一种癌症相关SNP、基因、miRNA和蛋白质相互作用的多层网络模型构建方法和应用与流程
本发明属于癌症的生物信息学分析技术领域,尤其涉及一种癌症相关snp、基因、mirna和蛋白质相互作用的多层网络模型构建方法和应用。
背景技术:
癌症是严重威胁人类生存和社会发展的重大疾病和严重的公共卫生问题之一,癌症控制已成为世界各国政府的卫生战略重点。随着基因测序技术日益成熟和对基因组学的深入研究,生物信息科学家通过全基因组关联分析gwas技术挖掘鉴定了多个与复杂疾病有关的遗传变异。gwas技术依托单个snp位点为遗传标志,对全基因组进行对照或相关性的探究,以期发现显著影响表型的snp位点。gwas在探寻基因与疾病关联方面取得了巨大成效。自首例年龄相关性视网膜黄斑变性全基因组研究之后,生物科学家陆续展开了一系列其他疾病的全基因组分析,并确定了易感区域的致病基因和snp变异情况。当前gwas分析的研究已经持续了10余年,发现了一批与各类复杂疾病有关的遗传变异。在方法研究上主要集中在统计学方法、生物学实验验证、生物学信号通路等。虽然单个snp的统计检验分别能够获得主要遗传效应,对于识别疾病风险或复杂相互作用的snp是必要的。
疾病的复杂性状受到多基因的微小影响所导致的,snp位点通过作用于基因的表达量间接影响表型,所以在分析遗传变异时需要考虑基因编码区域和调控区域。由于大部分疾病属于多基因疾病,同时相邻的遗传变异可能存在连锁不平衡关系,确定基因型与表型的因果关系存在一些困难。gwas方法着重于测试疾病与单个snp在基因组上的关联,仅报道具有显著统计学意义的snp。因此,gwas不足以检测具有小边际效应的遗传变异体,无法获得snp位点-基因表达数据-mirna数据-蛋白质数据之间的相互关联关系。
技术实现要素:
有鉴于此,本发明的目的在于提供一种癌症相关snp、基因、mirna和蛋白质相互作用的多层网络的研究方法和应用。
为了实现上述目的,本发明提供了以下技术方案:一种癌症相关snp、基因、mirna和蛋白质相互作用的多层网络模型构建方法,包括以下步骤:1)将癌症组织样本与正常组织样本的全基因组数据进行全基因组关联分析获得snp位点数据,并从snp位点数据中筛选差异显著的snp位点数据;2)用xgboost法分别分析癌症组织样本与正常组织样本的基因表达数据、mirna数据和蛋白质数据获得差异显著的基因表达数据、mirna数据和蛋白质数据;3)以步骤2)获得的差异显著的基因表达数据、mirna数据和蛋白质数据分别为一层,用最大信息系数法分别分析各层层内数据的关联关系以及任意两层间数据的关联关系;4)以步骤1)中获得的差异显著的snp位点数据为一层,将所述差异显著的snp位点数据与步骤3)中的差异显著的基因表达数据层进行关联获得snp位点与基因表达数据层之间的关联关系;从而获得由snp位点-基因-mirna-蛋白质之间的多层网络关联关系;步骤3)中所述关联关系的确定为计算关联系数,若关联系数mic≥0.5则确定关联关系;步骤4)中所述差异显著的snp位点数据与步骤3)中差异显著的基因表达数据层关联关系的确定为当所述差异显著的snp位点位于差异显著的基因内部时,确定关联关系;步骤1)与步骤2)之间无时间顺序限定。
优选的,所述癌症组织样本与正常组织样本的全基因组数据为经过主成分分析筛选的基因背景相同的全基因组数据。
优选的,所述筛选差异显著的snp位点数据的方法为χ2检验、fisher检验分析法、逻辑回归法和xgboost算法中的一种。
优选的,步骤3)中当所述关联系数mic≥0.6时,确定关联关系。
优选的,步骤2)中所述差异显著的基因表达数据、mirna数据和蛋白质数据为xgboost法计算重要度排序前2~8的数据。
优选的,所述癌症包括乳腺癌。
本发明提供了所述多层网络模型构建方法获得的多层网络模型在筛选肿瘤标志物中的应用。
优选的,选择上述方法获得的多层网络模型中,能够自snp位点-基因-mirna-蛋白质构成一条连通的通路中的snp位点、基因、mirna或蛋白质作为肿瘤标志物。
本发明提供的癌症相关snp、基因、mirna和蛋白质相互作用的多层网络的模型构建方法,通过全基因组关联分析gwas获得癌症组织样本和正常组织样本差异显著的snp位点数据,并通过gxboost法获得癌症组织样本和正常组织样本差异显著的基因表达数据、mirna数据和蛋白质数据,然后用最大信息系数法mic分析差异显著的基因表达数据、mirna数据和蛋白质数据层内和层间两两之间的关联关系,获得由snp位点数据-基因表达数据-mirna数据-蛋白质数据之间的多层网络关联关系,构建出由snp位点-基因-mirna-蛋白质的病变通路。本发明所述的方法从snp位点、基因、mirna和蛋白质四个层面分析了癌症组织样本和正常组织样本间的差异,并从中获得对癌症病变影响较大的因素,同时确定由snp位点-基因-mirna-蛋白质的病变通路,进而分析出差异显著的snp位点是如何影响基因、mirna和蛋白质的表达量,从而影响病变。在此基础上,可以通过阻断上述病变通路而达到抑制癌变的目的。
附图说明
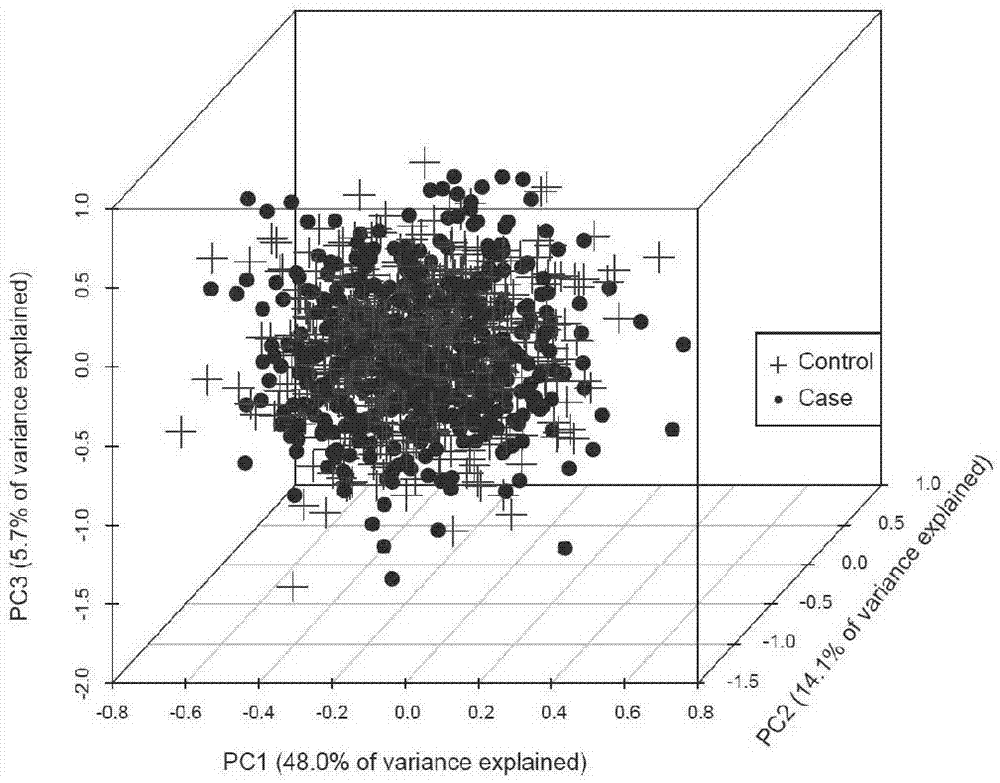
图1乳腺癌人群case-control主成分三维分层图;
图2乳腺癌snp位点曼哈顿图;
图3基因表达数据错误率及逻辑损失值迭代图;
图4基因分类识别重要度排名图;
图5mirna表达数据错误率及逻辑损失值迭代图;
图6mirna分类识别重要度排名图;
图7蛋白质表达数据错误率及逻辑损失值迭代图;
图8蛋白质分类识别重要度排名图;
图9基于mic基因表达量网络图;
图10基于micmirna表达量网络图;
图11基于mic蛋白质表达量网络图;
图12生物分子标志物互作多层网络示意图;
图13肿瘤组织snp、基因、mirna、蛋白质多层网络图;
图14正常组织snp、基因、mirna、蛋白质多层网络图;
图15肿瘤组织snp、基因、蛋白质多层网络子图;
图16正常组织snp、基因、蛋白质多层网络子图;
图17基因pfkfb3在正常组织及肿瘤组织中表达量情况;
图18蛋白质14.3.3_zeta在正常组织及肿瘤组织中表达量情况;
图19蛋白质bax在正常组织及肿瘤组织中表达量情况;
图20基因tmem132e在正常组织及肿瘤组织中表达量情况;
图21蛋白质msh6在正常组织及肿瘤组织中表达量情况;
图22基因prc1在正常组织及肿瘤组织中表达量情况;
图23基因ebf1在正常组织及肿瘤组织中表达量情况;
图24基因tgfbr2在正常组织及肿瘤组织中表达量情况;
图25基因tmem132c在正常组织及肿瘤组织中表达量情况;
图26蛋白质ku80在正常组织及肿瘤组织中表达量情况;
图27蛋白质pi3k.p85在正常组织及肿瘤组织中表达量情况;
图28蛋白质s6_ps240_s244在正常组织及肿瘤组织中表达量情况。
具体实施方式
本发明提供了一种癌症相关snp、基因、mirna和蛋白质相互作用的多层网络研究方法,包括以下步骤:1)将癌症组织样本与正常组织样本的全基因组数据进行全基因组关联分析获得snp位点数据,并从snp位点数据中筛选差异显著的snp位点数据;2)用xgboost法分别分析癌症组织样本与正常组织样本的基因表达数据、mirna数据和蛋白质数据获得差异显著的基因表达数据、mirna数据和蛋白质数据;3)以步骤2)获得的差异显著的基因表达数据、mirna数据和蛋白质数据分别为一层,用最大信息系数法分别分析各层层内数据的关联关系以及任意两层间数据的关联关系;4)以步骤1)中获得的差异显著的snp位点数据为一层,将所述差异显著的snp位点数据与步骤3)中的差异显著的基因表达数据层进行关联获得snp位点与基因表达数据层之间的关联关系;从而获得由snp位点-基因-mirna-蛋白质之间的多层网络关联关系;步骤3)中所述关联关系的确定为计算关联系数,若关联系数mic≥0.5则确定关联关系;步骤4)中所述差异显著的snp位点数据与步骤3)中差异显著的基因表达数据层关联关系的确定为当所述差异显著的snp位点位于差异显著的基因内部时,确定关联关系;步骤1)与步骤2)之间无时间顺序限定。
本发明将癌症组织样本与正常组织样本的全基因组数据分别进行全基因组关联分析gwas获得差异显著的snp位点数据。本发明对所述癌症组织样本的种类没有特殊限定,常规癌症组织样本均可,在本发明具体实施过程中,以乳腺癌为例进行。在本发明中,所述乳腺癌的全基因组数据(vcf)来自剑桥大学乳腺癌实验室数据库,所下载的数据共有1011例,其中包括640名乳腺癌患者和371名健康对照乳腺癌。本发明在获得所述乳腺癌全基因组数据后,将癌症组织样本与正常组织样本的全基因组数据进行全基因组关联分析gwas获得snp位点数据。本发明为保证基因背景相同,对所获得的snp位点数据进行主成分分析,筛选基因背景相同的数据,过滤基因背景不同的数据;本发明所述基因背景相同主要是指乳腺癌目标人群分层分类相同,排除种族、地域等因素的影响。本发明对所述主成分分析的方法没有特殊限定采用本领域常规的组成分分析方法即可。在发明具体实施过程中,主成分分析包括与以下步骤和参数:原始变量涉及多维指标,分别用p表示,p个snp位点构成p维随机变量x=(x1,x2,...,xp)′。设随机变量x均值为μ,协方差矩阵为σ。将x1,x2,...,xpx线性转化为新的综合变量,用y表示。即:
为取得更理想的效果,在下列约束下保证yi=μ′ix的方差尽量大且各yi之间线性无关:
(1)μ′iμi=1(i=1,2,...,p);
(2)yi与yj相互无关μ′iμi=1(i≠j;i,j=1,2,...,p);
(3)y1是x1,x2,...,xp符合条件(1)情形下方差最大者;…,yp是与y1,y2,...,yp-1均线性无关的方差p次大者。
通过计算样本随机变量x=(x1,x2,...,xp)′的协方差矩阵σ,进而计算特征值和特征向量。
设矩阵α′=α,将a的特征根λ1,λ2,...,λn值从大到小排列,设λ1>λ2>...>λn,γ1,γ2,...,γn为矩阵a的特征根λ1,λ2,...,λn所对应的标准正交向量,则对任意向量x,有:
则第i个主成分:
yi=γ1ix1+γ2ix2+…+γpixp,i=1,2,...,p(3-3)
此时有:
称
本发明用xgboost法分别分析癌症组织样本与正常组织样本的基因表达数据、mirna数据和蛋白质数据获得差异显著的基因表达数据、mirna数据和蛋白质数据。在本发明中,所述基因表达数据、mirna数据和蛋白质数据来源于tcga癌症数据库。在本发明中所述基因表达数据为rna-sequencing数据,主要包括数据库中mrna微阵列、外显子芯片或rna测序基因表达;所述mirna数据为mirna-seqcing数据,主要包括数据库中的microrna微阵列芯片或mirna测序表达数据;本发明中所述蛋白质数据主要包括数据库中reverse-phase蛋白质阵列测序得到的蛋白质表达数据。在本发明中采用xgboost法分别分析癌症组织样本与正常组织样本的上述数据获得差异显著的基因表达数据、mirna数据和蛋白质数据。
在本发明中,所述xgboost法监督学习模型指的是待训练的样本数据集的决策函数或条件概率。假设γ表示模型空间的决策函数集合,即γ={f|y=f(x)}或γ={p|p(y|x)}。其中,x和y。此处γ与一个或多个参数向量有关的函数簇:γ={f|y=fθ(x),θ∈rn}或γ={p|pθ(y|x),θ∈rn},参数向量θ属于n维欧氏空间rn。
xgboost算法模型每次是在保留原模型的基础上,将一个新函数加入模型。选择加入新函数是为了尽可能降低损失目标函数。
使用泰勒级数展开的方法近似表示目标函数:
构建xgboost算法损失函数:有监督学习在假设空间γ中选择模型f作为最佳判别函数,通过慢性乙肝的snp位点或乳腺癌肿瘤标志物的数据,由f(x)判断分类结果。经过假设空间、损失函数和乳腺癌训练数据集,可以唯一确定经验损失函数。一般认为经验损失最小的决策模型即为该数据集的最佳模
型。即:
为了防止过拟合的情况出现,往往基于经验损失函数加入拉格朗日算子,即:
在本发明中,以差异显著的基因表达数据、mirna数据和蛋白质数据分别为一层,用最大信息系数法mic分析差异显著的基因表达数据、mirna数据和蛋白质数据层内和层间两两之间的关联关系。在本发明具体实施过程中,定义多层网络的表达式m=(g,c),其中g={gα;α∈{1,2,...,m}}是一组单层复杂网络图gα=(xα,eα)的集中表现形式,单层复杂网络图称为m的层。同时
层间eαβ的邻接矩阵表示为:
m的投影网络表示为:
proj(m)=(xm,em)(5-8)
其中:
识别发挥中心结构角色的节点的问题是复杂网络研究重点之一。在单层复杂网络中,有许多参数可以衡量每个节点的结构性质:节点度数,聚集系数等。接下来将讨论将这些指标扩展到多层网络。其中一个主要的指标是每个节点的度:节点拥有的连接边越多,它就越有存在的意义。多层网络m=(g,c)的节点i∈x的度用向量表示
更复杂的方法是考虑不同层次的网络的不同程度的重要性,并将这些信息包含在层间相互影响的矩阵中。如果考虑层内三个基因i、j和k,i和j之间以及i和k之间存在互作关系,则基因i的聚类系数表示j和k关联的可能性。显然局部聚类系数是传递性的度量,可以解释为节点邻域的密度。对于每个节点i∈x,ni表示投影网络proj(m)中的i的所有邻居的集合。对于每个α∈{1,2,...,m},满足:nα(i)=ni∩xα和
相似的,定义
本发明中,用最大信息系数法mic分析差异显著的基因表达数据、mirna数据和蛋白质数据层内和层间两两之间的关联关系,所述关联关系的确定为计算关联系数,若关联系数mic≥0.6则确定关联关系。
本发明利用所述差异显著的snp数据、差异显著的基因表达数据、mirna数据和蛋白质数据层内和层间两两之间的关联关系绘制多层网络图。由于snp位点是位于基因内部或基因与基因之间,snp位点数据与基因表达数据是被包含的关系,因此snp位点数据与基因表达数据的关联是单向的,并且snp位点数据与mirna数据和蛋白质数据之间没有关联。在本发明中,以snp位点数据、基因表达数据、mirna数据和蛋白质数据分别为一层,以最大新信息系数法mic分别计算基因表达数据、mirna数据和蛋白质数据层内和层间的两两关联关系,若关联系数mic≥0.6确定关联关系,在两个节点之间连边。
本发明中,若snp位点数据、基因表达数据、mirna数据和蛋白质数据的连边能够连成一条通路,则获得由snp位点-基因表达数据-mirna数据-蛋白质数据之间的多层网络关联关系,构建出由snp位点-基因-mirna-蛋白质的病变通路。
本发明还提供了多层网络模型构建方法获得的多层网络模型在筛选肿瘤标志物中的应用。本发明中,所述应用为选择上述方法获得的多层网络模型中,能够自snp位点-基因-mirna-蛋白质构成一条连通的通路中的snp位点、基因、mirna或蛋白质作为肿瘤标志物。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
乳腺癌的全基因组vcf数据来自剑桥大学乳腺癌实验室数据库,所下载的数据共有1011例,其中包括640名乳腺癌患者和371名健康对照。用于构建网络的乳腺癌表达水平数据来自tcga癌症数据库。下载乳腺癌病人rna-sequencing(下称基因表达量数据)、mirnaseq(下称mirna表达量数据)和蛋白质表达量数据。具体说明见表1.
表1肿瘤标志物数据说明表
基因表达量数据量1214×24991(其中肿瘤组织样本数为1101,正常组织样本数为113;24991为基因种类);mirna表达数据量为1200×1882(其中肿瘤组织样本数为1096,正常组织样本数为104;1882为mirna种类);蛋白质表达数据量为925×285(其中肿瘤组织样本数为882,正常组织样本数为43;285为蛋白质种类)。下载的mirna表达水平数据中较多mirna表达率低,蛋白质表达水平数据中较多缺失数据,删除mirna表达水平低于5%的变量和蛋白质缺失数据,另外由于蛋白质表达数据变量间的数量级差别较大,考虑对其做z-score标准化处理,最终用于分析的mirna和蛋白质表达量数据维度为1200×320和925×147。
在对乳腺癌进行全基因组关联分析前,为了研究人口分层的混杂效应,采用主成分分析(principalcomponentsanalysis,pca)对多元数据关联结构进行分类和排序。
pca方法被用于验证人群分层、检测人群亚结构等方面。pca通过保留较多样本信息,通过降维方法将多变量变换为几个有代表性的综合变量的多元统计方法。每个主成分是样本变量的线性组合(综合变量),同时各个主成分之间正交。
原始变量涉及多维指标,分别用表示,p个snp位点构成p维随机变量x=(x1,x2,...,xp)′。设随机变量x均值为μ,协方差矩阵为σ。将x1,x2,...,xpx线性转化为新的综合变量,用y表示。即:
为取得更理想的效果,在下列约束下保证yi=μ′ix的方差尽量大且各yi之间线性无关:
(1)μ′iμi=1(i=1,2,...,p);
(2)yi与yj相互无关μ′iμi=1(i≠j;i,j=1,2,...,p);
(3)y1是x1,x2,...,xp符合条件(1)情形下方差最大者;…,yp是与y1,y2,...,yp-1均线性无关的方差p次大者。
通过计算样本随机变量x=(x1,x2,...,xp)′的协方差矩阵σ,进而计算特征值和特征向量。
设矩阵α′=α,将a的特征根λ1,λ2,...,λn值从大到小排列,设λ1>λ2>...>λn,γ1,γ2,...,γn为矩阵a的特征根λ1,λ2,...,λn所对应的标准正交向量,则对任意向量x,有:
则第i个主成分:
yi=γ1ix1+γ2ix2+…+γpixp,i=1,2,...,p(3-3)
此时有:
称
应用fisher精确检验对慢性乙肝和乳腺癌的全基因组数据进行修正调整。
fisher精确检验是一种基于超几何分布的检验方法,作用于离散变量,用来检验列联表分类单元出现是否有显著差别,见表2。根据每个位点的频数计算统计量及超几何分布下我们观测列联表的p(x)信息。
表2基因型与表型数据统计表
fisher精确检验有两种基于样本大小的检验方法:
(1)样本量少于20,查询统计表。
(2)样本量足够大时,使用标准正态分布统计量进行检验。
公式(3-7)中
枚举相同行和和及列和的矩阵集,所有小于超几何分布概率的矩阵的概率之和即为p值[38,39]。如下面描述所示:有一个snp位点的r×c的列联表矩阵x,定义i行j列的元素xij,第i行的和
公式(3-8)中
矩阵x的精确p值可以定义为:p=∑y∈γp(y),其中:p(y)≤p(x)
采用fisher精确检验对乳腺癌的全基因组数据进行显著性分析,结果见表3。同时根据p值信息,绘制关于乳腺癌的曼哈顿图,结果见图2。
表3乳腺癌局部显著性p值信息表
表3(续表)
表3展示了部分乳腺癌的snp显著位点信息,根据fisher精确检验的p值可以看出1、16、10、5、3等号染色体上的某些snp位点较为显著,p≤10-10的位点数有6个,属于较为显著。其中位于lmod1基因上的rs2819348位点最为显著,p值达到1.84×10-11。图2展示了乳腺癌各snp的p值情况,可以看出chr1,chr2,chr3,chr5,chr6,chr10和chr16染色体上的某些snp位点较为显著,同时可直观的看出显著snp位点所在的基因。
显著snp位点所在基因功能分析
根据表3中展示的是统计学意义上显著的snp位点,结合已发表的文献文献查看snp位点所在基因的功能,从文献角度验证筛选的位点的可靠性。
lmod1:通过谷歌学术和ncbi数据库对lmod1基因进行文献检索,目前在乳腺癌的研究上没有关于lmod1基因与之相关的报道。miller等在冠状动脉疾病研究中发现lmod1基因与冠状动脉有特异性结合和表达。halim等在巨囊虫微肠道低通气综合征研究中表明lmod1和mylk中的变体影响蛋白质表达,并采用体外数据证实变体对平滑肌收缩力受损的致病性。
cdh13:钙粘蛋白13(cdh13)基因属于钙粘蛋白家族。xu和yang等收集乳腺肿瘤样本数据,发现cdh13启动子甲基化与乳腺癌有显著关联。
pfkfb3:novellasdemunt等发现pfkfb3基因广泛参与细胞增殖,pfkfb3蛋白和基因调控的双重机制运作,以确保乳腺癌细胞中的糖酵解。imbert等研究表明pfkfb3表达在相对于正常乳腺组织的iii期淋巴结转移中最高,并且将人mcf-7乳腺癌细胞暴露于e2导致葡萄糖摄取和糖酵解的快速增加。
ebf1:purrington等通过gwas和富集分析,确定了包含ebf1在内的25个基因座与三阴性乳腺癌有关。基于已知乳腺癌风险变量的多基因风险评分显示,最高和最低值之间的风险差异为4倍。ghoussaini等通过gwas分析方法发现包含ebf1在内的72个基因座与er-乳腺癌相关,但不清楚参与肿瘤发生的机制。
tgfbr2:ma等发现tgfbr2的一个变体在两个研究阶段的参与者之间,与乳腺癌风险具有高度一致和显著相关性,在乳腺癌易感性中起遗传变异的作用。
ccdc170:jiang等发现ccdc170的遗传改变与gwas相关的乳腺癌风险有关,并提供证据表明ccdc170的失调影响癌细胞迁移,可能有助于乳腺癌细胞极性和运动性的标志性变化。
xgboost算法分析乳腺癌癌症组织样本与正常组织样本差异显著的基因表达数据
基因表达水平描述的是通过提取生物组织测量的基因的转录产物mrna-seq在生物组织细胞中的丰富水平,该数据展示了生物组织的基因表达水平的变化程度和基因之间的关联性。研究基因表达水平数据可以挖掘基因功能和基因表达调控的信息通路情况,这在生物信息学的研究上属于难点之一。
基因表达数据包含1214×24991,1214为样本个数(其中肿瘤组织样本为1101例,正常组织样本为113例),24991为基因的个数。由于正负样本存在不平衡的情况,所以考虑采用上取样的方法扩增正样本,将正样本随机抽样扩大10倍以此使得正负样本保持均衡。在本次数据二分类中,将负样本肿瘤组织赋值为0,正样本正常组织赋值为1。乳腺癌的基因表达数据分类情况下的错误率及logisticloss值变化趋势见图3,重要变量排名图见图4.
在图3中可以看出,训练集及测试集的错误率、逻辑损失值、amse等指标在模型训练中逐渐减小,auc逐渐增大,在经过27次迭代之后逐渐趋于稳定。训练集分类错误率为0,逻辑损失值为0.007847,auc值为1,rmse值为0.022062;测试集的分类错误率为0.004545,逻辑损失值为0.016866,auc值为1,rmse值为0.061639。可以看出xgboost算法在基因表达量数据上对于分类识别同样具有很高的准确率。
从图4中可以看出lmod1,tgfbr2,bcl9,tmem132c,col1a1,tcf7l2,pfkfb3,mtx1,cadps,klf4等基因组合对乳腺癌的肿瘤组织与正常组织分类准确率的贡献非常大。
xgboost算法分析乳腺癌癌症组织样本与正常组织样本差异显著的mirna数据
选择的乳腺癌的mirna数据包含1200×320,1200为样本个数(其中肿瘤组织样本为1096例,正常组织样本为104例),320为mirna的个数。由于正负样本存在不平衡的情况,所以考虑采用上取样的方法扩增正样本,仍然选择将正样本随机抽样扩大10倍以此使得正负样本保持均衡。同样将负样本肿瘤组织赋值为0,正样本正常组织赋值为1。乳腺癌的mirna表达数据分类情况下的错误率及logisticloss值变化趋势见图5,重要变量排名图见图6
在图5中可以看出,训练集及测试集的错误率及逻辑损失值在模型训练中逐渐减小,在经过35次迭代之后逐渐趋于稳定。训练集分类错误率为0.000517,逻辑损失值为0.009773;测试集的分类错误率为0.005,逻辑损失值为0.027423。可以看出xgboost算法在mirna表达数变异数量对于分类识别具有很高的准确率。
xgboost算法分析乳腺癌癌症组织样本与正常组织样本差异显著的蛋白质表达数据
选择的乳腺癌的蛋白质表达数据量为925×147,925为样本个数(其中肿瘤组织样本为882例,正常组织样本为43例),147为所要研究的蛋白质数量。由于正负样本存在不平衡的情况,所以考虑采用上取样的方法扩增正样本,仍然选择将正样本随机抽样扩大20倍以此使得正负样本保持均衡。同样将负样本肿瘤样组织赋值为0,正样本正常组织赋值为1。乳腺癌的蛋白质表达数据分类情况下的错误率及logisticloss值变化趋势见图7,重要变量排名图见图8。
在图7中可以看出,训练集及测试集的错误率、逻辑损失值、amse等指标在模型训练中逐渐减小,auc逐渐增大,在经过30次迭代之后逐渐趋于稳定。训练集分类错误率为0,逻辑损失值为0.007403,auc值为1,rmse值为0.012621;测试集的分类错误率为0.011765,逻辑损失值为0.040086,auc值为0.999031,rmse值为0.094079。可以看出xgboost算法在蛋白质表达数变异数量对于分类识别同样具有很高的准确率。
从图8中可以看出bax,gsk3.alpha.beta,e.cadherin,rab11,caveolin.1,collagen_vi,c.myc,pkc.alpha,gapdh,p.cadherin等蛋白质对乳腺癌的肿瘤组织与正常组织分类准确率的贡献非常大。
最大信息系数模型研究
通过基因表达数、mirna和蛋白质表达量数据研究其在乳腺癌病人正常组织和肿瘤组织之间的关系规律。首先定义两个联合随机变量(x,y)特征矩阵的分布规律的性质,用g(k,l)表示。其中x=(x1,x2,...,xm),式中xi代表基因表达水平(或mirna或蛋白质表达水平)x在病人i中的表达量。y=(y1,y2,...,ym),yi代表基因表达水平(或mirna或蛋白质表达水平)y在病人i中的表达量,共m个病人。k,l均为正整数。联合随机变量(x,y)分布在[0,1]×[0,1]的区间中。下面我们定义网格g,使得(x,y)g=(colg(x),rowg(y)),此处colg(x)表示网格g的x轴上的网格列数,rowg(y)表示网格g的y轴上的网格行数。
公式(5-1)中的x,y——基因表达水平(或mirna或蛋白质表达水平);
p(x,y)——联合概率分布;
p(x)p(y)——边际概率分布;
定义5.1m(x,y)表示x,y的群体特征矩阵,
i*((x,y),s,t)=max(x,y)g(5-3)
公式(5-4)中的i(x,y)——x,y的交互信息;
s,t——x轴和y轴分区的数量,s·t<b(n)=n0.6;
n——样本的数量;
mic不依赖于测量数据的假设分布,与之前提到的方法相比能精确的识别更多的函数关系。mic=maxs·t<b(n)m(x,y)s,t∈[0,1]。
基因(或mirna、蛋白质)网络中的节点i的度ki定义为与其他基因(或mirna、蛋白质)节点直接相连的边的数量。所有节点的度的均值记为网络的平均度
聚类系数可刻画基因(或mirna、蛋白质)网络中节点的紧密程度,是指与某基因(或mirna、蛋白质)节点相连的两个基因(或mirna、蛋白质)节点之间相连的概率平均值。用数学定义表示为:
将基因(或mirna、蛋白质)网络的邻接矩阵表示为α,aij=0表示基因(或mirna、蛋白质)节点i和j之间没有边,aij=1表示有边。定义sij=1表示i和j属于同一类别,sij=0表示不在同一类别。在随机网络下,任意两点i和j连接边数的期望值是:
公式(5-5)中的
evv——类别v内部连边数占网络连边数的比例,
av——其中一端连接类别v的边数占比,
根据mic方法,分别计算乳腺癌病人正常组织和肿瘤组织的基因表达水平(或mirna、蛋白质表达水平)的相关强度。选择阈值为≥0.5,当基因表达水平(或mirna、蛋白质表达水平)之间的关系值≥0.5时,确定两个基因(或mirna、蛋白质)有连边。绘制基因、mirna与蛋白质的单层复杂网络图及网络图的基本度量统计,见图9,10,11及表4。其中图9,10,11中左图为肿瘤组织,有图为正常组织。
表4复杂网络基本度量属性表
由网络图9,10,11的连通结构及表4可知,正常组织和肿瘤组织的基因(或mirna、蛋白质)网络结构差异较大,正常组织的基因(或mirna、蛋白质)网络结构比肿瘤网络的结构复杂,说明正常组织中的某些基因(或mirna、蛋白质)功能在肿瘤组织中有所减弱或消失,基因(或mirna、蛋白质)应该在这两种网络结构差异中起到了重要作用。
结合对乳腺癌的肿瘤组织和正常组织分类分析结果,根据基因表达量数据分类效果,lmod1贡献度最大,tgfbr2次之;根据mirna数据的分类结果,hsa.mir.139贡献度最大,hsa.mir.21次之;根据蛋白质表达量分类效果,bax贡献度最大,gsk3.alpha.beta次之。对比肿瘤组织和正常组织网络图贡献度最大的节点度情况,统计结果如表5所示:
表5重要变量在网络中基本度量属性表
对比表4,5中数据可以发现,肿瘤组织的复杂网络结构小于正常组织。但是较为重要的基因(或mirna、蛋白质)节点,在肿瘤组织和正常组织的差异性较小。
考虑使用snp、基因、mirna、蛋白质之间的相互作用多层网络来进一步分析显著性snp。我们引入一个张量多层网络,讨论几个重要的网络描述符包括中心度、聚类系数等,确定其与疾病的真实关联性。多层网络的表达式m=(g,c),其中g={gα;α∈{1,2,...,m}}是一组单层复杂网络图gα=(xα,eα)的集中表现形式,单层复杂网络图称为m的层。同时
层间eαβ的邻接矩阵表示为:
m的投影网络表示为:
proj(m)=(xm,em)(5-8)
其中:
多层网络m=(g,c)的节点i∈x的度用向量表示
更复杂的方法是考虑不同层次的网络的不同程度的重要性,并将这些信息包含在层间相互影响的矩阵中。例如考虑层内三个基因i、j和k,i和j之间以及i和k之间存在互作关系,则基因i的聚类系数表示j和k关联的可能性。显然局部聚类系数是传递性的度量,可以解释为节点邻域的密度。为了将聚类的概念扩展到生物snp、基因、mirna和蛋白质的多层网络,不仅要考虑层内连接,还要考虑层间连接。生物分子标志物互作多层网络示意图见图12。
对于每个节点i∈x,ni表示投影网络proj(m)中的i的所有邻居的集合。对于每个α∈{1,2,...,m},满足:nα(i)=ni∩xα和
相似的,定义
为表示由snp、基因、mirna、蛋白质之间的相互作用多层网络组成的系统,我们允许每个生物分子节点属于任何层的子集。根据乳腺癌snp位点的分析结果,选择p<5×10-10的显著snp位点用于探究方法的可行性及可靠性。通过ncbi数据库查询显著snp所在的基因,结合基因、mirna、蛋白质表达量数据构建多层网络。选择mic≥0.6作为连边阈值,分析其影响的生物网络通路。构建肿瘤组织及正常组织的多层网络图,结果见图13,14。
图13,14第一层为snp位点层,由于未考虑乳腺癌snp位点之间的连锁不平衡情况,所以snp位点是独立的;snp与基因之间的连接取决snp∈gene;当基因与基因(mirna,蛋白质)、mirna与mirna(基因,蛋白质)、蛋白质与蛋白质(基因、mirna)之间的mic≥0.6时,确定连边关系。图中每层节点使用不同的颜色表示,节点大小表示度的情况;层内之间的使用红色线段连接,层间关系使用不同颜色表示。
由肿瘤组织和正常组织多层网络图,统计节点的度及聚类系数等,见表6。比较图13,14与表6,与单层复杂网络相类似,正常组织的网络规模远远大于肿瘤组织的网络规模。合理的解释是:在正常组织中某些关联度较强基因、mirna和蛋白质的表达量在肿瘤组织发生了过表达或下调变化,与其他基因、mirna和蛋白质表达量不能保持线性(或非线性)一致。
表6肿瘤组织和正常组织多层网络中基本度量属性表
为了更清晰明了寻找snp可能影响蛋白质的表达量继而影响表型的通路情况,过滤与snp位点不连通的冗余位点,抽出包含snp、基因、mirna和蛋白质有连通关系的多层子网络,如15,16所示。
图15,16表示的是在肿瘤和正常组织中显著的snp位点与基因节点和蛋白质节点多层网络通路图,由于mirna层连通性较差,因此暂不对其进行比较分析。对图15与16比较发现,正常组织所有的网络结构复杂度及通路结构要远大于肿瘤组织。可能是由于不同组织中的肿瘤标志物拥有特异性结构、功能的原因。结合图15,16及蛋白质分类结果,在正常组织中,重要变量在网络图中显示的度较小,例如bax(importance=0.46,rank=1,degree=1),说明并没有对其他蛋白质的调节起主要影响作用;度较大的节点拥有的重要度排名较低,例如c.kit(importance=0.0047,rank=14,degree=18),但是能通过他们与其他的节点联系起来,在多层复杂网络的通路中对其他节点的功能调控起至关重要的作用。
探讨在正常组织和肿瘤组织中通路子图中保留的snp位点。
(1)共同出现的snp位点(仅有rs11257188)。在肿瘤组织中,rs11257188(p=2.54×10-11,rank=3,属于pfkfb3基因)通过pfkfb3(importance=0.0077,rank=7,degree=2)与14.3.3_zeta(importance=0.002,rank=23)建立了通道关系;在正常组织中,rs11257188节点通过pfkfb3基因与bax(importance=0.46,rank=1)关联在了一起。说明pfkfb3直接作用的蛋白质发生了转移。
根据图17,18,19发现pfkfb3基因表达量在肿瘤组织和正常组织中有明显的区别,癌症组织较正常组织有明显的下调;可见rs11257188位点通过pfkfb3作用于14.3.3_zeta蛋白质,使其表达量在多数乳腺癌病人中有过表达趋势;同时阻断bax蛋白质,使其表达量在多数乳腺癌病人中同样有过表达趋势,从而引起表型改变。
(2)肿瘤组织中出现的snp位点(仅rs11654964)。在肿瘤组织中,rs11654964(p=1.86×10-10,rank=12,介于tmem132e和loc105371740基因间区)通过tmem132e(importance=0.0032,rank=14,degree=2)基因和msh6(importance=0.002,rank=22)蛋白质建立了通道关系。
由图20,21可知:tmem132e和msh6表达量在肿瘤组织和正常组织中并没有太明显的区别,可见rs11654964位点通过tmem132e作用msh6导致乳腺癌发生并不可靠,同时符合在基因间区的snp位点可靠性不强的特征。
(3)正常组织中出现的snp位点。此处从节点较大的基因开始分析。prc1(importance=0.0036,rank=11,degree=14),rs2290203(p=4.59×10-10,rank=21)位点在此基因上;ebf1(importance=0.0024,rank=18,degree=11),rs1432679(p=4.11×10-11,rank=4)在此基因上;tgfbr2(importance=0.082,rank=2,degree=10),rs3773651(p=5.04×10-11,rank=5)在此基因片段上;tmem132c(importance=0.010,rank=4,degree=8),rs11059635(p=3.89×10-10,rank=17)在此基因片段上。首先对此4个基因在乳腺癌病人的正常组织和癌症组织的表达量进行分析。对prc1、ebf1、tgfbr2和tmem132c基因画散点图,见图22至图25。根据表达水平,可知prc1基因表达水平在肿瘤组织中过表达;ebf1、tgfbr2和tmem132c基因表达水平在肿瘤组织中明显偏低。根据文献证实所筛选基因的功能及snp位点是否与乳腺癌有关,见表7。
表7度较大基因位点功能分析表
针对prc1,ebf1,tgfbr2,tmem132c四个基因所连接的蛋白质进行分析,由于所连接的蛋白质较多,现选择ku80(importance=0.002,rank=20,degree=4),pi3k.p85(importance=0.0019,rank=24,degree=3),s6_ps240_s244(importance=0.0012,rank=29,degree=5)进行正常与癌症组织之间的表达量差异分析,见图26至图28。根据图26至28的ku80、pi3k.p85、s6_ps240_s244在肿瘤及正常组织中的表达量有显著差异。其中ku80、pi3k.p85表达量在肿瘤组织中发生量明显的上调,s6_ps240_s244表达量在肿瘤组织中发生量明显的下降。
可以发现根据多层网络的分析方法(在肿瘤组织及正常组织中)筛选出的基因或蛋白质节点连通关系及度有明显的差异,同时筛选的基因或蛋白质的表达量在肿瘤组织及正常组织中亦有显著差异,本发明所述方法可有效地识别全基因组关联分析中显著的候选位点,并根据多层网络的连通关系网络确定其所影响的蛋白质,以此达到改变表型的结果。rs11059635位点通过tmem132c基因作用了一批蛋白质,并起到了调控作用。
本发明以乳腺癌显著致病的肿瘤标志物数据为基础,通过mic算法计算基因-基因,mirna-mirna,蛋白质-蛋白质之间的关系强度,确定阈值构建单层复杂网络,对比肿瘤和正常组织中的节点的度及聚类系数的差异,发现正常组织的网络结构及密度明显大于肿瘤组织,说明正常组织中的某些基因(或mirna、蛋白质)功能在肿瘤组织中有所减弱或消失。为研究snp位点与肿瘤标志物之间的通路关系,通过mic算法计算基因-mirna,基因-蛋白质,mirna-蛋白质之间的最大信息系数,确定阈值为0.6构建乳腺癌的snp、基因、mirna和蛋白质的多层网络关系图,对比肿瘤和正常组织中的节点的度及聚类系数等指标,发现:
(1)肿瘤组织和正常组织中同时出现的rs11257188位点通过pfkfb3作用于14.3.3_zeta蛋白质(及阻断调控bax蛋白质通路),使其表达量在多数乳腺癌病人中有明显的过表达,从而引起表型改变。
(2)正常组织中基因层度值较大的基因节点,在肿瘤组织与正常组织的表达量方面显著过表达或欠表达,对于其共同作用的蛋白质层中度值较大的蛋白质节点,在肿瘤组织与正常组织的表达量方面显著过表达或欠表达。结合文献资料,发现prc1、ebf1和tgfbr2基因均有与乳腺癌有关的报道,因此通过此方法可有效筛选致病基因和验证snp位点的可靠性,tmem132c基因没有文献支持关于与乳腺癌的关联,可推断其可能是有效的致病基因。
本发明所述方法证实了snp位点将对基因产生影响,间接作用于蛋白质表达量,导致表型发生改变。结合文献资料,发现prc1、ebf1和tgfbr2基因均有与乳腺癌有关的报道,因此推断rs11059635是显著的遗传变异位点,tmem132c是有效的致病基因。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 腈水解酶突变体及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白AsTMK1及其编码基因和应用的制造方法与工艺
- 植物抗旱相关蛋白EtSnRK2.2及其编码基因和应用的制造方法与工艺
- OsSAPK7蛋白及其编码基因在提高水稻白叶枯病抗性中的应用的制造方法与工艺
- 一种抗除草剂抗虫融合基因、编码蛋白及应用的制造方法与工艺
- 毛果杨PtrZFP103基因及其编码蛋白和应用的制造方法与工艺
- 大豆bHLH转录因子基因GmFER及其编码蛋白和应用的制造方法与工艺
- 一种花狭口蛙的基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种泽陆蛙基因及其编码的多肽和该多肽的用途的制造方法与工艺
- 一种水稻MIT1基因、其编码蛋白及应用的制造方法与工艺