一种脂质包裹固体药物纳米颗粒的制备方法与流程
本发明属于生物医药领域,更具体地,涉及一种脂质包裹固体药物纳米颗粒的制备方法。
背景技术
纳米化疗药物输送体系虽然在一定程度上提高了疗效、延长了病人的存活时间,但是癌症仍然是目前对人类健康威胁最大的疾病之一。到目前为止,虽然已有纳米药物制剂正在开发和生产,甚至已经在被批准于临床使用,但是大多数纳米药物都存在一个致命的缺陷,就是其载药量较低,通常的载药量小于10%;另外纳米药物中采用的一些载体本身没有药效,甚至会产生毒副作用;而且纳米药物的制备过程往往涉及繁琐的工艺或者复杂的化学合成,导致不同批次的重复性差,难以大规模工业化生产。
快速纳米沉淀是一种微流控技术,可以通过动力学控制分子聚集过程连续生产药物纳米颗粒。快速纳米沉淀方法制备的纳米颗粒粒径可控、分散窄、重复性好、可连续大规模化生产,适合工业化生产。它的主要机制是依靠高湍流混合器装置(例如,同轴湍流混合器,四通道涡流混合器等)实现溶剂(含药物)与非溶剂(含稳定剂)的快速交换,通过调控溶质成核与增长速率控制纳米颗粒的粒径及分散性。但是快速纳米沉淀方法制备的纳米药物同样存在载药量不高的缺陷。
因此,规模化生产、降低生产成本、提高载药量、减少毒副作用以及药物智能释放,是目前纳米药剂临床转化所急需解决的问题。
技术实现要素:
本发明的目的在于针对现有技术中的缺陷和不足,提供一种脂质包裹固体药物纳米颗粒的制备方法。本发明所述方法通过结合快速纳米沉淀方法和脂质挤出技术,先在不使用稳定剂的条件下将两亲性小分子药物制备成为纳米颗粒,然后再利用脂质挤出技术对药物纳米颗粒进行包裹,所述方法对于药物本身的抗癌作用无任何不良影响,制备得到载药量高,且粒径小、分散窄、稳定性好、毒副作用小的脂质包裹固体药物纳米颗粒。
本发明的上述目的是通过以下方案予以实现的:
一种脂质包裹固体药物纳米颗粒的制备方法,在不使用稳定剂的条件下先将的两亲性小分子药物利用快速纳米沉淀方法得到药物纳米颗粒溶液;然后将其与脂质通过挤出的方法制备得到脂质包裹固体药物纳米颗粒。
本发明所述方法适用于两亲性小分子药物,当药物为完全疏水性药物时,则经过快速纳米沉淀方法制备得到的药物纳米颗粒溶液,在几秒钟之内很快析出肉眼可见的沉淀,无法进行第二步的脂质挤出处理步骤;而当药物为完全亲水性药物时,则经过快速纳米沉淀方法制备的溶液中,药物不会以纳米颗粒的形式存在溶液中,而是溶解在溶液中,同样无法进行脂质挤出处理。
优选地,所述两亲性小分子药物为甲氨蝶呤、苯丁酸氮芥或阿霉素。
优选地,所述快速纳米沉淀的具体过程包括:将溶有两亲性小分子药物的溶液与水按照体积比为1:3~11分别引入四通道涡流混合器中的不同通道,通过高速湍流混合制备得到药物纳米颗粒溶液。具体为将两亲性性小分子药物的有机溶液与超纯水分别引入四通道涡流混合器的第1和2、3、4通道,在涡流混合器中高速湍流混合,快速形成药物纳米颗粒溶液。
本发明所述快速纳米沉淀的过程中,未添加稳定剂,直接将两亲性小分子药物制备成为药物纳米颗粒溶液,由于未添加稳定剂,溶液中的药物纳米颗粒完全是100%的药物,从而保证了最后制备得到的脂质包裹固体药物纳米颗粒的高载药量。
优选地,溶解两亲性小分子药物的溶剂包括但不限于n,n-二甲基甲酰胺、n-甲基吡咯烷酮或二甲基亚砜中的一种、两种或两种以上的混合液;所述高速湍流混合过程中溶有两亲性小分子药物的溶液的流速为1ml/min~20ml/min。
优选地,当两亲性小分子药物分别为甲氨蝶呤、苯丁酸氮芥或阿霉素时,溶有药物的溶液中药物的浓度分别为1~6mg/ml、0.5~4mg/ml或0.1~2mg/ml。当溶液中药物浓度过大时,经过快速纳米沉淀法处理后的溶液中,会析出肉眼可见沉淀,而不是形成纳米颗粒;但当浓度过低时,则药物分子又难以形成纳米颗粒。
优选地,当两亲性小分子药物分别为甲氨蝶呤、苯丁酸氮芥或阿霉素时,溶有药物的溶液中药物的浓度分别为3~5mg/ml、0.5~2mg/ml或0.3~1mg/ml。
更优选地,当两亲性小分子药物分别为甲氨蝶呤、苯丁酸氮芥或阿霉素时,溶有药物的溶液中药物的浓度分别为4mg/ml、1mg/ml或0.5mg/ml。
优选地,所述挤出的具体过程包括:将溶有脂质的乙醇溶液与药物纳米颗粒溶液混合后,通过孔径为50~200nm的聚偏二氟乙烯膜重复挤出至少7次,即可得到脂质包裹固体药物纳米颗粒。更优选地,所述聚偏二氟乙烯膜的孔径为100nm,溶解脂质采用的是质量浓度为4%的乙醇。
优选地,所述脂质包括但不限于1,2-二油酰-sn-甘油-3-磷酰乙醇胺、胆固醇琥珀酸单酯、1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇(2000)dspe-peg或1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇-叶酸中至少两种。
优选地,所述脂质为1,2-二油酰-sn-甘油-3-磷酰乙醇胺和胆固醇琥珀酸单酯,以及1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇(2000)或1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇-叶酸,三者的摩尔比为10~15:4~9:1~5;更优选地,三者的摩尔比为10:9:1。
优选地,溶有脂质的乙醇溶液与药物纳米颗粒溶液混合后,溶液中两亲性小分子药物和脂质的质量比为1~4:1;更优选地,两亲性小分子药物和脂质的质量比为4:1。
当脂质用量较少时,虽然最后制备的药物纳米颗粒的载药量更高,但药物纳米颗粒的稳定将显著性下降。
与现有技术相比,本发明具有以下有益效果:
与传统的药物纳米颗粒相比,本发明所得脂质包裹固体药物纳米颗粒的一个最大优势就是具有超高的载药量,可达到60%以上,显著性地高于传统的药物纳米颗粒;另外快速纳米沉淀方法可连续化生产,批次间的差异小,适用于工业化生产;药物颗粒的表面包裹着脂质,这一保护层赋予药物纳米颗粒可长期储存的稳定性、肿瘤组织主动靶向性和ph响应性。本发明所述方法制得的脂质包裹固体药物纳米颗粒在药物输送方面具有较大的应用前景。
附图说明
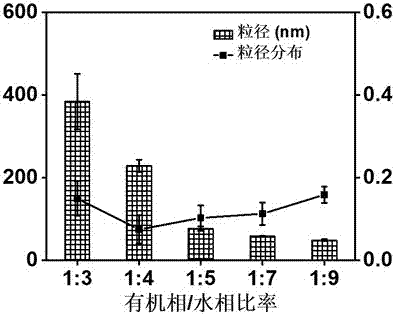
图1为有机相/水相比率对甲氨蝶呤纳米颗粒的粒径和粒径分布的影响。
图2为甲氨蝶呤药物纳米颗粒在脂质包裹前后的粒径和表面电位的变化情况。
图3为甲氨蝶呤药物纳米颗粒在脂质包裹前后的透射电镜图。
图4为有机相/水相比率苯丁酸氮芥和阿霉素药物纳米颗粒的粒径(a图)和粒径分布(b图)的影响,苯丁酸氮芥(c图)和阿霉素(d图)药物纳米颗粒脂质包裹前后的粒径和表面电位的变化情况。
图5为甲氨蝶呤药物纳米颗粒在脂质包裹前后的体外稳定性。
图6为甲氨蝶呤、脂质包裹甲氨蝶呤纳米颗粒的体外毒性。
图7为甲氨蝶呤、脂质包裹甲氨蝶呤纳米颗粒抑制裸鼠肿瘤的试验结果。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。。
实施例1甲氨蝶呤纳米颗粒溶液的制备
称取甲氨蝶呤,溶于n,n-二甲基甲酰胺和n-甲基吡咯烷酮的混合溶剂(体积比7:3),配成4mg/ml的甲氨蝶呤有机溶液,将其引入四通道涡流混合器的第1通道,其它三个通道引入的则是超纯水,实现快速湍流混合以制备甲氨蝶呤纳米颗粒溶液。所述甲氨蝶呤有机溶液和水相的体积比分别为1:3~9,有机相流速为6ml/min。
当改变甲氨蝶呤有机溶液(有机相)和水相的体积比时,可制备得到的粒径不同的甲氨蝶呤纳米颗粒,具体的比例和粒径关系如图1所示,从图中可知,当有机相和水相的体积比越小时,制备得到的甲氨蝶呤纳米颗粒的粒径越小。
当有机相和水相的体积比为1:3~9时,制备的甲氨蝶呤纳米颗粒的粒径范围在49nm~384nm之间,粒径分布在0.1~0.2之间,表示甲氨蝶呤纳米颗粒溶液中甲氨蝶呤纳米颗粒的粒径分布很均一。
因为50nm左右的纳米颗粒在肿瘤部位的epr效应和组织穿透性较好,因此我们选用有机相和水相的体积比为1:9制备的甲氨蝶呤纳米颗粒,其粒径为49.1nm。
实施例2脂质包裹甲氨蝶呤的纳米颗粒的制备
称取1,2-二油酰-sn-甘油-3-磷酰乙醇胺(dope)、胆固醇琥珀酸单酯(chems)、1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇(2000)(dspe-peg),按摩尔比10:9:1的比例制成混合物,溶于二氯甲烷配成10mg/ml的混合脂质溶液备用。移液枪定量移取0.1ml的混合脂质二氯甲烷溶液加入10ml的4%的乙醇溶液中,溶液利用探头超声仪超声至澄清备用。将等体积的脂质乙醇溶液和实施例1制备的粒径为49.1nm的甲氨蝶呤纳米颗粒溶液混合后,混合溶液中甲氨蝶呤和混合脂质的质量比为4:1,混合液通过100纳米的聚偏二氟乙烯膜重复挤出7次,得到脂质包裹甲氨蝶呤纳米颗粒mtxnp@lipid。
同样采用上述制备过程,将脂质更换为1,2-二油酰-sn-甘油-3-磷酰乙醇胺(dope)、胆固醇琥珀酸单酯(chems)和1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-聚乙二醇-叶酸(dspe-peg-fa)按摩尔比为10:9:1混合的混合脂质,制备得到叶酸脂质包裹甲氨蝶呤纳米颗粒mtxnp@lipid-fa
检测3种甲氨蝶呤纳米颗粒的粒径、并观察其形态。结果如图2和图3所示,其中,mtxnp为甲氨蝶呤纳米颗粒,mtxnp@lipid和mtxnp@lipid-fa为采用不同的脂质的2种脂质包裹甲氨蝶呤纳米颗粒。
从图2中可知,相比于甲氨蝶呤纳米颗粒,脂质包裹甲氨蝶呤纳米颗粒的粒径增大至56nm,表面电位也由中性变为约-10mv,有利于提高脂质包裹甲氨蝶呤纳米颗粒的体内稳定性。
从图3中可知,甲氨蝶呤纳米颗粒颗粒(mtxnp)之间存在聚集,而2种脂质包裹甲氨蝶呤纳米颗粒(mtxnp@lipid和mtxnp@lipid-fa)则分散很好,证明其稳定性好。
制备的脂质包裹甲氨蝶呤纳米颗粒的粒径为56nm,分散度为0.187,包封率为84.5%,载药量为62.3%。
采用本发明制备的脂质包裹甲氨蝶呤纳米颗粒的载药量高达62.3%,是单独使用快速纳米沉淀法制备的纳米颗粒载药量的2倍,本发明所述方法显著性地提升了甲氨蝶呤纳米颗粒的载药量。
实施例3脂质包裹苯丁酸氮芥或阿霉素纳米颗粒的制备
称取苯丁酸氮芥或阿霉素分别溶于二甲基亚砜或n,n-二甲基甲酰胺,各自配成1mg/ml或0.5mg/ml的有机溶液,将其分别引入四通道涡流混合器的第1通道,其它三个通道引入的则是超纯水,实现快速湍流混合以制备苯丁酸氮芥纳米颗粒溶液和阿霉素纳米颗粒溶液。其中,苯丁酸氮芥纳米颗粒溶液(有机相)或阿霉素纳米颗粒溶液(有机相)和水相的体积比分别设为1:3~11,有机相流速为10ml/min,制备得到苯丁酸氮芥纳米颗粒溶液和阿霉素纳米颗粒溶液。
参照实施例2中的步骤,分别制备得到脂质包裹苯丁酸氮芥纳米颗粒和脂质包裹阿霉素纳米颗粒。
测试有机相与水相的体积比对制备的2种纳米颗粒的粒径和分散度的影响,结果如图4所示。从图4a和4b中可知,苯丁酸氮芥米颗粒和阿霉素纳米颗粒的粒径均随着有机相与水相的体积比的变小而减小。当有机相与水相的体积比为1:3~11时,苯丁酸氮芥米颗粒的粒径范围在48nm~263nm之间,粒径分布在0.06~0.18之间;阿霉素颗粒粒径范围在65nm~258nm之间,粒径分布在0.06~0.15之间。两种药物的纳米颗粒的粒径分布都很均一。
当有机相和水相的体积比为1:9时,苯丁酸氮芥和阿霉素纳米颗粒的粒径最接近于50nm,分别为48nm和65nm。分别选用48nm和65nm的苯丁酸氮芥和阿霉素纳米颗粒,保证药物和混合脂质的质量比为4:1,按实施例2的步骤对苯丁酸氮芥和阿霉素纳米颗粒进行包裹,制备得到脂质包裹苯丁酸氮芥纳米颗粒和脂质包裹阿霉素纳米颗粒。
包裹脂质前后,2中药物纳米颗粒的粒径大小和表面电位的变化情况如图4c和4d所示。从图4c和4d可知,药物纳米颗粒在脂质包裹后,粒径增大10nm左右,表面电位也由中性变为约-10mv,证明成功的对药物纳米颗粒完成脂质包裹。
制备的脂质包裹阿霉素纳米颗粒的粒径为66nm,分散度为0.124,包封率为89.4%,载药量为66.4%。
制备的脂质包裹苯丁酸氮芥纳米颗粒的粒径为55nm,分散度为0.145,包封率为87.4%,载药量为63.7%。
同样地,采用本发明所述方法制备的脂质包裹阿霉素纳米颗粒和脂质包裹苯丁酸氮芥纳米颗粒的载药量分别高达66.4%和63.7%,显著性地高于常规的药物纳米颗粒。
实施例4脂质包裹的药物纳米颗粒的体外稳定性
以实施例1制备的甲氨蝶呤纳米颗粒(mtxnp)和脂质包裹甲氨蝶呤纳米颗粒(mtxnp@lipid)为测试对象,测试2种纳米颗粒的体外稳定性。
测试步骤如下:分别将a组(mtxnp颗粒)、b组(mtxnp@lipid颗粒)和c组(透析后mtxnp@lipid颗粒)的pbs溶液在室温静置一周,在预设时间内,记录三组的粒径变化情况。
记录结果如图5所示。从图5中可知,mtxnp颗粒随着静止时间的延长,其粒径逐渐增大,静止时间超过4天时,粒径增大的幅度明显上升;而b组和c组的mtxnp@lipid颗粒在pbs缓冲溶液中静置一段时间后其粒径无明显变化,因此可知mtxnp@lipid颗粒具有良好的体外稳定性。
实施例5脂质包裹的药物纳米颗粒的体外细胞毒性
采用cck8方法评价甲氨蝶呤纳米颗粒的体外细胞毒性。具体步骤如下:mcf-7细胞以1×104个/孔的密度种植于96孔板内,待细胞过夜生长贴壁后,取100μl包含不同mtx(a组)、mtxnp@lipid(b组)和mtxnp@lipid-fa(c组)的完全培养基替换原有介质。共同孵育48h后,利用cck8试剂检测相应细胞的活力。
实验结果如图6所示,mtx、mtxnp@lipid和mtxnp@lipid-fa均表现出药物浓度依赖性,3组对于mcf-7细胞的杀伤力由大到小的顺序为mtxnp@lipid-fa>mtxnp@lipid>mtx,即mtxnp@lipid-fa对于mcf-7细胞的抑制作用最强;其中又以mtxnp@lipid-fa靶向为最。
实施例6脂质包裹的药物纳米颗粒抑制裸鼠肿瘤的试验
接种mcf-7细胞的裸鼠用于评估甲氨蝶呤纳米颗粒的体内抗肿瘤实验。当裸鼠肿瘤体积达到100mm3左右时随机分为4组,每组6只,分别静脉注射生理盐水(a组),甲氨蝶呤(b组),mtxnp@lipid(c组)和mtxnp@lipid-fa(d组),其中甲氨蝶呤的给药剂量都为10mg/kg。每组裸鼠三天给一次药,试验周期内共给药6次,每两天一次用游标卡尺测量肿瘤长短径,计算肿瘤体积。
实验结果如图7所示,从图中可知,b、c、d三组对于裸鼠的肿瘤均有抑制作用,,且与a组相比,抑制效果较为明显;但b、c、d三组之间相比较是,d组对于裸鼠肿瘤的抑制作用最佳,其次是c组,c组和d组对于裸鼠肿瘤的抑制作用显著性由于b组作用;即脂质包裹后的甲氨蝶呤药物纳米颗粒对于裸鼠肿瘤的抑制作用明显优于甲氨蝶呤小分子药物。
采用本发明所述方法制备的脂质包裹药物纳米颗粒,不仅显著性的提升了药物的载药量,同时对于药物本身的性能和作用并没有丝毫的影响,药物的效果反而因为载药量的提高而有显著性的改善。
对比例1
在试验过程中,除了甲氨蝶呤、苯丁酸氮芥或阿霉素这3种药物外,还测试过其他多种药物,但是并非全部都能成功制备得到脂质包裹的药物纳米颗粒,其中以疏水性药物姜黄素和紫杉醇为例,其具体的制备过程如下:
1、称取姜黄素,溶于乙醇,配成1mg/ml的姜黄素有机溶液,将其引入四通道涡流混合器的第1通道,其它三个通道引入的则是超纯水,用以制备姜黄素纳米颗粒。所述姜黄素有机溶液和水相的体积比为1:3~9,有机相流速为10ml/min。但是姜黄素纳米颗粒溶液在几秒钟之内均析出肉眼可见沉淀,无法进行第二步脂质挤出操作。
2、称取紫杉醇,溶于二甲基亚砜,配成1mg/ml的紫杉醇有机溶液,将其引入四通道涡流混合器的第1通道,其它三个通道引入的则是超纯水,用以制备紫杉醇纳米颗粒。所述紫杉醇有机溶液和水相的体积比为1:3~9,有机相流速为10ml/min。但是紫杉醇纳米颗粒溶液在几秒钟之内均析出肉眼可见沉淀,无法进行第二步脂质挤出操作。
通过对上述试验失败原因的分析,发现姜黄素和紫杉醇均未能成功制备得到脂质包裹的药物纳米颗粒的原因均是由于药物的疏水性太强,经快速纳米沉淀处理后形成的纳米颗粒因为ostwald熟化过程而聚集析出沉淀,进而无法进行脂质挤出操作。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!