一种帕瑞昔布钠与盐酸曲马多组成的复合物及其用途的制作方法
本发明属于治疗疼痛、抗炎的药物领域,特别涉及一种帕瑞昔布钠与盐酸曲马多组成的复合物及其用途。
背景技术:
疼痛是已经被功能上分类为感觉性、自发性、运动性、和感情性成分的复杂应答。总体上,疼痛可以被分成慢性和急性的。慢性疼痛包括神经性疼痛和慢性炎症性疼痛,例如关节炎,或未知来源的疼痛,如纤维肌痛。急性疼痛通常跟随非神经组织损害,例如来自外科手术或发炎的组织损伤,或偏头痛。
目前,有许多用于治疗或处理疼痛的药物,例如阿片样物质、吗啡衍生物等。吗啡衍生物被指定用于对人的急性疼痛的缓和治疗,通过它们对吗啡受体,优选μ-受体的作用获得镇痛效果。在吗啡的这些衍生物之中,可以提到吗啡、可待因、哌替啶、右旋丙氧吩美沙酮等。
经口服给药显示良好疼痛治疗效果的一种吗啡衍生物为曲马多,其也可以生理盐的形式使用,尤其盐酸盐,曲马多为中枢作用止疼药,其通过活化类鸦片受体及增强神经元单胺突触浓度来发挥作用,曲马多的化学名称为2-(二甲胺基甲基)-1-(3-甲氧基苯基)环己醇,结构如下:
目前常用的治疗疼痛的药物还包括非甾体抗炎药,例如昔布类,其中帕瑞昔布钠是伐地昔布的前体药物,伐地昔布在临床剂量范围是选择性环氧合酶-2(cox-2)抑制剂,研究显示cox-2作为环氧化酶异构体由前-炎症刺激诱导生成,因此cox-2与疼痛、炎症和发热有关。帕瑞昔布钠由于其水溶性好,可制成注射剂,用于手术后疼痛的短期治疗,临床上可用于中度或重度术后急性疼痛的治疗。
目前单一药物治疗疼痛的疗程长,所以经常采用混合药物治疗疼痛,例如现有技术公开了采用曲马多和帕瑞昔布钠联合治疗食管癌根治术后的疼痛,曲马多联合帕瑞昔布钠用于治疗妇科腹腔镜手术术后疼痛等。但是从现有技术公开的联合治疗效果来看,曲马多和帕瑞昔布钠联合治疗疼痛的效果并不理想,并且二者联合后的剂型选择也存在很大的问题。
技术实现要素:
为了解决上述技术问题,本发明提供一种帕瑞昔布钠与盐酸曲马多组成的复合物,该复合物对强直性脊柱炎所引起的疼痛具有很好的治疗效果,用药周期短,起效时间长。
本发明提供一种帕瑞昔布钠与盐酸曲马多组成的复合物,该复合物包括共晶体水合物,所述共晶体水合物中帕瑞昔布钠、盐酸曲马多和水的分子个数比为1:2:1。
该共晶体水合物的制备方法为:将1mol帕瑞昔布钠和1mol盐酸曲马多均溶解于28ml水中,向水中滴入14.5ml的正丁醇,于-5℃下放置析晶。
进一步的改进,共晶体水合物以2θ表示的x-射线衍射在8.2°、10.5°、17.3°、20.4°、22.7°和25.8°处有特征峰。
进一步的改进,共晶体水合物以2θ表示的x-射线衍射在4.3°、13.7°、19.4°、28.6°、30.4°和36.5°处有特征峰。
进一步的改进,该复合物还包括阿司匹林,所述阿司匹林和共晶体水合物的质量比为1.5:1。
本发明另一方面提供一种微球冻干制剂,其包括本发明提供的复合物和辅料,其中,复合物和辅料的重量份数比为1:17-30,所述辅料主要由如下重量份的成分制备而成:
本发明提供的微球冻干制剂可显著提高微球冻干制剂的稳定性和包封率。
进一步的改进,该辅料还包括重量份数为2.6-4.3份的冻干保护剂。
进一步的改进,冻干保护剂由如下重量份的成分制备而成:
甘露醇复合膜2.3-3.5
海藻酸钠0.3-0.8。
进一步的改进,所述所述甘露醇复合膜是由如下重量份的成分制备而成:
甘露醇1.4-2
磷脂酰丝氨酸0.8-1.1
羟乙基-β-环糊精0.1-0.4。
本发明提供将常规的冻干保护剂制成复合膜后,可显著提高微球冻干制剂的载药量和包封率。并且可以进一步提高复合物的渗透性。
本发明提供的复合物可用于治疗强直性脊柱炎所引起的疼痛。
本发明提供的复合物还可以用于治疗类风湿性关节炎。
本发明另一方面提供了甘露醇复合膜的制备方法,该方法包括如下步骤:将1.4-2g甘露醇、0.8-1.1g磷脂酰丝氨酸和0.1-0.4g羟乙基-β-环糊精溶于50ml乙醇中,于旋转蒸发仪上减压蒸发成膜,将成的膜溶于水中,经水化后,过滤,干燥,制得甘露醇复合膜。
本发明另一方面提供了微球冻干制剂的制备方法,该方法包括如下步骤:
s1:将0.3-0.6g乳酸钠溶于10ml水中,制得水相;
s2:将1.2-1.8g辛烯基琥珀酸淀粉钠溶于25ml乙醇中,制得油相;
s3:将步骤s2制得的油相加入到步骤s1中制得水相中,混合均匀,于4600r·min-1搅拌,形成乳液;
s4:向步骤s13制得的乳液中加入3.7%的甲醛溶液,固化,离心分离,蒸馏水洗至无甲醛味,抽滤,干燥,制得微粒;
s5:将10.6-15.7g聚醋酸乙烯酯、5-8.6g聚丙烯葡聚糖和1g复合物、步骤s4制得的微粒溶于20ml水和65ml乙醇中,制得混合液;
s6:将混合液于3600r·min-1搅拌,形成乳液,控制乳液温度低于5℃下,继续搅拌,使乙醇挥发至微球固化,制得微球;
s7:将制得的微球与冻干保护剂混合均匀,冷冻,灭菌,制得微球冻干制剂。
本发明的有益效果在于,本发明提供一种帕瑞昔布钠与盐酸曲马多组成的复合物及其用途,通过将帕瑞昔布钠与盐酸曲马多组合在一起,组合方式通过氢键键合,通过氢键连接形成的共晶体水合物性质更稳定,并且药代动力学性质显著提供;虽然两者的作用机制不一样,但是两者形成的复合物具有意料之外的协同作用,在强直性脊柱炎引起的疼痛的治疗领域具有积极的应用前景。
附图说明
图1为共晶体水合物的x-射线衍射图;
图2为微球冻干制剂的体外释放度图。
具体实施例方式
实施例1一种帕瑞昔布钠与盐酸曲马多组成的复合物
该复合物为共晶体水合物,该共晶体水合物中帕瑞昔布钠、盐酸曲马多和水的分子个数比为1:2:1,该共晶体水合物以2θ表示的x-射线衍射在8.2°、10.5°、17.3°、20.4°、22.7°和25.8°处有特征峰。
实施例2一种帕瑞昔布钠与盐酸曲马多组成的复合物
该复合物为共晶体水合物,该共晶体水合物中帕瑞昔布钠、盐酸曲马多和水的分子个数比为1:2:1,该共晶体水合物以2θ表示的x-射线衍射在4.3°、8.2°、10.5°、13.7°、17.3°、19.4°、20.4°、22.7°、25.8°、28.6°、30.4°和36.5°处有特征峰,如图1所示,以上各特征峰的相对强度如下所示:
实施例3一种帕瑞昔布钠与盐酸曲马多组成的复合物
该复合物由1.5g阿司匹林和1g共晶体水合物组成。
实施例4微球冻干制剂
微球冻干制剂各成分的用量为:
实施例5微球冻干制剂
微球冻干制剂各成分的用量为:
实施例6微球冻干制剂
微球冻干制剂各成分的用量为:
该微球冻干制剂的制备方法为:
s1:将0.5g乳酸钠溶于10ml水中,制得水相;
s2:将1.6g辛烯基琥珀酸淀粉钠溶于25ml乙醇中,制得油相;
s3:将步骤s2制得的油相加入到步骤s1中制得水相中,混合均匀,于4600r·min-1搅拌,形成乳液;
s4:向步骤s13制得的乳液中加入3.7%的甲醛溶液,固化,离心分离,蒸馏水洗至无甲醛味,抽滤,干燥,制得微粒;
s5:将14.6g聚醋酸乙烯酯、7.5g聚丙烯葡聚糖、1g复合物、步骤s4制得的微粒溶于20ml水和65ml乙醇中,制得混合液;
s6:将混合液于3600r·min-1搅拌,形成乳液,控制乳液温度低于5℃下,继续搅拌,使乙醇挥发至微球固化,制得微球;
s7:将制得的微球冷冻,灭菌,制得微球冻干制剂。
实施例7微球冻干制剂
微球冻干制剂各成分的用量为:
甘露醇复合膜
实施例8微球冻干制剂
微球冻干制剂各成分的用量为:
甘露醇复合膜
甘露醇复合膜是通过如下方法制备得到的::将1.7g甘露醇、0.9g磷脂酰丝氨酸和0.2g羟乙基-β-环糊精溶于50ml乙醇中,于旋转蒸发仪上减压蒸发成膜,将成的膜溶于水中,经水化后,过滤,干燥,制得甘露醇复合膜。
实施例9微球冻干制剂
微球冻干制剂各成分的用量为:
甘露醇复合膜
该微球冻干制剂的制备方法为:
s1:将0.6g乳酸钠溶于10ml水中,制得水相;
s2:将1.8g辛烯基琥珀酸淀粉钠溶于25ml乙醇中,制得油相;
s3:将步骤s2制得的油相加入到步骤s1中制得水相中,混合均匀,于4600r·min-1搅拌,形成乳液;
s4:向步骤s13制得的乳液中加入3.7%的甲醛溶液,固化,离心分离,蒸馏水洗至无甲醛味,抽滤,干燥,制得微粒;
s5:将15.7g聚醋酸乙烯酯、8.6g聚丙烯葡聚糖、1g复合物步骤s4制得的微粒溶于20ml水和65ml乙醇中,制得混合液;
s6:将混合液于3600r·min-1搅拌,形成乳液,控制乳液温度低于5℃下,继续搅拌,使乙醇挥发至微球固化,制得微球;
s7:将制得的微球与甘露醇复合膜和0.8g混合均匀,冷冻,灭菌,制得微球冻干制剂。
对照例1
微球冻干制剂各成分的用量为:
对照例2
微球冻干制剂各成分的用量为:
实验例1复合物稳定性实验
1.高温影响因素实验
取实施例2的复合物,在温度60℃,相对湿度75%的条件下放置10天,分别在第0、5、10天取样观察其外观、色泽并测定总杂质(盐酸曲马多和帕瑞昔布钠)含量,盐酸曲马多标示量,结果见表1。
表1高温影响因素实验结果
从表中可以看出,本发明提供的复合物在高温条件下放置10天,从性状、外观上看没有变化,包封率和含量的变化稳定。
2.高湿影响因素实验
取实施例2的复合物,在温度25℃,相对湿度92.5%的条件下放置10天,分别在第0、5、10天取样观察其外观、色泽并测定总杂质(盐酸曲马多和帕瑞昔布钠)含量,盐酸曲马多标示量,结果见表2。
表2高湿影响因素实验结果
从表中可以看出,本发明提供的复合物在高湿条件下放置10天,从性状、外观上看没有变化,包封率和含量的变化稳定。
3.强光影响因素实验
取实施例2的复合物,在照度为4000-5000lx的日光灯的光照箱内放置10天,分别在第0、5、10天取样观察其外观、色泽并并测定总杂质(盐酸曲马多和帕瑞昔布钠)含量,盐酸曲马多标示量,结果见表3。
表3强光影响因素实验结果
从表中可以看出,本发明提供的复合物在强光条件下放置10天,从性状、外观上看没有变化,包封率和含量的变化稳定。
实验例2微球冻干制剂稳定性实验
1加速试验
取本发明实施例5、及对照例1和2的微球冻干制剂,均在40℃±2℃下,相对湿度为75%±5%的条件下放置6个月,在试验期间1个月、2个月、3个月、6个月末分别取样一次,按中国药典中的规定,检测微球冻干制剂的性状、帕瑞昔布钠的含量(标示量%),结果见表4。
表4微球冻干制剂的加速试验结果
从表中可看出本发明实施例5所提供的微球冻干制剂,经加速试验结果可知,放置6个月后,所述微球冻干制剂的性状、含帕瑞昔布钠的量均未发生明显的变化;而对照例1-2的微球冻干制剂放置6个月后,微球冻干制剂发黄,且帕瑞昔布钠的含量显著下降;表明本发明的微球冻干制剂与对照例1-2相比,稳定性显著提高。
1.2长期试验
取本发明实施例5及对照例1-2的微球冻干制剂,均于温度25℃±2℃下,相对湿度为60%±10%的条件下放置12个月,每3个月取样一次,分别于0个月、3个月、6个月、9个月、12个月、24个月取样,检测微球冻干制剂的性状、帕瑞昔布钠的含量(标示量%),结果见表5。
表5微球冻干制剂的长期试验结果
从表中可看出本发明实施例5所提供的微球冻干制剂,经长期试验结果可知,放置24个月后,所述微球冻干制剂的性状、含帕瑞昔布钠的量均未发生明显的变化;而对照例1-2的微球冻干制剂放置24个月后,微球冻干制剂发黄,并且帕瑞昔布钠的含量显著下降;表明本发明的微球冻干制剂与对照例1-2的微球冻干制剂相比,稳定性显著提高。
实验例3不同冻干保护剂对微球冻干制剂的影响
本试验选择不同的冻干保护剂,其余的成分与实施例8的相同,制成不同的微球冻干制剂,对微球冻干制剂的包封率、外观、再分散时间等因素进行考察,考察结果见表6。
表6不同冻干保护剂对微球冻干制剂的影响结果
注意:a表示甘露醇复合膜由重量份比为1.7:0.9:0.2的甘露醇、磷脂酰丝氨酸和羟乙基-β-环糊精组成,制备方法见本发明说明书,b表示甘露醇复合膜由重量份比为1.7:0.9的甘露醇和磷脂酰丝氨酸组成,制备方法见本发明说明书,c表示甘露醇复合膜由重量份比为1.7:0.9:0.2的甘露醇、甘氨酸和羟乙基-β-环糊精组成,制备方法见本发明说明书,d表示甘露醇复合膜由重量份比为1.7:0.9:0.2甘露醇、磷脂酰丝氨酸和羟乙基-β-环糊精组成,但是制备方法为该三个物质均匀混合,压成膜。
从表中可以看出,选择本发明的甘露醇复合膜和海藻酸钠作为混合冻干保护剂,使得制备的微球冻干制剂的外观为白色、饱满,包封率和再分散时间好,其余的冻干保护剂外观不好,且有萎缩现象,分散时间长,包封率低。
实验例4体外释放度测定试验
药物释放度检测:参照2015版《中国药典》附录xixd体外药物释放度检查。
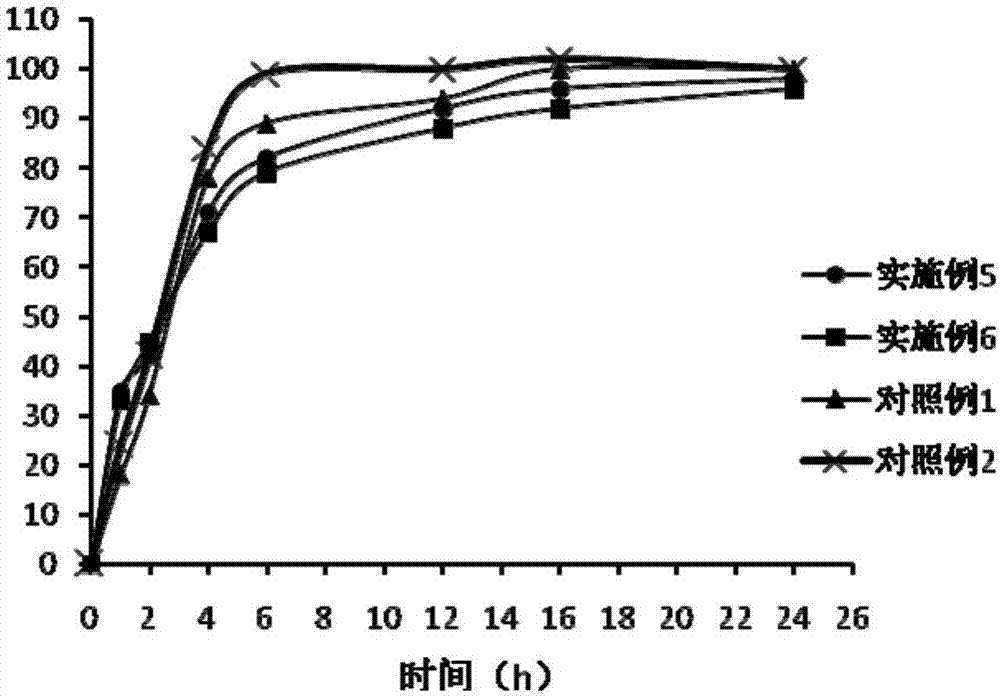
分别取以上实施例5、实施例6和对照例1和对照例2的微球冻干制剂,置药物溶出仪中,于1h、2h、4h、6h、12h、16h、24h分别取样,用高效液相色谱法检测溶出百分率,并计算药物的累积释放百分率,结果见图2。
从图中可以看出实施例5和实施例6的微球冻干制剂,在24h内缓慢释放,且实施例6的释放过程更加平稳,而对照例存在明显的峰谷现象。
- 还没有人留言评论。精彩留言会获得点赞!