一种药物复合物及其制备方法与应用与流程
本发明属于医学领域,涉及一种载药系统及其制备方法与应用,特别涉及一种自组装表没食子儿茶素没食子酸酯(egcg)纳米粒子载药系统制备及协同用药的应用。
背景技术
表没食子儿茶素没食子酸酯(epigallocatechingallate,egcg)属于儿茶酚素家族,主要是来源于绿茶中的茶多酚化合物。egcg是一种典型的多酚类化合物,因结构中拥有活性的没食子儿茶酚基团和没食子儿茶酸酯基团,所以egcg具有强抗氧化性,可作为强抗氧化剂清除体内自由基。近期研究发现egcg可抑制cbr1蛋白从而阻断阿霉素(dox)转化成阿霉素醇(doxol),从而提高dox的抗癌药效,同时egcg被誉为广谱性疾病干预补充物用于辅助治疗一些与年龄相关的疾病,例如癌症、糖尿病和组织退让性疾病。
由于egcg结构稳定性较差,在体内生物利用度低,单独使用egcg作为抗氧化、抗炎症和抗癌药具有用量大,效率低等缺点,将egcg与小分子抗癌药物如阿霉素dox协同用药不仅能提高小分子抗癌药物的疗效还可以大大降低其毒副作用。目前研究主要使用两种技术方法实现egcg协同小分子抗癌药物的功能。一种用纳米脂质体包裹egcg和小分子抗癌药物构成纳米脂质体载药系统;另一种将egcg共价偶联peg形成两亲性嵌段共聚合物后自组装包裹小分子抗癌药物提高egcg的稳定性,从而实现egcg和小分子抗癌药物协同用药并提高小分子药物药效的目的。
将脂质体作为egcg和dox共担载的药物载体,利用脂质体亲水性和疏水性的原理将水溶性较好的egcg和dox包裹在脂质体内,实现egcg和dox的协同治疗,旨在提高egcg的稳定性和体内生物利用度,从而提高dox的药效。但传统型脂质体剂型存在药物包封率低,纳米药物载体稳定性差和粒子尺寸较大等缺点,且释放egcg单体后egcg仍存在稳定性差和生物利用度低等缺点,因此并没有较好的实现egcg辅助dox协同用药的作用。
利用“加特曼-科赫反应”,将peg-cooh与多巴胺的a环发应生成peg-多酚两亲性嵌段共聚合物,进一步在与fe3+离子水中自组装成共同担载egcg和dox的纳米胶束载药系统,旨在实现egcg和dox的协同作用,提高小分子抗癌药物dox的药效。但由两亲性嵌段共聚合物所包裹小分子药物自组装成的纳米胶束药物载体仍存在在体内分散成大分子的两亲性嵌段共聚合物,很难释放出功能性egcg,实现egcg与小分子抗癌药物的协同作用。
技术实现要素:
为了解决现有技术的不足,本发明应用金属-多酚配位化学合成技术制备含多酚小分子药物阿霉素fe3+-dox/fe3+-小分子药物-多酚作为纳米药物载体的内核,fe3+-egcg为纳米粒子载药系统的表面,fe3+-peg-多酚/peg-荧光染料-多酚作为纳米粒子载药系统外围,自组装新型纳米粒子载药系统,以fe3+-多酚配位为纳米粒子诊疗剂骨架,共担载dox/小分子药物、荧光染料和egcg,从而得到新型纳米粒子载药系统dfepnps。利用“荧光染料→监测药物分布及代谢”、“egcg和fe3+清除h2o2→氧自由基ros,egcg抑制cbr1蛋白”和“增强小分药物/化疗药物疗效”相结合,杀死肿瘤细胞,实现分子影像与化疗的精准治疗。
本发明技术利用金属-多酚配位法,通过dox与fe3+配位,egcg与fe3+配位,peg-荧光染料-多巴胺与fe3+配位,从而自组装成dfepnps纳米粒子载药系统,利用“荧光染料→监测药物分布及肿瘤位置”、“egcg和fe3+清除h2o2→氧自由基ros,egcg抑制cbr1蛋白”和“增强小分子药物/化疗药物疗效”相结合,并利用肿瘤细胞比正常细胞代谢旺盛的特点,可被动靶向肿瘤细胞,杀死肿瘤细胞,实现分子影像与化疗的精准治疗。
本发明专利利用化学配位法,通过第一步小分子药物如阿霉素,或是多酚-小分子药物(紫杉醇,多西紫杉醇等)与三价铁配位作为在药物载体的内核fe3+-dox,第二步将具有活性部位的没食子儿茶酚基团和没食子儿茶酸酯基团的eccg与过量的三价铁离子形成配位化合物作为小分子药物载药系统的表面fe3+-egcg,第三步peg-多酚/peg-多酚-荧光染料进一步与过量铁离子配位形成纳米载药系统的外围fe3+-peg-多酚/peg-荧光染料-多酚,增加其生物相容性/标记功能,从而自组装成dfepnps纳米粒子载药系统,利用“荧光染料→监测药物分布及肿瘤位置”、“egcg和fe3+清除h2o2→氧自由基ros,ecgc抑制cbr1蛋白”和“增强小分子药物/化疗药物疗效”相结合,杀死肿瘤细胞,实现分子影像与化疗的精准治疗。
1.金属-多酚配位化学合成技术制备含多酚小分子药物阿霉素dox-fe3+/小分子药物-多酚作为内核,egcg-fe3+为纳米粒子载药系统的表面,peg-多酚/peg-荧光染料-多酚-fe3+作为纳米粒子载药系统外围,自组装新型纳米粒子载药系统,以fe3+-多酚配位为纳米粒子诊疗剂骨架,共担载dox/小分子药物、荧光染料和egcg,从而得到新型纳米粒子载药系统dfepnps。克服传统脂质体剂型药物包封率低,稳定性差等缺点,同时避免两亲性嵌段聚合物自组成包裹dox的纳米胶束载药系统在体内分解成大分子两亲性嵌段聚合物peg-egcg,很难释放自由egcg,从而降低egcg抗氧化性,抗癌等功能,不能实现egcg+dox协同用药和提高dox药效的目的。dfepnps具有载药量高,稳定性好,能实现egcg和小分子药物协同用药及提高小分子药物药效等优势。
2.dox作为一线小分子抗癌药物,但存在dox在cbr1蛋白催化下转化成doxol,降低dox抗癌药效,增加了dox的心脏毒性。egcg可抑制cbr1蛋白从而阻断dox转化成doxol,增强dox的药效,减少其心脏毒性。
3.egcg作为强还原剂具有稳定性差,生物利用度差等缺点,利用fe3+-多酚-peg为平台可增加egcg的稳定性,提高在体内的生物利用度,从而增强小分子药物的药效。
4.利用两端羧基或是一端羧基一端氨基peg分别共价偶联荧光染料及多酚(多巴胺)制备peg-荧光染料-多酚(多巴胺)与fe3+配位形成纳米载药系统的外围,“荧光染料→监测药物分布及肿瘤位置”、“egcg和fe3+清除h2o2→氧自由基ros,egcg抑制cbr1蛋白”和“增强小分子药物/化疗药物疗效”相结合,杀死肿瘤细胞,实现分子影像与化疗的精准治疗。
5.利用肿瘤细胞比正常细胞新陈代谢旺盛,多摄取纳米药物载体,使纳米药物载体滞留在肿瘤细胞如图3,4或是肿瘤组织如图5,实现纳米药物载体被动靶向肿瘤细胞和肿瘤组织,取得较好的肿瘤抑制率如图6。
较为具体地,本发明第一方面提供了一种药物复合物,所述药物复合物的组分包括:具有配位能力的金属离子、小分子药物,茶多酚和外围分子,所述外围分子包括可溶性骨架高分子和多酚,或可溶性骨架高分子、多酚和荧光分子。
在一些实施方式中,所述具有配位能力的金属离子选自三价铁离子、二价锌离子或其组合;
可选地,所述小分子药物是具有酚羟基的小分子药物;
可选地,所述小分子药物选自阿霉素,柔红霉素,紫杉醇,多西紫杉醇,吉西他滨,培美曲塞,铂类药物,恩曲他滨,替诺福韦二吡呋酯或其中任意种的组合;
可选地,所述茶多酚选自表没食子儿茶素没食子酸酯,儿茶素、表儿茶素,没食子儿茶素,表没食子儿茶素,表儿茶素没食子酸酯或其中任意种的组合;
可选地,所述可溶性骨架高分子为聚乙二醇分子;
可选地,所述多酚选自多巴胺、5-羟基多巴胺或其组合;
可选地,所述荧光分子选自alexafluor488,icg,ce6,亚甲蓝,荧光素纳,5-ala,fitc,罗丹明;
可选地,所述可溶性骨架高分子和所述多酚共价连接,或所述可溶性骨架高分子分别与所述多酚和所述荧光分子共价连接;
优选地,所述具有配位能力的金属离子选自三价铁离子;所述小分子药物选自阿霉素;所述茶多酚选自表没食子儿茶素没食子酸酯,所述可溶性骨架高分子为聚乙二醇分子;所述多酚选自多巴胺;所述荧光分子选自alexafluor488。
在一些实施方式中,在所述药物复合物的微观结构中,所述具有配位能力的金属离子为配位离子中心,单个所述具有配位能力的金属离子分别单独或同时与所述茶多酚和所述外围分子中的所述多酚配位,所述小分子药物为所述药物复合物的内核,所述茶多酚为所述药物复合物的表面,所述外围分子位于所述药物复合物的外围。
本发明第二方面提供了一种药物运载体,所述药物运载体的组分包括:具有配位能力的金属离子、茶多酚和外围分子,所述外围分子包括可溶性骨架高分子和多酚,或可溶性骨架高分子、多酚和荧光分子;
可选地,所述药物运载体运载的小分子药物是具有酚羟基的小分子药物;
可选地,所述药物运载体运载的小分子药物选自阿霉素,柔红霉素,紫杉醇,多西紫杉醇,吉西他滨,培美曲塞,铂类药物,恩曲他滨,替诺福韦二吡呋酯或其中任意种的组合;
可选地,所述具有配位能力的金属离子选自三价铁离子、二价锌离子或其组合;
可选地,所述茶多酚选自表没食子儿茶素没食子酸酯,儿茶素、表儿茶素,没食子儿茶素,表没食子儿茶素,表儿茶素没食子酸酯或其中任意种的组合;
可选地,所述可溶性骨架高分子为聚乙二醇分子;
可选地,所述多酚选自多巴胺、5-羟基多巴胺或其组合;
可选地,所述荧光分子选自alexafluor488,icg,ce6,亚甲蓝,荧光素纳,5-ala,fitc,罗丹明;
所述荧光分子选自用于活体成像的icg,ce6,亚甲蓝,用于细胞检测的荧光素纳,alexafluor488,5-ala,fitc,罗丹明。
可选地,所述可溶性骨架高分子和所述多酚共价连接,或所述可溶性骨架高分子分别与所述多酚和所述荧光分子共价连接;
优选地,所述具有配位能力的金属离子选自三价铁离子;所述药物运载体运载的小分子药物是阿霉素;所述茶多酚选自表没食子儿茶素没食子酸酯,所述可溶性骨架高分子为聚乙二醇分子;所述多酚选自多巴胺;所述荧光分子选自alexafluor488。
本发明第三方面提供了一种本发明第一方面的药物复合物的制备方法,所述制备方法包括如下步骤:
a)将所述小分子药物溶液加入所述具有配位能力的金属离子的可溶性金属盐溶液,形成含有第一配位复合物的第一溶液;
b)将所述茶多酚溶液加入所述第一溶液,形成含有第二配位复合物的第二溶液;
c)将所述外壳分子溶液加入所述第二溶液,形成含有所述药物复合物的第三溶液。
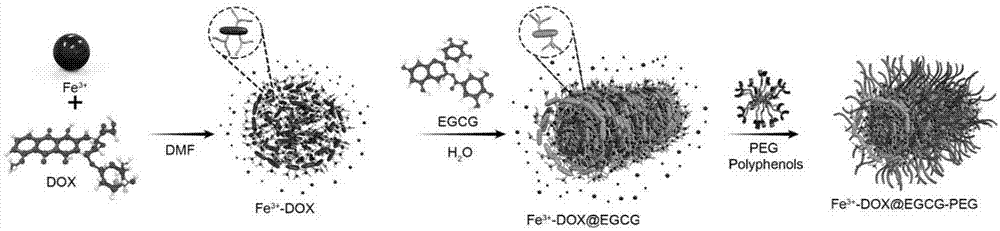
选用具有酚羟基结构的小分子抗癌药物阿霉素(dox)、茶多酚如表没食子儿茶素没食子酸酯(egcg)和5-羟基多巴胺共价偶联可溶性骨架高分子peg(5-羟基多巴胺-peg),dox、egcg和5-羟基多巴胺-peg结构中的多个邻位酚羟基可作为一种多基配位体与金属离子fe3+发生配位反应,在fe3+过量的条件下,可逐步由里向外从而形成fe3+-dox@egcg-peg(dfepnps)稳定的纳米粒子配位化合物。其中fe3+-dox作为纳米粒子的内核,fe3+-egcg为纳米粒子的表面,5-羟基多巴胺-peg/5-羟基多巴胺-peg-荧光染料为纳米粒子的外围。
利用金属-多酚依次配位的原理,在fe3+过量的条件下,首先含有酚羟基的小分子药物dox或多酚共价偶联的小分子药与fe3+配位形成纳米药物复合物的内核,其次利用过量fe3+与茶多酚如egcg配位构成纳米药物复合物的表面,最后多酚-可溶性骨架高分子或是多酚-可溶性骨架高分子-荧光染料共价偶联化合物与fe3+配位组成药物复合物的外围,从而构成纳米粒子药物复合物(dfepnps)。所述纳米药物复合物为网状结构,所述小分子药物化合物位于所述网状结构内核,所述表没食子儿茶素没食子酸酯egcg位于药物复合物的表面,所述多酚-peg/多酚-peg-荧光分子为药物复合物的外围分子如图1所示。
在一些实施方式中,所述制备方法还包括如下步骤:
d)纯化所述第三溶液,得到所述药物复合物;
可选地,所述小分子药物为疏水性阿霉素,
可选地,所述疏水性阿霉素的制备步骤为:以二甲基甲酰胺作为盐酸阿霉素的溶剂,浓度为1-10mg/ml,优选3mg/ml,滴加三氟乙胺,盐酸阿霉素与三氟乙胺摩尔比为1:520优选1:10,反应过夜,除去盐酸,制备疏水性阿霉素;
可选地,所述小分子药物溶液的溶剂为二甲基甲酰胺或dmso;
可选地,所述小分子药物溶液为所述小分子药物的二甲基甲酰胺溶液,浓度为1-10mg/ml,优选3mg/ml;
可选地,所述具有配位能力的可溶性金属离子的金属盐溶液为卤化物水溶液,更优选,氯化物水溶液,浓度为1-10mg/ml,优选3mg/ml。
可选地,所述具有配位能力的可溶性金属离子的金属盐溶液为0.554mmol/l,0.925mmol/l和1.479mmol/l的氯化铁溶液;
可选地,所述茶多酚溶液为水溶液,浓度为0.1-5mg/ml,优选1mg/ml;
可选地,所述聚乙二醇分子为两头端羧基聚乙二醇分子或一端羧基一端氨基聚乙二醇分子,更优先地,聚乙二醇分子羧基为nhs,所述聚乙二醇分子的分子量为2000-200000,优选地100000-200000,更优选地,150000或200000,
可选地,所述外围分子的制备步骤为:利用所述聚乙二醇-cooh与所述多酚在edc/nhs或ddc/nhs催化下反应制备聚乙二醇-多酚或nh2-聚乙二醇-多酚,更优选地,nh2-聚乙二醇-多酚进一步共价偶联荧光分子;
可选地,所述外壳分子溶液为水溶液,浓度50-500mg/ml,优选150mg/ml;
可选地,在步骤a)中,将所述小分子药物溶液加入所述具有配位能力的金属离子的可溶性金属盐溶液后进行涡旋,可选1-10分钟,优选,2分钟;
可选地,在步骤b)中,将所述茶多酚溶液加入所述第一溶液后进行涡旋,可选1-10分钟,优选,2分钟;
可选地,在步骤c)中,将所述外壳分子溶液加入所述第二溶液后进行涡旋,可选1-10分钟,优选,2分钟;
可选地,所述小分子药物溶液,所述具有配位能力的金属离子的可溶性金属盐溶液,所述茶多酚溶液与所述外壳分子溶液的用量体积比为:100:20-300:50-500:10-100,优选100:40-200:100-300:15-50,更优选100:60-160:200:40;
可选地,纯化所述第三混合物的步骤为将所述第三混合物放入透析袋中在水溶液中透析;
可选地,将所述第三溶液置于分子量为5000-10000的透析袋中透析过夜,其转子转速为200-500rpm/分钟,更优选,透析袋分子量为8000,转速为300rpm/分钟。
本发明第四方面提供了一种肿瘤细胞或肿瘤组织的被动定向药物递送系统,所述药物递送系统是本发明第一方面所述的药物复合物或根据本发明第三方面所述的制备方法制备的药物复合物。
本发明第五方面提供了一种联合用药系统,所述联合用药系统是本发明第一方面所述的药物复合物或根据本发明第三方面所述的制备方法制备的药物复合物。
本发明第六方面提供了本发明第一方面所述的药物复合物或本发明第二方面所述的药物运载体或如本发明第三方面所述的制备方法或如根据本发明第三方面所述的制备方法所制备的药物复合物在药物递送,药物体内成像与跟踪,监测药物分布及代谢,制备肿瘤诊断制剂,或制备精准治疗制剂中的应用或用途。
本发明第七方面提供了一种试剂盒,所述试剂盒中含有本发明第一方面所述的药物复合物或本发明第二方面所述的药物运载体或如本发明第三方面所述的制备方法所制备的药物复合物。
附图说明
图1为载药系统的合成步骤示意图。
图2为用三种不同摩尔浓度的三氯化铁制备不同尺寸的egcg-fe@dox自组装纳米药物载体系统(dfepnps)示意图。
图3为dfep-anps在u87mg肿瘤细胞的摄取情况照片。
图4为dfep-bnps在u87mg肿瘤细胞的摄取情况照片。
图5为dfep-anps和dfep-bnps在u87mg肿瘤模型鼠中组织分布。
图6为dfep-anps和dfep-bnps对u87mg肿瘤模型鼠的抑制率。
具体实施方式
为了更好的解释本发明的技术方案,下面结合附图详细介绍本发明的实施例。以下实施例用于进一步说明本发明,但不应理解为对本发明的固定或限制。若未特别指明,实施例中所用的技术特征可以替换为具有在不背离发明构思前提下等同或相似功能或效果的其他本领域已知的技术特征。
目前传统脂质体剂型包裹egcg和小分子药物药物传递系统存在药物传递系统不稳定,不能准确将小分子药物输送到病灶部位发挥药效,同时其药物载体所包裹药物量较少,降低其药效等缺点。而egcg-peg两亲性嵌段聚合物自组成包裹dox的纳米胶束载药系统也存在在体内分解成大分子两亲性嵌段聚合物egcg-peg,很难释放自由egcg,从而降低egcg实施抗氧化和抗癌等功能,不能实现egcg+dox协同用药和提高dox的药效的目的。
针对目前所存在如何利用egcg提高小分子药物药效的问题,本发明提出利用金属-多酚配位化学合成技术制备含有多酚小分子药物阿霉素dox-fe3+/小分子药物-多酚作为内核,egcg-fe3+为纳米粒子载药系统的表面,peg-多酚/peg-荧光染料-多酚-fe3+作为纳米粒子载药系统外围,自组装新型纳米粒子载药系统,以fe3+-多酚配位为纳米粒子诊疗剂骨架,共担载dox/小分子药物、荧光染料和egcg,从而得到新型纳米粒子载药系统dfepnps。利用“荧光染料→监测药物分布及肿瘤位置”、“egcg和fe3+清除氧自由基ros,egcg抑制cbr1蛋白”和“增强小分子药物/化疗药物疗效”相结合,杀死肿瘤细胞,实现分子影像与化疗的精准治疗。
1.egcg与三价铁配位所形成的配位化合物,可稳定egcg结构,提高egcg在体内实施抗氧化和抗肿瘤的功能。
2.利用egcg清除氧自由基/抑制cbr1蛋白的功能,阻断dox转化成doxol,从而实现egcg+dox的协同用药,提高抗肿瘤小分子化疗药物dox的药效。
3.利用peg-多酚-荧光染料/peg-多酚进一步修饰纳米粒子载药系统,在增加纳米粒子载药系统的生物相容性的同时,可实现监测药物在体内的代谢途径。
实施例1:载药系统的制备方法
参见图1,载药系统的制备步骤如下:
1.以dmf(二甲基甲酰胺)作为盐酸阿霉素dox-hcl的溶剂,浓度为3mg/ml,滴加三氟乙胺,盐酸阿霉素与三氟乙胺摩尔比为1:10,反应过夜,除去盐酸,制备疏水性阿霉素dmf溶液(过量的三氟乙胺会挥发掉)。
2.利用多巴胺的氨基与两端羧基peg-cooh(分子量0.5-20万)羧基在edc/nhs或dcc/nhs催化下进行酰胺反应,生成peg-多巴胺,进一步peg-多巴胺上peg另一端羧基/氨基与荧光染料的氨基/羧基在edc/nhs或dcc/nhs催化下进行酰胺反应,制备浓度为100-200mg/ml荧光染料-peg-多巴胺水溶液,如制备alexafluor488-peg-多巴胺水溶液。
3.配制浓度为3mg/ml的三氯化铁水溶液,浓度为1mg/ml的egcg水溶液。
4.取50μl以二甲基甲酰胺为溶剂的浓度为3mg/ml疏水性dox,并加入30-80μl的浓度为3mg/ml的三氯化铁溶液,涡旋2分钟(2000次/分)。
5.在4中缓慢滴加100μl浓度为1mg/ml的egcg水溶液并涡旋2分钟(2000次/分)。
6.在5中缓慢滴加20μl浓度为150mg/mlpeg-荧光染料-多巴胺水溶液如alexafluor488-peg-多巴胺,并涡旋2分钟(2000次/分),形成。
7.在分子量为8000的透析袋中在水溶液中透析过夜,其转子转速为300次/分钟。
本专利发明主要是利用金属-多酚配位形成纳米粒子,第一步fe3+与dox结构上三个结合位点形成配位组成fe3+-dox化合物如图1第一步,红色代表fe3+-dox化合物,左上角为dox结合位点示意图,第二步过量的fe3+与egcg有二个结合位点形成配位,进一步形成fe3+-dox@egcg,第三步,过量的铁与peg-多巴胺/荧光染料-peg-多巴胺配位形成fe3+-dox@egcg-peg(dfepnps)纳米粒子载药系统。
目前临床上广泛使用的小分子和高分子药物存在各自缺点:(1)小分子药物通过口服或注射给药,短时间内体内药物浓度远超过实际需求量,且缺乏进入人体的选择性;新陈代谢快、半衰期短、体内浓度很快降低而影响疗效,故需要大剂量给药,而过高的药物浓度又会增强药物的副作用;(2)生物高分子药物在体内易被酶降解或失活,生物半衰期短,需重复给药;还受到如免疫系统、组织、细胞膜等的限制,多数不易通过这些生物屏障,因而高分子药物的生物利用度较低。纳米粒子载药系统由于颗粒度极小,表面积非常大,药物可高密度地负载在纳米载体中,从而增强局部的浓度,提高药物的利用率。此外,纳米粒子载体系统跨细胞膜的作用机制不同于游离药物,主要以内吞形式进入细胞,不受细胞膜上相应泵的作用,因此可以增加药物对生物膜的透过性,有利于药物透皮吸收与细胞内药效发挥。另外纳米粒子载药系统可实现被动靶向途策略:具有的纳米级粒径的结构具有由其粒径决定的“粒径效应”使其具有一些特殊的功能,如可以增加这些与细胞膜以及蛋白质的相互作用而肿瘤组织中新生的血管内皮细胞不完整,没有完整的内皮结构,缺乏有效的淋巴回流,这样不仅会增加了对这些的通透性,而且易使它们在肿瘤内部滞留,使纳米药物在肿瘤组织的分布多于正常组织,这种现象被称为epr效应目前被动靶向策略是研究的主要方向。
实施例2:载药系统制备的优化与表征
采用实施例1的方法,分别采用三个不同摩尔浓度的三氯化铁0.554mmol/l,0.925mmol/l和1.479mmol/l,制备了三种载药系统,并命名为dfep-a,dfep-b和dfep-c,结果如图2所示,其中上部为显微照片,下部为直径分布情况。
从图2中可以看出随着浓度提高,粒径变大。三种纳米载药系统的粒径分别为:dfep-a:77.4±13nm,dfep-b:126±24nm,dfep-c:296±19.7nm。
实施例3:载药系统在肿瘤细胞中的摄取
材料:人胶质瘤细胞u87mg肿瘤细胞,购买于中科院细胞库。
dfep-anps、dfep-bnps来自本发明实施例2。
试验步骤:将2*105u87mg细胞铺于共聚焦4孔板,置于37℃、5%co2、90%湿度的恒温培养箱中培养过夜。将100μl0.554mmol/l的dfep-a,0.925mmol/ldfep-b分别加入4孔板u87mg细胞中,分别孵育1、4和8小时后弃去培养液,加入1mlpbs清洗,重复2-3次,用4%甲醛固定液固定20min,滴加dapi染液染细胞核5min,盖上盖玻片,在激光共聚焦扫描显微镜,放大倍数为200倍下观察,可清楚看到由dfep-a和dfep-b所孵育的肿瘤细胞u87mg的绿色荧光和红色荧光亮度随着时间增加而增强,实验结果证明dfepnps利用肿瘤细胞旺盛的新陈代谢摄取dfepnps且滞留在肿瘤细胞中,说明dfepnps可被动靶向肿瘤细胞。实验结果如图3和图4所示。,图中所示蓝色荧光为dapi(试剂盒染色),绿色为染料(alexafluor488-peg-多巴胺与fe3+配位形成纳米粒子载体的外围),红色是dox的荧光。
实施例4:应用试验
将0.2ml5mg/mldfep-anps和dfep-bnps水溶液的纳米载药系统分别尾静脉注射到不同u87mg肿瘤模型小鼠体内,24h后将小鼠处死并将其多种组织取出,在近红外成像仪(济南显微智能科技有限公司,yc-1)下观察载药系统在体内组织中的分布,成像结果参见图5。
从图5中可以看到dfep-a和dfep-b在注射24小时后,多集中在肿瘤部位,其肿瘤部位的荧光强度明显高于其他组织,用近红外roi软件分析其肿瘤部位荧光强度相对值为31±1.8和28±2.6,而对照组游离dox在注射24小时后在肿瘤部位荧光强度为5±0.68,实验结果证明dfepnps载药系统可被动靶向肿瘤组织且看可滞留在肿瘤部位。
实施例5:药效试验
将u87mg肿瘤模型的裸鼠随机分为六组每组5只:(1)对照组,(2)游离dox(200μl/小鼠);(3)游离egcg(200μl/mouse);(4)游离dox+游离egcg(200μl/小鼠);(5)dfep-a(200μl/小鼠);(6)dfep-b(200μl/小鼠).用u87mg肿瘤模型,隔天注射尾静脉共三次,观察肿瘤鼠体重,肿瘤大小的变化,并计算死亡率。实验结果显示dfep-a肿瘤抑制率为75%,dfep-bnps肿瘤抑制率为69.75%,高于其他几组的肿瘤抑制率dox(40.08%),dox+egcg(44.13%)和egcg(18.02%),如图6所示。肿瘤抑制率=w(对照组)-w(治疗组)/w(对照组)x100%,w为肿瘤重量。
由此可知,本发明的载药系统中,dox与egcg在体内发生了协同作用,可以良好地用于抑制肿瘤。
以上各个实施例只是用于进一步说明本发明,并不是用来限制本发明的保护范围,凡是基于本发明的构思所作出的等同变换及对本发明的各个技术方案显而易见的改进,均落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!