一种聚合物/相变微胶囊复合纤维及其制备方法与流程
本发明属于复合纤维领域,涉及一种聚合物/相变微胶囊复合纤维及其制备方法。
背景技术:
相变微胶囊是利用胶囊技术将相变材料,如石蜡,包裹在微胶囊壁材,如聚甲基丙烯酸,内部制成的相变复合材料,相变微胶囊直径一般在微米级别,其常可做为添加剂加入到基体材料中制成相变功能性复合材料,相变功能性复合材料被广泛应用于服装和医疗用品等领域。
利用相变微胶囊制备相变纤维,相变纤维在30℃左右具有高温吸热,低温放热的现象,用作织物具有实时调温的作用。
已有的相变纤维有腈纶,纤维素纤维体系,因熔纺温度高,易导致微胶囊破损,因此溶液纺是制备相变纤维的主要方法,而纤维素的资源丰富,易于自然降解等特性也使其功能化具有更高的价值。
研究人员常在粘胶纤维或其它复合纤维中添加相变微胶囊,添加量为20%~40%时,相变焓可达10~20j/g,可以极大的提高复合纤维的品质。但在制备相变微胶囊复合纤维的过程中,往往将相变微胶囊直接与纺丝液混合,此时,由于纺丝液粘度高,使相变微胶囊难以在纤维素溶液中均匀分散,对混合设备的要求也较为苛刻,在制备过程中容易产生相变微胶囊分散不均匀和易破损流失等问题,例如文献dissolutionandformingofcellulosewithionicliquids中纤维素聚合度为569,浓度为12.9wt%的纤维素/离子液体溶液,温度为85℃时的零切粘度为8670pa·s;纤维素聚合度为569,浓度为10.4wt%的纤维素/1-丁基-3-甲基咪唑氯盐([bmim]cl)溶液,温度为85℃时的零切粘度达到7920pa·s。
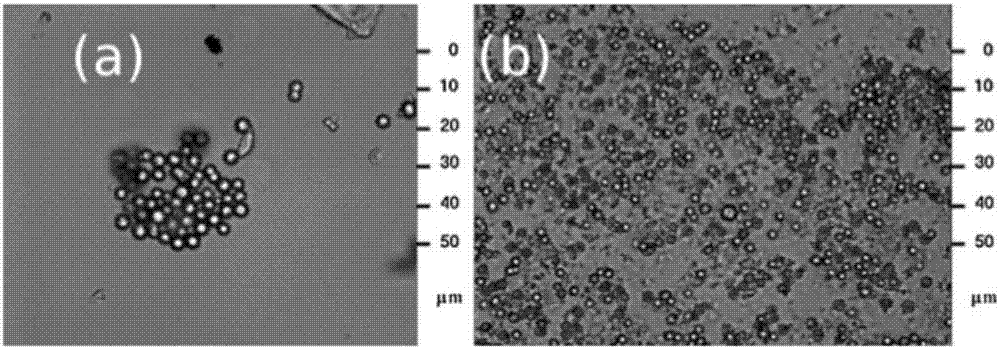
对于这一问题,也曾有其他学者指出,例如文献fabricationandmorphologicalcharacterizationofmicroencapsulatedphasechangematerials(micropcms)andmacrocapsulescontainingmicropcmsforthermalenergystorage曾指出在相变微胶囊的制备过程中容易产生破损的情况,如图1所示,其在制备过程中便存在一定的破损率,破损后的微胶囊中相变物质流失,失去相变功能,且小分子相变物质进入纤维中易使纤维的力学性能变差,因此,在后续制备纤维的过程中高温和真空搅拌更易使微胶囊出现破损的情况,最终导致微胶囊的添加效率变低;文献thermoregulatingresponseofcottonfabriccontainningmicroencapsulatedphasechangematerials中通过微胶囊的技术制备了纤维素纤维相变材料,添加过程中采用高速搅拌使微胶囊在高粘溶液中分散,如图2所示,搅拌速率达到10000rpm时尚不能使微胶囊良好的分散,搅拌速率达13500rpm时微胶囊才可以达到分散状态,在高搅拌速率保证其分散性良好的条件下,相变微胶囊本身的完整性则不能够得到保证。
以上相变纤维的制备过程中存在的问题为:
(1)相变粘胶纤维强度较低;
(2)离子液体体系没有实现工业化,且溶剂本身粘度大,给微胶囊的分散造成困难;
(3)常规分散微胶囊通常需要用分散剂,影响氮甲基吗啉氮氧化物的回收利用;
(4)强剪切分散和高真空度作用不会影响到溶剂的回收利用,但容易使微胶囊破裂而降低添加效率。
因此,研究一种分散性好且不易发生破损流失的聚合物/相变微胶囊复合纤维及其制备方法具有十分重要的意义。
技术实现要素:
本发明先将相变微胶囊在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中分散均匀,然后再采用低真空脱水的方式,克服了传统制备方法中直接在高真空状态下将相变微胶囊直接置于高粘度纺丝溶液中而导致其分散性差和易破损的缺点,提供了一种分散性好且不易发生破损流失的聚合物/相变微胶囊复合纤维及其制备方法,从而提高了相变微胶囊的添加效率和最终得到的复合纤维的力学性能。
为了达到上述目的,本发明采用的技术方案为:
一种聚合物/相变微胶囊复合纤维的制备方法,步骤如下:
(1)将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,现有技术中常将相变微胶囊直接置于高粘度溶液中,由于溶液粘度较大,故而相变微胶囊在其中的分散性较差;
(2)加入聚合物搅拌混合后进行脱水处理得到混合分散液,所述聚合物是指能够溶解在浓度为87wt%的氮甲基吗啉氮氧化物水溶液中且可进行纺丝的物质,所述脱水处理是在低真空状态下进行的,真空压力为-0.05~0.08mpa,此时溶液粘度为最终溶液粘度的70%;现有技术采用的真空压力为-0.1mpa,开始抽真空过程中,微胶囊并未被固定在高粘的聚合物溶液中,在高真空度下易被搅拌和真空共同作用而破损;
(3)在高真空状态下搅拌脱水,制得功能性纺丝液,所述高真空状态的真空压力的绝对值大于等于0.095mpa;
(4)纺丝制得聚合物/相变微胶囊复合纤维。
本发明先将微胶囊与溶剂氮甲基吗啉氮氧化物混合,在低剪切条件下制备微胶囊/氮甲基吗啉氮氧化物分散液,再在低真空状态下进行溶解纤维素,待溶液达到一定的粘度,再用高真空脱水使纤维素完全溶解,此时溶液粘度高微胶囊在溶液中难以运动,因此高真空状态不会破坏微胶囊的结构。
作为优选的技术方案:
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,所述聚合物为纤维素、醋酸纤维素或聚芳砜酰胺。本发明中的聚合物的材料包括但不限于此,能够满足溶解在浓度为87wt%的氮甲基吗啉氮氧化物水溶液中且可进行纺丝的物质皆可作为本发明中聚合物的备选材料。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,步骤(1)中,所述相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,所述分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为0.2~1wt%。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,步骤(1)中,将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中采用的方式为在线添加,即在生产线上的浓度为50wt%的氮甲基吗啉氮氧化物水溶液输送管路上加装微胶囊在线添加装置。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,步骤(2)中,所述脱水处理采用的方式为减压蒸馏;所述脱水处理的温度为80~110℃;所述混合分散液中聚合物的含量为2~6wt%,聚合物与相变微胶囊的质量比为5~20:1。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,步骤(3)中,所述搅拌脱水采用的方式为减压蒸馏,所述搅拌脱水的温度为80~110℃,所述功能性纺丝液中聚合物的含量为6~12wt%,所述高真空状态的真空压力为-0.095~0.1mpa。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,步骤(4)中,所述纺丝采用的工艺为湿法纺丝工艺或干喷湿法纺丝工艺。
如上所述的一种聚合物/相变微胶囊复合纤维的制备方法,所述纺丝的工艺流程为挤出、凝固和牵伸,其中凝固是在浓度为0~10wt%nmmo水溶液中进行的,纺丝工艺参数为:卷绕速率5~50m/min,牵伸倍数5~10。
本发明还提供了与上述制备方法相对应的一种聚合物/相变微胶囊复合纤维,相变微胶囊在纤维中分散均匀,纤维可以是长丝,也可以是短纤维,具体表现为复合纤维中相变微胶囊的平均粒径为10~15μm,复合纤维的干态断裂强度为1.7~4.5cn/dtex;相变微胶囊在加工过程中不容易发生破损流失,具体表现为相变微胶囊的添加效率为90~95%,所述添加效率的具体计算公式为:
常规方法一般为60%左右。
作为优选的技术方案:
如上所述的聚合物/相变微胶囊复合纤维,所述聚合物/相变微胶囊复合纤维的单丝纤度为0.8~4.8dtex。
发明原理:
本发明先将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中,浓度为50wt%的氮甲基吗啉氮氧化物水溶液更易与相变微胶囊均匀混合,保证相变微胶囊分散均匀后,再加入聚合物浆粕搅拌混合并进行低真空脱水,此时体系还未形成高粘溶液,微胶囊在混合分散液中仍处于自由分散状态,低真空度既能促进相变微胶囊与聚合物溶液的均匀混合又能有效防止将微胶囊直接置于高温真空状态下混合而导致其破损的现象,待溶液粘度逐渐升高,再进行高真空度脱水,此时微胶囊被固定包埋在高粘溶液当中,微胶囊不易破损。本发明克服了传统制备方法中直接在高真空状态下将相变微胶囊直接置于高粘度纺丝溶液中导致其分散性差和易破损的缺点,在保证相变微胶囊分散均匀的同时减少了相变微胶囊的破损流失现象,从而同时提高了相变微胶囊的添加效率和最终形成的纤维的力学性能。
有益效果:
(1)本发明的一种聚合物/相变微胶囊复合纤维的制备方法,仅需在聚合物/相变微胶囊复合纤维生产线上增加在线添加装置即可制得分散性良好的聚合物/相变微胶囊复合纤维,无需额外分散剂的加入,不增加溶剂与凝固浴回收的负担,成本低,方法简便灵活。
(2)本发明的一种聚合物/相变微胶囊复合纤维的制备方法,相变微胶囊同时具有了分散性良好和不易破损的优点,采用低粘度溶液与低真空脱水再配合高真空脱水的方式,克服了传统制备方法中高粘度溶液相变微胶囊分散性差和高真空状态下相变微胶囊易破损的缺点。
(3)本发明的一种聚合物/相变微胶囊复合纤维,复合纤维中相变微胶囊分散性好且破损率低,从而进一步提高了相变微胶囊的添加效率和最终得到的复合纤维的力学性能。
附图说明
图1为电镜下不同放大倍数下相变微胶囊的破损情况示意图,其中图a和图b为以苯乙烯-二乙烯基苯共聚物为壁材的微胶囊,图a的放大倍数为4000,图b的放大倍数为16000;图c和图d为以聚胺酯为壁材的微胶囊,图c的放大倍数为30000,图d的放大倍数为45000;
图2为相变微胶囊在高粘溶液中的分散状态示意图,其中图a为转速为10000rpm的分散状态示意图,图b为转速13500rpm的分散状态示意图。
具体实施方式
下面结合具体实施方式,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1
一种纤维素/相变微胶囊复合纤维的制备方法,步骤如下:
(1)在纤维素/相变微胶囊复合纤维的浓度为50wt%的氮甲基吗啉氮氧化物水溶液的输送管路上加装微胶囊在线添加装置,通过在线添加的方式将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为0.8wt%;
(2)加入纤维素搅拌混合后在真空压力为-0.07mpa的条件下通过减压蒸馏的方式进行脱水处理得到混合分散液,脱水处理的温度为80℃,混合分散液中纤维素的含量为6wt%,纤维素与相变微胶囊的质量比为7:1;
(3)在真空压力为-0.095mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为80℃,功能性纺丝溶液中纤维素的含量为8wt%;
(4)经湿法纺丝工艺的挤出、凝固和牵伸后制得纤维素/相变微胶囊复合纤维,其中凝固在水中进行,卷绕速率为10m/min,牵伸倍数为7。
最终制得的纤维素/相变微胶囊复合纤维的干态断裂强度为3cn/dtex,单丝纤度为3dtex,其中相变微胶囊的平均粒径为12μm,添加效率为92%。
实施例2
一种纤维素/相变微胶囊复合纤维的制备方法,步骤如下:
(1)在纤维素/相变微胶囊复合纤维的浓度为50wt%的氮甲基吗啉氮氧化物氮甲基吗啉氮氧化物水溶液的输送管路上加装微胶囊在线添加装置,通过在线添加的方式将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为1wt%;
(2)加入纤维素搅拌混合后在真空压力为-0.05mpa的条件下通过减压蒸馏的方式进行脱水处理得到混合分散液,脱水处理的温度为110℃,混合分散液中纤维素的含量为3wt%,纤维素与相变微胶囊的质量比为5:1;
(3)在真空压力为-0.1mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为110℃,功能性纺丝溶液中纤维素的含量为6wt%;
(4)经湿法纺丝工艺的挤出、凝固和牵伸后制得纤维素/相变微胶囊复合纤维,其中凝固在浓度为10wt%的氮甲基吗啉氮氧化物水溶液中进行,卷绕速率为5m/min,牵伸倍数为10。
最终制得的纤维素/相变微胶囊复合纤维的干态断裂强度为4.5cn/dtex,单丝纤度为4.8dtex,其中相变微胶囊的平均粒径为15μm,添加效率为93%。
对比例1
将与实施例2相同的相变微胶囊分散在与实施2相同的纤维素的氮甲基吗啉氮氧化物水溶液搅拌混合均匀得到分散液,分散液中相变微胶囊、纤维素、氮甲基吗啉氮氧化物和水的比例关系同实施例1中步骤(1)制得的分散液,然后将分散液在真空压力为-0.1mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为110℃,功能性纺丝溶液中纤维素的含量为6wt%,然后采用与实施例1步骤(4)相同的纺丝工艺和纺丝参数纺丝制得纤维素/相变微胶囊复合纤维。
最终制得的纤维素/相变微胶囊复合纤维的干态断裂强度为1.6cn/dtex,单丝纤度为0.8dtex,其中相变微胶囊的平均粒径为5μm,添加效率为62%。将实施例2与对比例1相对比可以看出,本发明先将相变微胶囊在浓度为50wt%的氮甲基吗啉氮氧化物中分散均匀,然后再采用低真空脱水的方式,克服了对比例1中直接在高真空状态下将相变微胶囊直接置于高粘度纺丝溶液中而导致其分散性差和易破损的缺点,极大的改善了相变微胶囊的分散均匀性,同时,待溶液粘度升高后,微胶囊被固定包埋在高粘溶液当中,微胶囊破损率低,因此,使得最终制得的复合纤维的相变微胶囊的添加效率和纤维本身的力学性能相对于比对比例1有了明显提高。
实施例3
一种聚芳砜酰胺/相变微胶囊复合纤维的制备方法,步骤如下:
(1)在聚芳砜酰胺/相变微胶囊复合纤维的浓度为50wt%的氮甲基吗啉氮氧化物水溶液的输送管路上加装微胶囊在线添加装置,通过在线添加的方式将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为0.2wt%;
(2)加入聚芳砜酰胺搅拌混合后在真空压力为-0.08mpa的条件下通过减压蒸馏的方式进行脱水处理得到混合分散液,脱水处理的温度为90℃,混合分散液中聚芳砜的含量为6wt%,聚芳砜与相变微胶囊的质量比为20:1;
(3)在真空压力为-0.1mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为90℃,功能性纺丝溶液中聚芳砜酰胺的含量为15wt%;
(4)经干喷湿法纺丝工艺的挤出、凝固和牵伸后制得聚芳砜酰胺/相变微胶囊复合纤维,其中凝固在浓度为5wt%的氮甲基吗啉氮氧化物水溶液中进行,卷绕速率为50m/min,牵伸倍数为5。
最终制得的聚芳砜酰胺/相变微胶囊复合纤维的干态断裂强度为1.7cn/dtex,单丝纤度为0.8dtex,其中相变微胶囊的平均粒径为10μm,添加效率为90%。
实施例4
一种醋酸纤维素/相变微胶囊复合纤维的制备方法,步骤如下:
(1)在醋酸纤维素/相变微胶囊复合纤维的浓度为50wt%的氮甲基吗啉氮氧化物水溶液的输送管路上加装微胶囊在线添加装置,通过在线添加的方式将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为0.6wt%;
(2)加入醋酸纤维素搅拌混合后在真空压力为-0.065mpa的条件下通过减压蒸馏的方式进行脱水处理得到混合分散液,脱水处理的温度为98℃,混合分散液中醋酸纤维素的含量为7wt%,醋酸纤维素与相变微胶囊的质量比为7.5:1;
(3)在真空压力为-0.096mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为98℃,功能性纺丝溶液中醋酸纤维素的含量为9wt%;
(4)经干喷湿法纺丝工艺的挤出、凝固和牵伸后制得醋酸纤维素/相变微胶囊复合纤维,其中凝固在浓度为8wt%的氮甲基吗啉氮氧化物水溶液中进行,卷绕速率为15m/min,牵伸倍数为10。
最终制得的醋酸纤维素/相变微胶囊复合纤维的干态断裂强度为3.0cn/dtex,单丝纤度为2.8dtex,其中相变微胶囊的平均粒径为13.5μm,添加效率为94%。
实施例5
一种纤维素/相变微胶囊复合纤维的制备方法,步骤如下:
(1)在纤维素/相变微胶囊复合纤维的浓度为50wt%的氮甲基吗啉氮氧化物水溶液的输送管路上加装微胶囊在线添加装置,通过在线添加的方式将相变微胶囊分散在浓度为50wt%的氮甲基吗啉氮氧化物水溶液中得到分散液,相变微胶囊的直径为10μm,相变微胶囊中壁材与芯材的质量比为1:1,壁材为苯乙烯与甲基丙烯酸的共聚物,芯材为质量比为1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相变微胶囊相对于浓度为50wt%的氮甲基吗啉氮氧化物水溶液的含量为0.4wt%;
(2)加入纤维素搅拌混合后在真空压力为-0.07mpa的条件下通过减压蒸馏的方式进行脱水处理得到混合分散液,脱水处理的温度为105℃,混合分散液中纤维素的含量为10wt%,纤维素与相变微胶囊的质量比为13:1;
(3)在真空压力为-0.098mpa的条件下通过减压蒸馏的方式进行搅拌脱水制得功能性纺丝液,搅拌脱水的温度为105℃,功能性纺丝溶液中纤维素的含量为12.5wt%;
(4)经干喷湿法纺丝工艺的挤出、凝固和牵伸后制得纤维素/相变微胶囊复合纤维,其中凝固在浓度为9wt%的氮甲基吗啉氮氧化物水溶液中进行,卷绕速率为20m/min,牵伸倍数为8。
最终制得的纤维素/相变微胶囊复合纤维的干态断裂强度为3.2cn/dtex,单丝纤度为3.0dtex,其中相变微胶囊的平均粒径为11μm,添加效率为90%。
- 还没有人留言评论。精彩留言会获得点赞!