明所用术语C1-4烷基磺酰基包括甲磺酰基、乙磺酰基等。
本发明所用术语“异构体”包括本发明式I所示的苦豆碱衍生物的所有可能的异构体(例如对映异构体、非对映异构体、几何异构体、构象异构体、差向异构体和旋转异构体等)形式。例如,不对称中心各自的R和S构型、(Z)和(E)双键异构体和(Z)和(E)构象异构体均在本发明的保护范围内。
本发明式Ⅰ所示的苦豆碱衍生物可以形成溶剂合物,例如水合物、醇合物等。一般来说,与药学可接受的溶剂如水、乙醇等形成的溶剂合物形式与非溶剂合物形式相当。
本发明式Ⅰ所示的苦豆碱衍生物还可以是前药或可在体内代谢变化后释放出所述活性成分的形式。选择和制备适当的前药衍生物是本领域技术人员公知技术,本发明对此不做限制。
本发明式Ⅰ化合物或其药学可接受的盐还可以以晶体存在,本发明包含式I所示的苦豆碱衍生物或其药学可接受的盐的任意晶型。
本发明式I所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐可以通过以下途径给药:胃肠外、局部、静脉内、口服、皮下、动脉内、真皮内、经皮、直肠、颅内、腹膜内、鼻内、肌内途径或作为吸入剂。
本发明式I所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐可以药物制剂的形式给药,该药物组合物包括式I所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐及药学可接受的载体或辅料。
本发明的式I所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐可根据给药途径配成各种适宜的剂型。
当口服用药时,本发明化合物可制成任意口服可接受的制剂形式,包括但不限于片剂、胶囊、水溶液或水悬浮液。其中,片剂使用的载体一般包括乳糖和玉米淀粉,另外也可加入润滑剂如硬脂酸镁。胶囊制剂使用的稀释剂一般包括乳糖和干燥玉米淀粉。水悬浮液制剂则通常是将活性成分与适宜的乳化剂和悬浮剂混合使用。任选地,以上口服制剂形式中还可加入一些甜味剂、芳香剂或着色剂。
当皮肤局部施用时,本发明化合物可制成适当的软膏、洗剂或霜剂制剂形式,其中将活性成分悬浮或溶解于一种或多种载体中。软膏制剂可使用的载体包括但不限于:矿物油、液体凡士林、白凡士林、丙二醇、聚氧化乙烯、聚氧化丙烯、乳化蜡和水;洗剂或霜剂可使用的载体包括但不限于:矿物油、脱水山梨糖醇单硬脂酸酯、吐温60、十六烷酯蜡、十六碳烯芳醇、2-辛基十二烷醇、苄醇和水。
本发明式I所示的苦豆碱衍生物、其光学异构体、溶剂合物或药学可接受的盐还可以无菌注射制剂形式用药,包括无菌注射水或油悬浮液或无菌注射溶液,也可以是冻干形式。其中,可使用的载体和溶剂包括水、林格氏溶液和等渗氯化钠溶液。另外,灭菌的非挥发油也可用作溶剂或悬浮介质,如单甘油酯或二甘油酯。
本发明的药物制剂包括药学上可实施的任何制剂,例如口服制剂、肠胃外给药制剂等。
本发明的有益效果在于:本发明提供了苦豆碱衍生物的新用途,该类化合物通过降低肿瘤细胞表面的PD-L1来治疗肿瘤,同时可以增强免疫细胞的杀伤活性,且具有很好的体内活性且毒性低。
附图说明
图1a为免疫印迹分析给予NKD-49不同时间后,H157细胞中PD-L1总蛋白的表达结果;其中,GAPDH为参照;
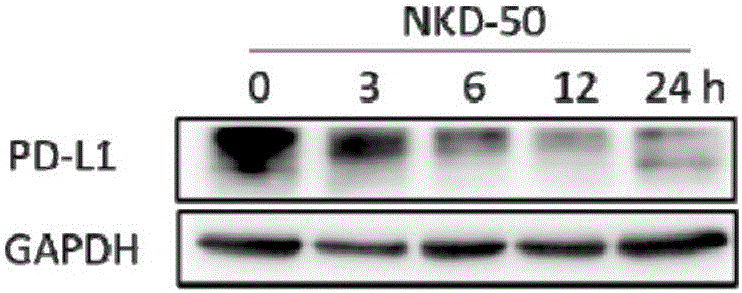
图1b为免疫印迹分析给予NKD-50不同时间后,H157细胞中PD-L1总蛋白的表达结果;其中,GAPDH为参照;
图2a为免疫印迹分析给予不同浓度NKD-49的H157细胞中PD-L1总蛋白的表达结果;其中,GAPDH为参照;
图2b为免疫印迹分析给予不同浓度NKD-50的H157细胞中PD-L1总蛋白的表达结果;其中,GAPDH为参照;
图3NKD-49、NKD-50处理后的H157细胞中PD-L1总蛋白的表达结果图,PD-L1+%表示PD-L1阳性细胞的比例;
图4a为肿瘤细胞与CIK细胞共培养示意图;
图4b将CIK细胞与用NKD-49预处理的A549细胞共培养,每孔的吸收值与最大LDH释放组的吸收值的比值图;
图4c将CIK细胞与用NKD-50预处理的A549细胞共培养,每孔的吸收值与最大LDH释放组的吸收值的比值图;
图5为NKD-49、NKD-50处理的A549细胞的存活率曲线图;
图6表示小鼠体内抗肿瘤活性,其中a为NKD-49处理后瘤重变化图,(上)空白对照,(中1)NKD-49,给药剂量为20mg/kg;(中2)NKD-49,给药剂量为50mg/kg;(下)NKD-49,给药剂量为100mg/kg;b为NKD-50处理后瘤重变化,(上)空白对照,(中)NKD-50,给药剂量为50mg/kg;(下)NKD-50,给药剂量为200mg/kg;C.NKD-49处理后小鼠体重变化,(上)空白对照,(中1)NKD-49,给药剂量为20mg/kg;(中2)NKD-49,给药剂量为50mg/kg;(下)NKD-49,给药剂量为100mg/kg;D.NKD-50处理后小鼠体重变化;(上)空白对照,(中)NKD-50,给药剂量为50mg/kg;(下)NKD-50,给药剂量为200mg/kg;图7为NKD-49、NKD-50处理后小鼠体内瘤重变化图,其中,A表示空白对照,B表示NKD-49,给药剂量为20mg/kg;C表示NKD-49,给药剂量为50mg/kg;D表示NKD-49,给药剂量为100mg/kg。
具体实施方式
试验例1 NKD-49、NKD-50时间-效应关系
1.1试验方法
1.H157细胞经0.25%胰酶消化后制成细胞悬液,以5×105/ml的浓度接种于6孔细胞培养板,每孔1ml,置37℃,5%CO2的无菌培养箱内培养24h;然后随机分成三组,分别为对照组、NKD-49组和NKD-50组。
2.NKD-49组和NKD-50组分别于加药孔中加入NKD-49和NKD-50至终浓度为20μM,对照组加入等体积DMSO,置37℃,5%CO2的无菌培养箱内培养3h,6h,9h,12h,24h。
3.培养结束后收获细胞,吸去细胞培养液,用1ml PBS清洗培养板2次,六孔板每孔加入50μl的裂解液,刮刀刮下细胞后,放置冰上裂解30min后在4℃,12000rpm离心15min。
4.离心后将上清液转移到新的EP管中,取2μl上清液加600μl Bradford,静置10min测蛋白浓度。
5.每个样本取总蛋白50μg至新的EP管中,按4:1加5×SDS Loading,混匀后于100℃孵育10min。
6.样本经10%及15%的SDS-PAGE胶电泳后,将蛋白转移至PVDF膜上,5%脱脂牛奶室温封闭1h,4℃孵育一抗过夜。
7.一抗孵育结束后,回收一抗,用TBS-T洗涤30min(每次10min),孵育二抗,二抗孵育结束,用TBS-T洗涤30min(每次10min),于成像仪中进行曝光,检测结果见图1a和图1b。
1.2试验结果
从图1a中可以看出,H157细胞于加药12h后,NKD-49可降低PD-L1总蛋白的表达,加药24h后,PD-L1总蛋白的表达显著降低;从图1b中可以看出H157细胞于加药3h后,NKD-50开始降低PD-L1总蛋白的表达,加药12h后,PD-L1总蛋白的表达显著降低。
1.3试验结论
NKD-49和NKD-50均能够降低细胞中PD-L1总蛋白的表达,并且呈时间依赖性。
试验例2 NKD-49、NKD-50剂量-效应关系
2.1试验方法
1.H157细胞经0.25%胰酶消化后制成细胞悬液,以5×105/ml的浓度接种于6孔细胞培养板,每孔1ml,置37℃,5%CO2的无菌培养箱内培养24h,然后随机分成三组,分别为对照组、NKD-49组和NKD-50组。
2.NKD-49组和NKD-50组分别于加药孔中加入NKD-49和NKD-50至终浓度为5μM,10μM,15μM,20μM,对照组加入等体积DMSO,置37℃,5%CO2的无菌培养箱内培养24h。
3.培养结束后收获细胞,吸去细胞培养液,用1ml PBS清洗培养板2次,六孔板每孔加入50μl的裂解液,刮刀刮下细胞后,放置冰上裂解30min后在4℃,12000rpm离心15min。
4.离心后将上清液转移到新的EP管中,取2μl上清液加600μl Bradford,静置10min测蛋白浓度。
5.每个样本取总蛋白50μg至新的EP管中,按4:1加5×SDS Loading。混匀后于100℃孵育10min。
6.样本经10%及15%的SDS-PAGE胶电泳后,将蛋白转移至PVDF膜上,5%脱脂牛奶室温封闭1h,4℃孵育一抗过夜。
7.一抗孵育结束后,回收一抗,用TBS-T洗涤30min(每次10min),孵育二抗,二抗孵育结束,用TBS-T洗涤30min(每次10min),于成像仪中进行曝光,检测结果见图2a和图2b。
2.2试验结果
从图2a中可以看出,H157细胞于加药24h后,NKD-49加药剂量为10μM,可显著降低PD-L1总蛋白的表达;从图2b中可以看出,H157细胞于加药24h后,NKD-50加药剂量为15μM,可降低PD-L1总蛋白的表达。
2.3试验结论
NKD-49和NKD-50均能够降低细胞中PD-L1总蛋白的表达,并且呈剂量依赖性。
试验例3 NKD-49、NKD-50降低肿瘤细胞中的PD-L1
3.1试验方法
1.H157细胞经0.25%胰酶消化后制成细胞悬液,以5×105/ml的浓度接种于6孔细胞培养板,每孔1ml,置37℃,5%CO2的无菌培养箱内培养24h;然后随机分成三组,分别为对照组、NKD-49组和NKD-50组。
2.NKD-49组和NKD-50组分别于加药孔中加入NKD-49和NKD-50,使其终浓度分别为15μM,20μM,25μM,对照组中加入等量DMSO,于无菌培养箱内培养24h。
3.培养结束后收获细胞,细胞经0.25%胰酶消化后,用PBS洗涤2次,1500rpm,5min离心收获细胞。
4.用200μl PBS重悬细胞,然后取出100μl加入5μl PE标记的IgG抗体,剩余100μl细胞悬液加入5μl PE标记的PD-L1抗体,于4℃孵育30min。
5.离心弃掉上清,用PBS洗涤两次,用500μl PBS重悬细胞,经300目筛网过滤至流式管中,上机检测,检测结果见图3。
3.2试验结果
从图3中可以看出,与对照组相比NKD-49和NKD-50使用浓度在20μM时可以显著降低H157细胞中PD-L1总蛋白的表达。
3.3试验结论
NKD-49和NKD-50可显著降低细胞中PD-L1总蛋白的表达。
试验例4 NKD-49、NKD-50增强免疫细胞的杀伤活性
4.1试验方法
1.A549细胞经胰酶消化后制成细胞悬液,以5×104/ml的浓度接种于96孔细胞培养板,每孔100μl,设置空白对照组、正常细胞组、最大LDH释放组、IFN-γ组及加药组,每组3个复孔,置37℃,5%CO2的无菌培养箱内培养24h。
2.A549细胞的加药组分别加入终浓度为20μM的NKD-49或NKD-50处理1h后加入IFN-γ,置37℃,5%CO2的无菌培养箱内再培养24h。
3.用100μl CIK细胞的完全培养基更换掉A549的培养基,之后CIK细胞以5×105/ml的浓度接种于已铺有A549细胞的96孔板中,每孔100μ,置37℃,5%CO2的无菌培养箱内培养3h。
4.于最大LDH释放组中加入20μl裂解液,置37℃,5%CO2的无菌培养箱内裂解45min。
5.取出96孔板,于1500rpm,离心5分钟,取50μl上清液于新的96孔板中,加入50μl底物避光反应30min。
6.再向每孔中加入50μl终止液,于492nm处测定吸收值。
7.每孔的吸收值与最大LDH释放组的吸收值的比值进行统计学计算,结果见图4a-图4c。
4.2试验结果
从图4a、图4b和图4c中可以看出,在IFN-γ存在的情况下,将A549细胞与CIK细胞以1:10的比例共培养,用NKD-49或NKD-50预处理A549细胞后,可以显著提高CIK细胞对A549细胞的杀伤活性。
4.3试验结论
NKD-49或NKD-50可增强免疫细胞的杀伤活性。
试验例5 NKD-49、NKD-50体外细胞毒性
5.1试验方法
1.A549细胞经胰酶消化后制成细胞悬液,以5×104/ml的浓度接种于96孔细胞培养板,每孔100μl,设置空白对照组、正常细胞组及加药组,每组3个复孔,置37℃,5%CO2的无菌培养箱内培养24h。
2.于加药组中分别加入NKD-49或NKD-50,使其终浓度分别为2μM,5μM,10μM,20μM,100μM,200μM,500μM,以培养基作为空白对照,于无菌培养箱内培养48h。
3.培养结束前4h,每孔加入5mg/ml MTT溶液20μl,结束培养后用2ml注射器吸去上清,每孔加入100μl二甲基亚砜,震荡10min后,于570nm处测定光吸收,结果见图5。
5.2试验结果
从图5中可以看出,NKD-49、NKD-50可降低A549细胞的存活率,当NKD-49或NKD-50的浓度达到500μM时,可使A549细胞的存活率降低40%左右。
5.3试验结论
NKD-49、NKD-50对肿瘤细胞具有较小的细胞毒性。
试验例6NKD-49、NKD-50体内活性
6.1试验方法
1.选择40只C57BL/6小鼠(雌性,6周龄,18-22g,北京维通利华实验动物技术有限公司),将鼠源肺癌(Lewis lung carcinoma)细胞接种于小鼠右侧腋下,随机分成实验1组、实验2组和空白对照组。
2.接种72h后,口服灌胃给药,实验组1组和实验2组分别给予NKD-49、NKD-50,给药剂量分别为20mg/kg,50mg/kg,100mg/kg,空白对照组给予200μl生理盐水,每日给药一次,共给药10天。
3.接种后,每两天测量一次小鼠体重;给药第三天开始,每隔2天测量一次瘤体积。
4.结束给药两天后,处理小鼠,取瘤组织拍照称重,检测结果见图6。
6.2试验结果
从图7中可以看出,与空白对照组相比,经NKD-49、NKD-50处理后的小鼠体内的肿瘤体积明显减少;从图6的第a图可以看出,给预剂量为20mg/kg的NKD-49,小鼠体内肿瘤的抑制率为57%,当NKD-49的给药剂量为50mg/kg,抑制率为56%;当NKD-49的给药剂量为100mg/kg,抑制率为65.8%;从第b图中可以看出,NKD-50的给药剂量为50mg/kg,抑制率为43%;NKD-50的给药剂量为200mg/kg,抑制率为57%;并且从第c和d图中可以看出,给予不同剂量的NKD-49和NKD-50,小鼠体重无明显变化。
6.3试验结论
NKD-49、NKD-50可抑制小鼠体内肿瘤的生长,起到治疗肿瘤的疗效,且无毒副作用。
- 还没有人留言评论。精彩留言会获得点赞!