制备范德华二维层状单晶的方法
本发明属于金属间化合物单晶制备的技术领域。具体地,本发明涉及制备范德华二维层状单晶的方法。更具体地,本发明涉及基于化学气相传输(cvt)原理,利用振荡温度场制备过渡金属硫属化合物和过渡金属卤素化合物单晶的方法。
背景技术:
过渡金属硫属化合物或过渡金属卤素化合物的层状化合物具有类似于石墨的层状结构,晶体内原子层与层之间依靠范德华力结合。这类材料的单层或少层薄膜由于具有优异的磁性、磁电输运和光学性质近年来重新引起材料学与物理学界的广泛关注。值得指出的是最高质量的单层或少层薄膜均是由单晶机械剥离单晶获得,而化学气相转输是合成这类化合物单晶的主要方法。
化学气相输运(cvt,chemicalvaportransport)是指在一个密闭的容器中,前驱体与传输介质发生可逆反应形成气态的中间产物,利用容器内部的温度梯度使化学平衡移动,某温区中原料分解并扩散至另一温区,然后形核结晶的技术。
化学气相输运是一种原本存在于自然界中的自然现象。早期德国化学家本生在研究火山喷发时发现,赤铁矿(fe2o3)的形成与火山气体的活动有关。火山口附近的高温气体中,包含有大量氯化氢。它们与岩浆中熔融的氧化铁反应并生成水汽和氯化铁蒸汽。当包含反应物的气体飘离火山口,遇冷则氯化铁与水汽反应重新生成赤铁矿。
人们最早应用化学气相输运的案例之一,是金属锆的提纯。一个密封含碘的容器中,插入纯度低的锆丝并通上电极加热。红热的锆丝与碘蒸气发生反应生成气化的化合物,扩散至温度较低的容器壁上,发生逆反应使得高纯度的金属锆沉积。而卤素灯的发明也是类似的原理,不同之处在于钨和碘的反应是放热反应。使用中升华的钨与碘反应,生成物碘化钨在高温处发生逆反应分解,使金属钨重新沉积在钨丝上,极大延长了灯丝寿命。
德国化学家schafer首先用热力学方法系统研究了化学气相输运,并发掘出该方法在晶体生长方面的巨大应用潜力。现在化学气相输运已经成为了一种多样的单晶生长技术,用于生长各种金属间化合物、卤素化合物、氧化物、硫化物等单晶。
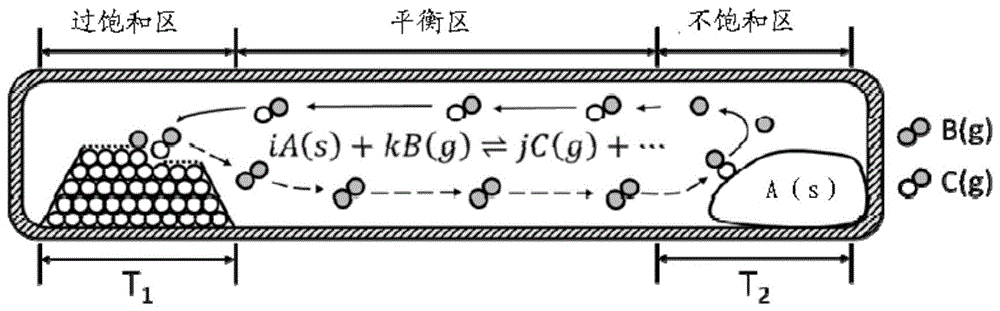
实验室典型的化学气相输运方法,是将一根装有前驱体与传输介质(如卤素、氯化氢、水汽等)的石英管,抽至高真空并密封,再放入高温炉里。前驱体与传输介质发生可逆反应生成气态产物,而不同温度下由于化学平衡不同,平衡状态下气态产物的浓度也不同。由于石英管内部的浓度梯度导致了扩散过程,而扩散趋于抹平浓度差,使不同温度下的实际气态组分偏离了平衡状态。原料端的气态产物处于不饱和状态,持续分解。形核端的气态产物处于过饱和状态,逆反应发生,实现单晶的形核与长大。
传统cvt生长晶体时使用恒定温度,原料端与形核端温度在晶体生长过程中保持不变,处于热力学平衡状态(参照图1a)。原料端与形核端的平衡状态相差越远,浓度差就越大,扩散速率增大,进而晶体生长的驱动力也大。为了配置合适的生长条件,理想情况下可采用的方法有两种:1.改变生长的平均温度,使平均温度下反应的吉布斯自由能变化尽可能接近零。此时溶解度-温度曲线斜率最大,很小的温差就能使形核端/原料端进入过饱和区/不饱和区(参照图1b);2.增大生长端与原料端的温差,使形核端/原料端进入过饱和/不饱和区(参照图1c)。传统的cvt存在以下缺点:一方面,过高的生长温度会增加晶体热缺陷;另一方面,过大的温差会使形核点密集而难以控制,近邻形核点的晶体会连成一片。也就是说,理想的反应温度可能高于单晶炉的使用温度。而且过高的生长温度,会使得晶体热缺陷增多。盲目增大温差会导致形核密度不可控,使得晶体形貌变差。因而发展一种可以在低温可控生长的技术,显得尤为重要。
技术实现要素:
本发明的目的是提供一种能在低温下高效制备高质量过渡金属硫属化合物和过渡金属卤素化合物单晶的制备方法。
本发明的上述目的是通过如下技术方案实现的。
本发明提供一种制备范德华二维层状单晶的方法,其包括如下步骤:
(1)在石英管的一端放置原料和传输介质,然后抽真空;其中,放置原料和传输介质的一端称为原料端,石英管的另一端称为形核端;
(2)使石英管的原料端的温度为t2,形核端的温度为材料的生长温度t1,并且使得原料端温度t2高于形核端温度t1,且控制原料端温度t2与形核端温度t1的温度差△t21为30-100℃;
(3)以相同的降温速率和升温速率,同步调控原料端温度t2与形核端温度t1,以实现降温幅度和升温幅度在1-30℃范围,从而制备范德华二维层状单晶。
优选地,在本发明所述的方法中,所述原料为用于制备过渡金属硫属化合物单晶或过渡金属卤素化合物单晶而使用的多晶粉末。
优选地,在本发明所述的方法中,所述过渡金属硫属化合物和过渡金属卤素化合物的过渡金属元素选自ti、zr、hf、v、nb、ta、cr、mo、w、ni、pt、pd中的一种或几种;所述过渡金属硫属化合物的硫属元素选自s、se、te中的一种或几种;所述过渡金属卤素化合物的卤素选自f、cl、br、i中的一种或几种。
优选地,在本发明所述的方法中,所述传输介质选自固态i2、液态br2、气态cl2中的一种或几种。
优选地,在本发明所述的方法中,所述降温速率和升温速率为1-40℃/min。
优选地,在本发明所述的方法中,升温速率大于降温速率,更有利于单晶生长。
优选地,在本发明所述的方法中,所述同步调控原料端温度t2与形核端温度t1的降温速率和升温速率是通过包括如下步骤的方法进行的:
原料端温度t2与形核端温度t1采用相同的降温速率和相同的降温时间降温后,在降温以后的温度下保持0-1h,然后,采用相同的升温速率和相同的升温时间升温,在升温以后的温度下保持0-1h;如此重复以上过程,使得原料端温度t2与形核端温度t1波形振荡。
优选地,在本发明所述的方法中,当所述过渡金属硫属化合物为nbse2、mos2、mose2或mote2,以及所述过渡金属卤素化合物为cri3或crbr3时,所述原料端温度t2为600-650℃,所述形核端温度t1为550-600℃;所述降温速率为1-10℃/min,所述升温速率为12-40℃/min;所述降温幅度和升温幅度在5-30℃范围。
优选地,在本发明所述的方法中,其还包括在所述步骤(1)之后和步骤(2)之前的反向控温步骤,该反向控温步骤包括以下步骤:
将石英管的原料端加热至材料的生长温度t1,将形核端加热至t3,使得形核端温度t3高于原料端温度t1,且控制形核端温度t3与原料端温度t1的温度差△t31为100-200℃。
在本发明中,增加反向控温步骤,具有以下优点:清洗成核端附着的杂质以及原料粉末,以减少正向控温生长时可能出现的形核点,确保每一个形核点都能发育成形貌完好的单晶。
优选地,在本发明所述的方法中,当所述过渡金属硫属化合物为nbse2、mos2、mose2或mote2,以及所述过渡金属卤素化合物为cri3或crbr3时,所述反向控温步骤是在如下条件下进行的:所述原料端温度t1为550-600℃,所述形核端温度t3为750-800℃。
优选地,在本发明所述的方法中,其还包括在所述步骤(1)之前对石英管采用丙酮或者稀盐酸以及无水乙醇依次超声清洗的步骤。
优选地,在本发明所述的方法中,所述步骤(1)中的抽真空是通过包括如下步骤的方法进行的:将石英管接在真空系统上,用保护性气体反复清洗三次以上,再抽至真空度10-5至10-4pa。
优选地,在本发明所述的方法中,所述保护性气体选自氩气和/或氮气。
本发明具有如下有益效果:
本发明的方法解决了传统cvt技术存在的单晶生长慢,生长缺陷多且质量不稳定的缺点,实现了在较低温度下合成高质量的单晶。
本发明的方法实现了在较低温度下合成单晶,比传统方法低200℃,本发明方法中的低温能够降低热缺陷,而本发明方法中的振荡温度场能够加快动量和质量传输。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
图1a为传统化学气相传输单晶生长原理示意图;
图1b为传统化学气相传输单晶生长改变平衡位置示意图;
图1c为传统化学气相传输单晶生长增大温差示意图;
图2a为传统化学气相传输中恒定温度梯度的生长原理示意图;
图2b为本发明的一个实施方案的振荡温度场的生长原理示意图;
图3a为本发明的一个实施方案的振荡温度场的生长方法示意图;
图3b为本发明的一个实施方案的振荡温度场的生长方法温度曲线示意图;
图4a为本发明实施例1-4制备的单晶图;
图4b为本发明实施例1制备的单晶的xrd谱图;
图4c为本发明对比例1制备的单晶图;
图4d为本发明实施例1制备的单晶图;
图5a为本发明实施例1制备的单晶的侧面结构的示意图;
图5b为本发明实施例1制备的单晶的ab面结构的示意图;
图5c为本发明实施例1制备的单晶的(001)方向球差电镜图;
图5d为本发明实施例1制备的单晶的(111)方向球差电镜图;
图6为本发明实施例1-4制备的单晶的变温电阻曲线;
图7为本发明实施例5制备的cri3单晶图。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。
参照图2a,对于恒定温度梯度的传统生长方法(吸热反应),石英管原料端应处于高温区t2,对应溶解度-温度图中的q点,气态组分处于过饱和状态,析出晶体。石英管形核端处于低温区t1,对应图中n点,气态组分处于不饱和状态,原料分解以补充形核端的消耗,使整个石英管内部处于动态平衡。
参照图2b,保持原料端t2与形核端t1的温差t2-t1不变,t1和t2同时升降温度来实现过饱和状态的变化。从而可以在不增加晶体形核密度的前提下,显著改善晶体生长。
如图2b所示,a-b过程:t1,t2同时升温。成核端的过饱和度降低,晶体生长被抑制;原料端进入不饱和区,原料加速分解。c-d过程:t1,t2同时降温。成核端的过饱和度增加,促进晶体生长;原料端的分解被暂时抑制。e-b-c-d-e构成循环过程,通过设置不同的降温速率,控制成核端晶体生长的过饱和度大小。即便在低的生长温度下,也可以实现高过饱和度的晶体生长。该方法可以控制晶体生长的形貌和质量,也可以拓宽生长温区,避开杂相,实现高质量单晶生长。
在本发明的一个具体的实施方案中,当制备nbse2单晶或者cri3单晶时,可以采用包括如下步骤的方法进行:
(1)在氩气手套箱内,将前驱体与传输介质装入内径16mm×长度200mm的石英管并密封;
(2)将准备好的石英管接在真空系统上,用氩气反复清洗三次,再抽至高真空(<10-4pa);
(3)在石英管合适的长度处,用火焰枪加热熔封石英管;
(4)预先校准双温区管式炉的温度分布,将石英管放置在两个温区之间;
(5)以5℃-10℃/分钟的速率,将靠近原料端的温度加热至550℃-600℃,靠近形核端的温度加热至750℃-800℃,维持1-2天;
(6)以5℃-10℃/分钟的速率,同步控制原料端的温度升温至600℃-650℃,靠近形核端的温度降至550℃-600℃。待温度稳定1-2小时后,将两个温区同步以1-10℃/小时速率降低5-30℃,再以12-40℃/小时速率升温5-30℃。重复以上过程,使原料端和形核端的温度波形振荡;
(7)待7-14天生长完成后,直接取出石英管,并将原料端浸入水中降温。
(8)在通风橱中敲碎石英管,用无水乙醇反复清洗单晶,然后放入真空烘箱中烘干。
实施例1
制备nbse2单晶
(1)利用丙酮和无水乙醇依次超声清洗内径16mm×长度200mm一端封闭的石英管内壁10分钟,并用去离子水冲洗反复两次,最后用鼓风干燥箱烘干。
(2)在手套箱内将总质量1g的nb、se粉末(纯度4n以上)以1:2的比例混合,以及5mg/cm3的碘(纯度3n以上)混合装入石英管并密封。
(3)将石英管开口端装入石英纤维棉,接到真空系统的汇流排上然后抽真空,当真空度达到10-4pa时,在离石英管底部高度(10cm)处,用火焰枪加热熔封石英管。
(4)将熔封后的石英管置入预先校准好温度分布的水平双温区管式炉中,如图3a所示。
(5)通电管式炉,以8℃/分钟的速率,原料端的温度加热至550℃,将靠近形核端的温度加热至750℃,并保持该温度1-2天,清理形核端可能附着的原料粉末。
(6)以10℃/分钟的速率,控制原料端的温度升温至600℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以1℃/小时速率降低12℃,再以12℃/小时速率升温24℃,再以1℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,再以1℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃处波形振荡。
(7)经过10天生长完成后,直接取出石英管,并将原料端蘸水降温;
(8)在通风橱中敲碎石英管,用无水乙醇反复清洗单晶,然后放入真空烘箱中烘干。
图3a示出了将熔封后的石英管置入预先校准好温度分布的水平双温区管式炉中。
图3b示出了原料端和形核端温度-时间设置曲线,s1是升温速率,s2是降温速率。
图4b示出了本实施制备的2h-nbse2的x射线衍射结果。单晶x射线衍射证明本实施制备的2h-nbse2是2h结构单晶体,无杂相。
图4d示出了本实施例制备的nbse2单晶表面光滑,形状规则,有的呈六角型,具有典型二维生长特征。
图5a-图5d的hrtem较明显地显示出了2h相的nbse2的原子排列。
实施例2
所有步骤的技术特征的参数均与实施例1相同,除了改变实施例1中的步骤(6)中的同步降温速率,即将实施例1中的步骤(6)修改为:以10℃/分钟的速率,控制原料端的温度升温至600℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以3℃/小时速率降低12℃,再以12℃/小时速率升温24℃,再以3℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,再以3℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃处波形振荡。
图4a示出了本实施例制备的单晶图。
实施例3
所有步骤的技术特征的参数均与实施例1相同,除了改变实施例1中的步骤(6)中的同步降温速率,即将实施例1中的步骤(6)修改为:以10℃/分钟的速率,控制原料端的温度升温至600℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以4℃/小时速率降低12℃,再以12℃/小时速率升温24℃,再以4℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,再以4℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃处波形振荡。
图4a示出了本实施例制备的单晶图。
实施例4
所有步骤的技术特征的参数均与实施例1相同,除了改变实施例1中的步骤(6)中的同步降温速率,即将实施例1中的步骤(6)修改为:以10℃/分钟的速率,控制原料端的温度升温至600℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以5℃/小时速率降低30℃,再以15℃/小时速率升温60℃,再以5℃/小时速率降低60℃。重复“再以15℃/小时速率升温60℃,再以5℃/小时速率降低60℃”过程,使原料端和形核端的温度在550℃处波形振荡。
图4a示出了本实施例制备的单晶图。
图4a示出了不同降温速率下生长的nbse2单晶形貌,随着降温速率增加,螺位错数目减少,单晶形貌变得规则。
图6为本发明实施例1-4制备的单晶的变温电阻曲线。图6示出了不同降温速率下生长的nbse2电输运性质对比。图6示出了随着降温速率增加,单晶剩余电阻率升高、超导转变温度升高,说明晶体质量变高。s2是降温速率,rrr是剩余电阻率。图6示出了在33k(-240℃)和7.2k(-265.8℃)附近出现电子结构相变的特征峰,分别对应着出现电荷密度波相变和超导转变,也证明是2h-nbse2。所测得的相变临界点与文献标准数据非常接近,表明所制备的样品为缺陷密度较少的高质量的2h-nbse2单晶。
实施例5
cri3的制备
(1)利用丙酮和无水乙醇依次超声清洗内径16mm×长度200mm一端封闭的石英管内壁10分钟,并用去离子水冲洗反复两次,最后用鼓风干燥箱烘干。
(2)在手套箱内将总质量0.3g的cr、i单质(纯度4n以上)以1:3的比例混合,装入石英管并密封。
(3)将石英管开口端装入石英纤维棉,接到真空系统的汇流排上然后抽真空,当真空度达到10-4pa时,在离石英管底部高度(8-10cm)处,用火焰枪加热熔封石英管。
(4)将熔封后的石英管置入预先校准好温度分布的水平双温区管式炉中。
(5)通电管式炉,以8℃/分钟的速率,原料端的温度加热至600℃,将靠近形核端的温度加热至700℃,并保持该温度1天,清理形核端可能附着的原料粉末。
(6)以10℃/分钟的速率,保持原料端温度600℃不变,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以4℃/小时速率降低12℃,再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃波形振荡。
(7)经过7天左右生长完成后,直接取出石英管,并将原料端蘸水降温;
(8)在手套箱中敲碎石英管,取出单晶并密封保存。
图7示出了本实施制备的cri3单晶。
实施例6
cri3的制备
(1)利用丙酮和无水乙醇依次超声清洗内径16mm×长度200mm一端封闭的石英管内壁10分钟,并用去离子水冲洗反复两次,最后用鼓风干燥箱烘干。
(2)在手套箱内将总质量0.3g的cr、i单质(纯度4n以上)以1:3的比例混合,装入石英管并密封。
(3)将石英管开口端装入石英纤维棉,接到真空系统的汇流排上然后抽真空,当真空度达到10-4pa时,在离石英管底部高度(9cm)处,用火焰枪加热熔封石英管。
(4)将熔封后的石英管置入预先校准好温度分布的水平双温区管式炉中。
(5)通电管式炉,以8℃/分钟的速率,原料端的温度加热至600℃,将靠近形核端的温度加热至700℃,并保持该温度1天,清理形核端可能附着的原料粉末。
(6)以10℃/分钟的速率,原料端温度降至580℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以4℃/小时速率降低12℃,再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃波形振荡。
(7)经过7天左右生长完成后,直接取出石英管,并将原料端蘸水降温;
(8)在手套箱中敲碎石英管,取出单晶并密封保存。
本实施制备的cri3单晶与实施例5类似。
实施例7
cri3的制备
(1)利用丙酮和无水乙醇依次超声清洗内径16mm×长度200mm一端封闭的石英管内壁10分钟,并用去离子水冲洗反复两次,最后用鼓风干燥箱烘干。
(2)在手套箱内将总质量0.3g的cr、i单质(纯度4n以上)以1:3的比例混合,装入石英管并密封。
(3)将石英管开口端装入石英纤维棉,接到真空系统的汇流排上然后抽真空,当真空度达到10-5pa时,在离石英管底部高度(9cm)处,用火焰枪加热熔封石英管。
(4)将熔封后的石英管置入预先校准好温度分布的水平双温区管式炉中。
(5)通电管式炉,以8℃/分钟的速率,原料端的温度加热至600℃,将靠近形核端的温度加热至700℃,并保持该温度1天,清理形核端可能附着的原料粉末。
(6)以10℃/分钟的速率,原料端温度升温至650℃,靠近形核端的温度降至550℃。待温度稳定2小时后,将两个温区同步以4℃/小时速率降低12℃,再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃。重复“再以12℃/小时速率升温24℃,在以4℃/小时速率降低24℃”过程,使原料端和形核端的温度在550℃波形振荡。
(7)经过7天左右生长完成后,直接取出石英管,并将原料端蘸水降温;
(8)在手套箱中敲碎石英管,取出单晶并密封保存。
本实施制备的cri3单晶与实施例5类似。
对比例1
所有步骤的技术特征的参数均与实施例1相同,除了采用传统化学气相传输中恒定温度梯度的进行单晶生长,即将形核端温度控制在550℃,原料端温度控制在730℃。
图4c示出了本对比例制备的单晶图。图4c示出了传统的单晶生长需要的温差比较大,生长出的晶体团聚。
- 还没有人留言评论。精彩留言会获得点赞!