一种丁苯酞药物活性组合物及其制备方法与流程
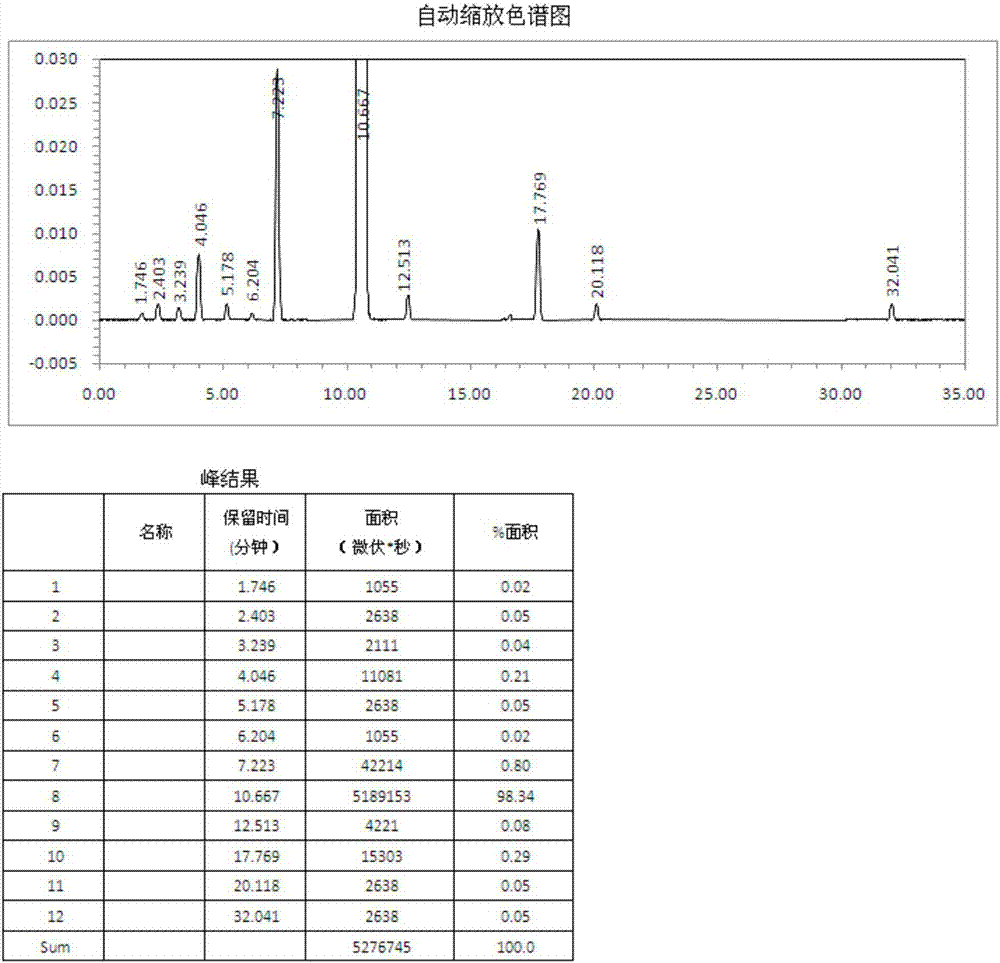
本申请是申请号为cn201210184391.8、申请日为2012年06月06日、发明名称为“一种丁苯酞药物活性组合物及其制备方法”的发明专利申请的分案申请。本发明属于医药
技术领域:
,具体涉及一种包含3-丁基-l(h)-异苯并呋喃酮(丁苯酞)的药物活性组合物及其制备方法。
背景技术:
:丁苯酞,化学名为3-丁基-l(h)-异苯并呋喃酮,又名芹菜甲素,是从芹菜籽中提取出来的消旋体,也可人工合成;在中国专利cn1100097中,公开了芹菜甲素在制备预防和治疗哺乳动物或人类脑缺血引起的疾病的药物中的应用,芹菜甲素即为无旋光活性的丁苯酞,丁苯酞为油状液体,具有浓烈的芹菜香味,化学结构式如式1所示:丁苯酞通过提高脑血管内皮no和pgi2的水平,降低细胞内钙浓度,抑制谷氨酸释放,降低花生四烯酸含量,抑制氧自由基和提高抗氧化酶活性等机制作用于脑缺血所致的多个病理环节,临床研究结果表明,丁苯酞对轻、中度急性缺血性脑卒中有显著改善作用,可促进患者功能恢复。关于丁苯酞产品,现有技术报道如下:李绍白等在《(±)芹菜甲素的合成》公开了一种丁苯酞产品及其制备方法,制备方法以式2所示的邻苯二甲酸酸酐为原料,与无水乙酸钠、戊酸酐在300℃下进行加热回流反应,乙醚萃取得到中间体丁烯苯酞3,中间体丁烯苯酞3溶于乙醚中,10%pd/c催化加氢,得到丁苯酞1。反应路线如下所示:采用上述方法制备得到的丁苯酞产品的缺点在于,(1)产品含量低,含量仅能达到95%左右,而杂质含量高达4%以上,影响临床疗效及用药安全;(2)产品稳定性差,在放置过程中,产品的含量显著降低,杂质显著升高,质量很不稳定,无法控制;因此该产品不能作为药品使用。随着以上问题的出现,现有技术出现了对丁苯酞产品的改进:中国专利cn101962374公开了一种丁苯酞产品及其制备方法,制备方法以邻苯二甲酸酸酐2为原料,通过与卤代丁烷的格氏试剂加成得到中间体邻戊酰基苯甲酸4,再经硼氢化钠还原、酸性环合得到丁苯酞1,反应路线如下所示:采用上述方法制备得到的丁苯酞产品含量、杂质等指标有所提高,如丁苯酞含量提高到约为97%,杂质含量降低到约3.0%,但该产品稳定性仍较差,在放置过程中,产品的含量明显降低,杂质明显升高,因此该产品也不能作为药品使用;(2)同时该方法在生产过程中使用了格式试剂,格式试剂需要无水无氧封闭保存,现用现做,操作繁琐,生产过程存在安全隐患,不适合工业化生产。鉴于以上产品的含量(或纯度)均较低,在放置过程中,质量不稳定,无法控制,不能作为药品使用,因此仍需要对现有技术进行改进,降低丁苯酞中各类杂质的含量,获得质量稳定的丁苯酞产品,将质量稳定的丁苯酞产品用于制备药物制剂,确保制剂的临床疗效和用药安全。技术实现要素:本发明人通过对现有技术的丁苯酞产品的杂质进行了系统的研究与分析,发现产品中的杂质主要有以下几类:①李绍白等在《(±)芹菜甲素的合成》公开的丁苯酞产品:(1)丁苯酞类似物,主要有两类,(a)烯基苯酞,起始原料戊酸酐中混有甲、乙、丙、丁、己等酸酐,与邻苯二甲酸酸酐反应,生成甲烯基、乙烯基、丙烯基、戊烯基等苯酞或苯酞,这些烯基苯酞或苯酞与丁烯苯酞性质相近,除去困难,会带入下一步反应直至丁苯酞中;(b)烷基苯酞,在催化氢化过程中,如果丁烯基苯酞中混有甲烯基、乙烯基、丙烯基、戊烯基等苯酞,同样被氢化而得到甲基、乙基、丙基、戊基等苯酞,这些苯酞与丁苯酞性质相近,除去困难,存在于最终产品丁苯酞中。(2)其他杂质,包括未反应的原料、酸酐、由原料带入的其他杂质等。(3)无机杂质,包括催化剂、重金属等。(4)残留溶剂。②cn101962374公开的丁苯酞产品:(1)丁苯酞类似物,主要有两类,(a)丁苯酞合环前中间体,邻戊酰基苯甲酸及其类似物邻乙酰基苯甲酸、邻丙酰基苯甲酸、邻丁酰基苯甲酸、邻己酰基苯甲酸(由格式试剂不纯带入);(b)烷基苯酞,在合环还原过程中,如果邻戊酰基苯甲酸中混有上述其类似物,同样被合环还原而得到甲基、乙基、丙基、戊基等苯酞及苯酞,这些丁苯酞类似物与丁苯酞性质相近,除去困难,存在于最终产品丁苯酞中。(2)其他杂质,包括未反应的原料、格式试剂、由原料带入的其他杂质等。(3)无机杂质,包括催化剂、重金属等。(4)残留溶剂。本发明人经过大量的试验研究发现:在第①种丁苯酞产品中,丁苯酞产品的质量稳定性主要与其中含有的烯基苯酞如甲烯苯酞、乙烯苯酞、丙烯苯酞、丁烯苯酞、戊烯苯酞组分的含量有关,当它们任一成分含量>0.5%时,将显著影响丁苯酞药物活性组合物的质量稳定性;在第②种丁苯酞产品中,丁苯酞产品的质量稳定性主要与其中含有的丁苯酞合环前中间体如邻乙酰基苯甲酸、邻丙酰基苯甲酸、邻丁酰基苯甲酸、邻戊酰基苯甲酸、邻己酰基苯甲酸组分的含量有关,当它们任一成分含量>0.5%时,将显著影响丁苯酞药物活性组合物的质量稳定性。另外,丁苯酞药物活性组合物中含有的其它烷基苯酞,虽然对组合物的质量稳定性影响较小,但由于它们的活性远远低于丁苯酞,当其中任一成分含量>1.0%时,将对丁苯酞药效产生较大影响;其他残余量的杂质的存在将会加剧丁苯酞产品的不稳定性。无论是烯基苯酞、丁苯酞合环前中间体还是其它烷基苯酞,均为丁苯酞类似物,与丁苯酞性质相近,去除困难。本发明人经过长期大量的丁苯酞产品质量优化工作,终于找到了一种丁苯酞药物活性组合物的制备方法,将相应组分控制到显著影响含量及稳定性以下,从而得到了一种质量稳定的丁苯酞药物活性组合物,从源头上保证丁苯酞制剂的临床疗效及用药安全。因此,本发明一方面提供了一种丁苯酞药物活性组合物,其特征在于,包含以下组分:组分ⅰ:丁苯酞,含量≥98.0%;组分ⅱ:选自甲烯苯酞、乙烯苯酞、丙烯苯酞、丁烯苯酞、戊烯苯酞、苯酞、甲苯酞、乙苯酞、丙苯酞、戊苯酞中的一种或几种,且组分ⅱ的含量>0且≤2.0%;且,当组分ⅱ包含甲烯苯酞、乙烯苯酞、丙烯苯酞、丁烯苯酞、戊烯苯酞任意之一时,所包含的任意之一成分的含量最高不超过0.5%;当组分ⅱ包含苯酞、甲苯酞、乙苯酞、丙苯酞、戊苯酞任意之一时,所包含的任意之一成分的含量最高不超过1.0%。优选的,上述丁苯酞药物活性组合物,所述的组分ⅰ:丁苯酞含量≥98.5%,组分ⅱ:含量>0且≤1.5%。优选的,上述丁苯酞药物活性组合物,所述的组分ⅰ:丁苯酞含量≥99.0%;组分ⅱ:含量>0且≤1.0%,且,当组分ⅱ包含甲烯苯酞、乙烯苯酞、丙烯苯酞、丁烯苯酞、戊烯苯酞任意之一时,所包含的任意之一成分的含量最高不超过0.3%;,当组分ⅱ包含苯酞、甲苯酞、乙苯酞、丙苯酞、戊苯酞任意之一时,所包含的任意之一成分的含量最高不超过0.5%。优选的,所述组分ⅱ选自苯酞、丁烯苯酞、丙苯酞中一种或几种。本发明的另一方面还提供上述丁苯酞药物活性组合物的制备方法,包括以下步骤:a.加料:将丁苯酞粗品加入到精馏塔中;b.任选的,常压蒸馏:将丁苯酞粗品升温至60~110℃,无回流,收集该温度下的馏分,弃去;c.减压蒸馏,控制真空度为1~5mmhg,将丁苯酞粗品升温至130~150℃,收集该温度下的馏分,弃去;继续升温至154~180℃,全回流0~300min,控制恒定的回流比,收集该温度下的馏分;d.任选的,根据所收集馏分中的组分含量,重复上述b、c步骤,得丁苯酞药物活性组合物。上述制备方法,所述的步骤c,控制真空度优选4~5mmhg;更优选5mmhg。上述制备方法,所述的步骤c,将丁苯酞粗品升温至优选140~150℃,更优选150℃。上述制备方法,所述的步骤c,继续升温至优选154~160℃,更优选154℃。上述制备方法,所述的步骤c,控制恒定的回流比优选1~10:1,更优选3~7:1。本发明所述制备方法中,升温、精馏塔的操作、温度的控制、馏分的收集等均按照常规方法进行。本发明所述方法有效将各种杂质控制在了相应指标以下,得到了质量稳定的丁苯酞药物活性组合物。本发明所述丁苯酞粗品是指含量达不到药用要求的丁苯酞。本发明所述的丁苯酞粗品可以按照李绍白等在《(±)芹菜甲素的合成》公开的制备方法,也可以按照其他方法制备。中国专利cn101962374公开的的制备方法由于反应条件苛刻,不适合工业化生产,本发明中不予采用。本发明另一方面还提供一种药物组合物,包含上述丁苯酞药物活性组合物及药学上可接受的载体,任选地,所述药物组合物还可存在其它治疗组分。当所述药物组合物包含上述丁苯酞药物活性组合物及药学上可接受的载体时,可将其制成口服制剂,优选为软胶囊、片剂、缓释片、滴丸;也可将其制成注射制剂,优选冻干粉针剂、静脉乳,这些制剂可采用本领域一般技术人员公知的相应辅料,采用相应公知的药物制剂的制备技术制得。所述其它治疗组分能与丁苯酞产生协同作用,在预防和治疗脑血管病时特别有利。本发明另一方面还提供上述丁苯酞药物活性组合物或包含其的药物组合物在制备脑缺血引起的疾病的药物中的应用。本发明所述丁苯酞药物活性组合物,各项指标均符合药用要求,在放置过程中,质量稳定,能够保证丁苯酞制剂的临床疗效及用药安全。附图说明图1:制备例1制备的丁苯酞的有关物质检测的液相图谱。图2:实施例1制备的丁苯酞药物活性组合物有关物质检测的液相图谱。图3:实施例5制备的丁苯酞药物活性组合物有关物质检测的液相图谱。图4:实施例6制备的丁苯酞药物活性组合物有关物质检测的液相图谱。图5:实施例7制备的丁苯酞药物活性组合物有关物质检测的液相图谱。组分i丁苯酞与组分ⅱ中各成分含量检测方法:照高效液相色谱法测定色谱条件及系统适用性试验:用十八烷基硅烷键合硅胶为填充剂,以甲醇-水(65:35)为流动相,检测波长280nm,理论板数按丁苯酞峰计算不低于1500;测定法:取丁苯酞药物活性组合物约50mg,精密称定,置100ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为供试品溶液,取供试品溶液20μl,注入液相色谱仪,记录色谱图至组分ⅰ峰保留时间的3倍,以面积归一化法计算组分ⅰ及组分ⅱ中各成分的含量。组分ⅱ中各成分的与组分i丁苯酞的相对保留时间为:1、苯酞峰:相对保留时间为0.35-0.39的峰;2、甲苯酞峰:相对保留时间为0.40-0.44的峰;3、乙苯酞峰:相对保留时间为0.46-0.50的峰;4、甲烯苯酞峰:相对保留时间为0.56-0.60的峰;5、丙苯酞峰:相对保留时间为0.66-0.69的峰;6、乙烯苯酞峰:相对保留时间为0.70-0.74的峰;7、丁苯酞峰:出峰时间为10.5~10.9分钟;8、丙烯苯酞峰:相对保留时间为1.05-1.09的峰;9、戊苯酞峰:相对保留时间为1.53-1.58的峰;10、丁烯苯酞峰:相对保留时间为1.63-1.67的峰;11、戊烯苯酞峰:相对保留时间为2.83-2.87的峰。具体实施方式下面结合具体实施例对本发明作进一步详细的说明。制备例1:丁苯酞,现有技术产品,按照李绍白等在《(±)芹菜甲素的合成》公开的制备方法制备(1)丁烯苯酞的制备邻苯二甲酸酸酐148.0kg、无水乙酸钠82.0kg和正戊酸酐300.0l在300℃加热回流4h,蒸除低沸点馏分(控制在150℃以下),残余物用热水溶解,再用nahco3中和至ph=6~7,用7×500l乙醚萃取,合并有机层,无水na2so4干燥,滤除干燥剂,蒸除乙醚,硅胶柱层析,氯仿-石油醚洗脱,得到丁烯苯酞45.0kg。(2)丁苯酞的制备3-丁烯苯酞45.0kg溶于乙醚中,加入4.5kg10%pd/c,用h2气置换6次,充入h2,搅拌,室温反应24h,滤除pd/c后浓缩,硅胶柱层析,氯仿-石油醚洗脱,得到丁苯酞43.0kg。我们对所得产品进行了质量研究和(40℃,75%rh)条件下的加速稳定性研究,结果见表1。制备例2:丁苯酞,现有技术产品,按照中国专利cn101962374公开的制备方法(1)溴丁烷格式试剂的制备氮气保护下,在配有搅拌、温度计和带有回流冷凝装置的反应罐中加入四氢呋喃200l、镁片6.0kg和碘0.1kg,升温至50℃,滴加溶于40l四氢呋喃的31.50kg溴代丁烷,控制温度不超过70℃,滴加完毕后,继续搅拌1h,得溴代丁烷的格氏试剂。(2)邻戊酰基苯甲酸的制备氮气保护下,加入300l四氢呋喃、30kg的邻苯二甲酸酐和2.5kg碘化铜,冷却至-10℃,滴加步骤(1)所得的溴代丁烷的格氏试剂,控制1h左右滴加完毕;滴加完毕后,再继续搅拌2h,加入1mol/l的盐酸溶液水解至ph为2,静置分去水层,水层用甲基叔丁基醚萃取两次,合并有机相,饱和氯化钠溶液洗涤至中性,无水硫酸镁干燥,再减压脱去溶剂得到邻戊酰基苯甲酸35kg。(3)丁苯酞的制备在反应罐中加入160kg5%的氢氧化钠溶液和上述步骤(2)所得的邻戊酰基苯甲酸,常温搅拌1h,冷却至0℃,缓慢加入6kg硼氢化钠,控制温度为0℃~10℃,加入完毕后在常温继续搅拌1h,之后加入6mol/lhcl酸化至ph为4.0,用甲基叔丁基醚萃取水层三次,合并有机相,分别用5%碳酸氢钠和饱和氯化钠溶液洗涤至中性,无水硫酸镁干燥,控制180-185℃/1mmhg条件进行减压蒸馏得丁苯酞19kg。我们对所得产品进行了质量研究和(40℃,75%rh)条件下的加速稳定性研究,结果见表2。实施例1:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中;b.控制真空度为5mmhg,加热升温至130℃左右,收集该温度下的馏分,弃去;继续升温至154℃,控制回流比为3:1,收集该温度下的馏分;c.将收集到的馏分重新加入精馏塔中,重复步骤b2次,得到丁苯酞2.7kg。实施例2:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中;b.升温至110℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为5mmhg,加热升温至130℃左右,收集该温度下的馏分;弃去;继续升温至154℃左右,控制回流比为4:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b3次,得到丁苯酞2.7kg。实施例3:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中;b.升温至110℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为5mmhg,加热升温至150℃左右,收集该温度下的馏分;弃去;继续升温至154℃左右,控制回流比为3:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b4次,得到丁苯酞2.6kg。实施例4:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中;b.升温至110℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为5mmhg,加热升温至140℃左右,收集该温度下的馏分;弃去;继续升温至154℃左右,控制回流比为6:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b7次,得到丁苯酞2.5kg。实施例5:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中,b.控制真空度为4mmhg,加热升温至135℃左右,收集该温度下的馏分,弃去;继续升温至160℃左右,控制回流比为5:1,收集该温度下的馏分;c.将收集到的馏分重新加入精馏塔中,重复步骤b3次,得到丁苯酞2.7kg。实施例6:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中;b.控制真空度为4mmhg,加热升温至140℃左右,收集该温度下的馏分,弃去;继续升温至160℃,控制回流比为1:1,收集该温度下的馏分,得到丁苯酞2.8kg。实施例7:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中,b.升温至90℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为4mmhg,加热升温至135℃左右,收集该温度下的馏分,弃去;继续升温至160℃左右,控制回流比为7:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b7次,得到丁苯酞2.6kg。实施例8:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中,b.升温至60℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为5mmhg,加热升温至150℃左右,收集该温度下的馏分,弃去;继续升温至154℃左右,控制回流比为4:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b4次,得到丁苯酞2.7kg。实施例9:丁苯酞药物活性组合物丁苯酞粗品:制备例1制备的丁苯酞a.将丁苯酞粗品3.0kg加入到精馏塔中,b.升温至90℃,无回流,收集该温度下的馏分,弃去;c.控制真空度为5mmhg,加热升温至150℃左右,收集该温度下的馏分,弃去;继续升温至154℃左右,控制回流比为6:1,收集该温度下的馏分;d.将收集到的馏分重新加入精馏塔中,重复步骤b7次,得到丁苯酞2.6kg。我们对实施例1~9所得产品进行了质量研究和(40℃,75%rh)条件下的加速稳定性研究,结果见表3-1,表3-2,表3-3,表3-4,表3-5,表3-6,表3-7,表3-8及表3-9。备注:40℃,75%rh的加速条件6个月相当于阴凉处保存2年。结论:从实施例6数据可以看出:组分ⅰ丁苯酞含量为98.0%;组分ⅱ的含量≤2.0%,甲烯苯酞、乙烯苯酞、丙烯苯酞、丁烯苯酞、戊烯苯酞(简称组分ⅱ中的烯基苯酞,下同)任意之一成分的含量最高不超过0.5%,苯酞、甲苯酞、乙苯酞、丙苯酞、戊苯酞(简称组分ⅱ中的烷基苯酞,下同)任意之一成分的含量最高不超过≤1.0%的丁苯酞药物活性组合物,在加速条件下放置6个月,组分ⅰ丁苯酞含量变化较小,为97.8%,组分ⅱ的含量较小,均≤2.0%,质量较稳定,基本能够作为药品使用。从实施例1数据可以看出:组分ⅰ丁苯酞含量>98.0%;组分ⅱ的含量≤2.0%,组分ⅱ中的烯基苯酞任意之一成分的含量最高不超过0.5%,组分ⅱ中的烷基苯酞任意之一成分的含量最高不超过1.0%的丁苯酞药物活性组合物,在加速条件下放置6个月,组分组分ⅰ丁苯酞含量变化较小,均≥98.0%,组分ⅱ的含量较小,均≤2.0%,质量较稳定,能够作为药品使用。从实施例2、实施例5、实施例8数据可以看出:组分ⅰ丁苯酞含量≥98.5%;组分ⅱ的含量≤1.5%,组分ⅱ中的烯基苯酞任意之一成分的含量最高不超过0.5%,组分ⅱ中的烷基苯酞任意之一成分的含量最高不超过1.0%的丁苯酞药物活性组合物,在加速条件下放置6个月,组分ⅰ丁苯酞含量变化小,均≥98.4%,组分ⅱ的含量变化小,均≤1.5%,质量稳定,能够作为药品使用。从实施例3、实施例4、实施例7、实施例9数据可以看出:组分ⅰ丁苯酞含量≥99.0%;组分ⅱ的含量≤1.0%、组分ⅱ中烯基苯酞任意之一成分含量最高不超过0.30%,组分ⅱ中烷基苯酞任意之一的成分含量最高不超过0.50%的丁苯酞药物活性组合物,在加速条件下放置6个月,组分ⅰ丁苯酞含量变化更小,均≥98.9%,组分ⅱ的含量变化更小,均≤1.0%,质量更稳定,能够为药品使用。综上所述,本发明提供的丁苯酞药物活性组合物,组分ⅰ丁苯酞含量≥98.0%;组分ⅱ的含量≤2.0%,组分ⅱ中烯基苯酞任意之一成分含量最高不超过0.5%,组分ⅱ中烷基苯酞任意之一成分含量最高不超过1.0%,其质量稳定,可以作为药品使用,进行制剂的制备。实施例10:丁苯酞软胶囊原料药:实施例6制备的丁苯酞药物活性组合物处方组成:组分用量(g)丁苯酞药物活性组合物100大豆油350明胶100甘油30水130尼泊金乙酯200mg制备方法:明胶液的制备:取明胶加入适量的水使其吸水膨胀。另将甘油、尼泊金乙酯及余下的水置溶胶锅中加热至70-80℃,混合均匀,加入膨胀的明胶搅拌,熔融,保温l~2小时,静置,使泡沫上浮,刮去上浮的泡沫,以洁净白布过滤,保温待用,配成胶液粘度一般为2.8~3.2度。药液油的制备:称取丁苯酞药物活性组合物100g与大豆油350g充分搅匀即得。压制软胶囊:将已制好的明胶甘油和丁苯酞药物活性组合物装入自动旋转轧囊机中,温度控制在40~50℃,压制出每囊含450mg药液油的软胶囊。实施例11:丁苯酞软胶囊原料药:实施例5制备的丁苯酞药物活性组合物。制备方法:同实施例10。实施例12:丁苯酞软胶囊原料药:实施例7制备的丁苯酞药物活性组合物。制备方法:同实施例10。实施例13:丁苯酞片原料药:实施5制备的丁苯酞药物活性组合物处方组成:片芯:制备方法:称取丁苯酞药物活性组合物和乳化剂泊洛沙姆,加少量水搅拌混合均匀,称取环糊精,加水溶解,将两溶液混合,快速搅拌l小时,置冰箱中冷藏(5℃)12小时,取出过滤,将所得固体于60℃干燥,得丁苯酞固体粉末。称取丁苯酞固体粉末适量,加入乳糖、部分交联羧甲基纤维素钠,混合均匀后,用羟丙基甲基纤维素(2%)水溶液制粒,干燥,整粒,外加交联羧甲基纤维素钠、硬脂酸镁、滑石粉等辅料,压片。包糖衣:称取片芯,加入适量的10%明胶包隔离层,干燥后,加入糖浆、滑石粉或滑石粉与淀粉的混合粉,包糖衣,出片干燥。实施例14:丁苯酞静脉乳原料药:实施4制备的丁苯酞药物活性组合物处方组成:组分用量丁苯酞药物活性组合物10g大豆卵磷脂12g大豆油100g维生素elg山梨醇25g注射用水加至l000ml制备方法:称取处方量的丁苯酞药物活性组合物、维生素e与大豆油混合,在60℃水浴中预热;称取处方量的大豆卵磷脂和山梨醇于水中,在60℃水浴中预热;将油相缓慢倒入水相,用高速剪切分散乳化机分散5min,l0000rpm,在高压均质机上循环5次,一级压力100mpa,二级压力10mpa,调整ph到8,过滤,121℃下灭菌15min。整个过程要充氮气保护。实施例15:丁苯酞冻干粉针剂原料药:实施4制备的丁苯酞药物活性组合物处方组成:组分用量丁苯酞药物活性组合物200g大豆磷脂(注射级)80g甘露醇2000g注射用水加至4000ml制备方法:1、按处方量分别称取丁苯酞药物活性组合物、大豆磷脂和甘露醇。2、丁苯酞药物活性组合物与大豆磷脂一起加入2000ml注射用水高速搅拌均匀,至透明溶液,无油滴。3、甘露醇加入1500ml注射用水溶解后,加总重量0.1%的活性炭100℃加热15分钟后脱炭。4、(2)与(3)溶液混合后,加入总重量0.01%的活性炭搅拌均匀后,过滤脱炭,补加注射用水至全量,测ph值、含量,精滤后,分装瓶中。5、将分装药液的瓶置冻干机中,冻干即得。对比例1:丁苯酞软胶囊原料药:制备例1制备的丁苯酞产品制备方法:同实施例10。对比例2:丁苯酞软胶囊原料药:制备例1制备的丁苯酞产品制备方法:同实施例10。本发明人长期致力于丁苯酞制剂的研究,为了获得质量稳定的丁苯酞制剂,发明人进行了加速试验的稳定性研究,检测结果如表4-1、4-2所示。表4-1:实施例10~15丁苯酞制剂稳定性结果表4-2:对比例1~2丁苯酞制剂稳定性结果备注:40℃,75%rh的加速条件6个月相当于阴凉处保存2年。结论:由表4-1、4-2数据可以看出:从实施例10数据可以看出:实施例6丁苯酞药物活性组合物(组分ⅰ丁苯酞含量为98.0%;组分ⅱ的含量≤2.0%,组分ⅱ中的烯基苯酞任意之一成分的含量最高不超过0.5%,组分ⅱ中的烷基苯酞任意之一成分的含量最高不超过≤1.0%)制备的丁苯酞软胶囊,在加速条件下放置6个月,杂质的变化较小,杂质总量≤2.0%,表明该丁苯酞药物活性组合物与辅料相容性较好,制剂质量较稳定。从实施例11数据可以看出:实施例5丁苯酞药物活性组合物(组分ⅰ丁苯酞含量≥98.5%;组分ⅱ的含量≤1.5%,组分ⅱ中的烯基苯酞任意之一成分的含量最高不超过0.5%,组分ⅱ中的烷基苯酞任意之一成分的含量最高不超过1.0%)制备的丁苯酞软胶囊,在加速条件下放置6个月,杂质的变化小,杂质总量≤1.5%,表明该丁苯酞药物活性组合物与辅料相容性好,制剂质量稳定。从实施例12数据可以看出:实施例7的丁苯酞药物活性组合物(组分ⅰ丁苯酞含量≥99.0%;组分ⅱ的含量≤1.0%、组分ⅱ中的烯基苯酞任意之一成分含量最高不超过0.30%,组分ⅱ中的烷基苯酞任意之一成分含量最高不超过0.50%)制备的丁苯酞软胶囊,在加速条件下放置6个月,杂质的变化更小,杂质总量≤1.0%,表明该丁苯酞药物活性组合物与辅料相容性更好,质量更稳定。从实施例10~12的数据可以看出:丁苯酞药物活性组合物的稳定性影响制剂的稳定性,丁苯酞药物活性组合物的稳定性提高,制剂的稳定性也随之提高。从实施例10~15的数据可以看出:将本发明的丁苯酞药物活性组合物制备制剂,在加速条件下放置6个月,制剂的各项指标变化很小,质量稳定,能够保证丁苯酞的临床疗效及用药安全。从对比例1和对比例2数据可以看出,现有技术的丁苯酞产品制备的丁苯酞软胶囊,在加速条件下放置6个月,杂质显著增加、含量显著降低,表明现有技术的丁苯酞产品与辅料相容性差,质量非常不稳定。由以上数据可以看出,现有技术中的丁苯酞产品杂质高、含量低,其与辅料相容性差,在存放过程中,其杂质均急剧升高,含量均急剧降低,无法保证临床疗效及用药安全,因此不能用于临床研究;本发明提供的丁苯酞药物活性组合物,组分ⅰ丁苯酞含量≥98.0%;组分ⅱ的含量>0且≤2.0%,组分ⅱ中的烯基苯酞任意之一成分的含量最高不超过0.5%,组分ⅱ中的烷基苯酞任意之一成分的含量最高不超过1.0%,与辅料相容性好,在加速条件下放置6个月,各组分均无明显变化,能够满足药品的安全有效、质量可控的特点,能够保证临床疗效及用药安全。实施例16:丁苯酞动物急性毒性试验试验目的:比较本发明提供的丁苯酞药物活性组合物与现有技术的丁苯酞产品在急性毒性的差异。试验动物:小鼠:健康昆明种,体重18~21g;wistar大鼠,体重120~130g。试验药品:实施例5~7制备的丁苯酞药物活性组合物、制备例1~2得到的现有技术丁苯酞产品。试验方法:将小鼠和大鼠两种动物,以口服给药途径进行急性毒性试验。口服给药者给药前禁食12小时,自由饮水,试验室温在22~24℃,给药后连续观察7天。小鼠0.1ml/10g,大鼠3~3.5ml/kg。试验结果:见表5表5:丁苯酞急性毒性结果表由上表可以看出:实施例5~7的ld50明显高于制备例1~2的ld50值,说明本发明提供的丁苯酞药物活性组合物与现有技术的丁苯酞产品相比,具有良好的安全性。实施例17:丁苯酞对大鼠脑动脉结扎脑水肿的作用试验目的:比较本发明提供的丁苯酞药物活性组合物与现有技术的丁苯酞产品在治疗脑缺血引起的疾病上药效的差异。试验动物:雄性wistar大鼠,体重300~350g。试验药品:实施例5~7制备的丁苯酞药物活性组合物、制备例1~2得到的现有技术丁苯酞产品。试验设计:脑组织缺氧缺血导致能量耗竭,细胞膜功能受损,不能维持离子梯度,引起脑组织水肿,na+堆积,k+浓度下降,严重损伤神经元功能或导致神经元死亡,本试验观察丁苯酞对局部脑缺血大鼠脑水肿的影响。试验方法:对大鼠实施右侧大脑中动脉结扎,导致脑水肿,15分钟后,口服实施例1~3、制备例1~2的丁苯酞(160mg、240mg/kg),术后24小时处死,取前脑,称左、右脑半球湿重;100℃烤24小时后称干重,用干湿法计算脑组织水重量;将组织硝化4小时,用hci调ph值,用离子选择性电极连接在氧组织分析仪上,测脑组织na+、k+浓度。试验结果:见表6、表7。表6:丁苯酞对大鼠脑水含量的影响表7:丁苯酞对大鼠脑阳离子浓度的影响备注:每组动物数10-14只;左侧为非缺血侧,右侧为缺血侧。由表6、表7可以看出:实施例5~7组(160mg、240mg/kg)降低脑水量和na+含量,增加k+含量非常显著,而且有剂量效应关系,说明本发明提供的丁苯酞药物活性组合物能明显减轻局部脑缺血引起的脑水肿;制备例1~2组(160、240mg/kg)可降低脑水量和na+含量,增加k+含量,但效果差于实施例5~7组。实施例16:丁苯酞软胶囊治疗轻中度急性缺血性脑卒中的临床研究药品:实施例10制备的丁苯酞软胶囊研究对象:(1)首次发病<72h的急性颈内动脉系统梗死。(2)美国国立卫生研究院卒中量表评分(nihss)神经功能缺损评分为5-25分。(3)符合1995年全国第四届脑血管病学术会议修订的诊断标准(4)发病年龄<75岁。(5)头部ct检查排除脑出血。(6)无意识障碍,检查合作且吞咽功能正常。试验设计:(1)随机对照试验,根据受试者就诊的先后顺序和随机数字表法确定人组情况,分为a,b两组;按照各组治疗方案对受试者进行治疗。(2)开放试验:所有受试者服药方法均为3次/d。给药方法:(1)随机对照试验:a组受试者药物剂量为200mg/次,3次/d(7:00,15:00,21:00);b组剂量为200mg/次,2次/d(7:00,21:00);疗程20d。基础药物为复方丹参注射液16ml加人0.9%盐水500ml中静脉滴注,1次/d,连续应用14d。(2)开放试验:药物剂量200mg/次,3次/d(7:00,15:00,21:00),疗程20d。基础药物与随机对照试验相同,1次/d,连续应用14d。疗效评价:标准神经功能缺损评分分为5个等级:基本痊愈,神经功能缺损评分减少91%-100%;显著进步,神经功能缺损评分减少46%-90%;进步,神经功能缺损评分减少18%-45%;无变化,神经功能缺损评分减少或增加<17%;恶化,神经功能缺损评分增加>18%。总有效率=(基本痊愈例数+显著进步例数)/总例数x100%。疗效观察:1、随机对照试验:(1)入选30例患者,按入院就诊顺序及随机数字表法分为两组,a组20例,b组10例。对两组受试者的一般临床资料,包括年龄、性别、伴发疾病评分、既往史评分,以及治疗前神经功能缺损评分及日常生活活动能力量表评分等进行均衡性检验,差异无统计学意义(p>0.05),具有可比性,具体见表8,表8:随机对照试验两组受试者治疗前临床资料的比较注:均p>0.05(2)疗效比较:两组受试者丁苯酞软胶囊治疗后,a组有效率为85.0%(17/20),b组有效率为40%(4/10),组间差异具有统计学意义(p<0.05)。(3)神经功能缺损评分的比较:两组受试者治疗前神经功能缺损评分差异无统计学意义(p>0.05);治疗第11天和第21天,a组评分均高于b组(均p<0.05),具体见表9。表9:随机对照试验两组受试者治疗前后神经功能缺损评分的比较注:*p<0.005;**p<0.001(4)日常生活活动能力量表评分的比较:两组受试者治疗前日常生活活动能力量表评分差异无统学意义(p>0.05);治疗第11天和第21天a组评分均高于b组(p<0.01),具有统计学意义,具体见表10。表10:随机对照试验两组受试者治疗前后日常生活活动能力量表评分的比较注:**p<0.012、开放试验:共入选34例患者,3次/d服药,200mg/次,有效率为76.47%(26/34)3、总体评估:(1)疗效观察:开放试验34例与随机对照试验30例合计64例,总体治疗有效率为73.44%(47/64)。(2)神经功能缺损评分(表4):与治疗前相比,治疗后第11天神经功能缺损评分明显减少,差异有高度统计学意义(q=5.575,p<0.01);治疗后第21天神经功能恢复程度优于第11天,二者之间差异有高度统计学意义(q=3.256,p<0.01)。(3)日常生活活动能力量表评分:与治疗前相比,治疗后第11天日常生活活动能力量表评分分明显增加,差异具有高度统计学意义(q=3.615,p<0.01);治疗后第21天日常生活活动能力量表评分与第11天相比差异亦有高度统计学意义(q=2.756,p<0.01)。具体见表11。表11:总体病例治疗前后神经功能缺损评分与日常生活活动能力量表评分的比较注:a为治疗前与治疗后第11天比较,b为治疗11天与治疗后第21天比较,c为为治疗前与治疗后第21天比较**p<0.01以上结果显示:丁苯酞软胶囊对轻中度急性缺血性脑卒中的治疗安全、疗效肯定,可作为急性缺血性脑卒中患者的早期治疗药物。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1