含烷氧基甲硅烷基的环氧化合物、该化合物的制备方法、包含该化合物的组合物、由该组合物制备的固化产物和该组合物的应用与流程
本发明涉及具有烷氧基甲硅烷基的环氧化合物(下文称为“烷氧基甲硅烷基化环氧化合物”),具有良好耐热性的所述化合物的复合材料和/或具有良好阻燃性的所述化合物的固化产物,制备所述化合物的方法,包含所述化合物的组合物,由所述组合物制得的固化产物,和所述组合物的应用。更具体地,本发明涉及烷氧基甲硅烷基化环氧化合物,具有良好耐热性的所述化合物的复合材料,制备所述化合物的方法,包含所述化合物的组合物,由所述组合物制得的固化产物,和所述组合物的应用,具体而言,所述良好的耐热性指具有低热膨胀系数(CTE)和高玻璃化转变温度(包括无玻璃化转变温度(无Tg)状态,其表示该复合材料不具有玻璃化转变温度)且无需单独的硅烷偶联剂。此外,还提供了在制备作为中间体的烷氧基甲硅烷基化环氧化合物的过程中得到的具有羟基和烯丙基的环氧化合物。
背景技术:
聚合物材料(特别是环氧树脂)的热膨胀系数(CTE)为约50-80ppm/℃,比陶瓷材料(如无机颗粒)或金属材料的CTE明显高了数倍至数十倍(例如,硅的CTE是3-5ppm/℃,铜的CTE是17ppm/℃)。因此,当在半导体、显示器等中将聚合物材料与无机材料或金属材料结合使用时,由于聚合物材料和无机材料或金属材料的CTE不匹配,聚合物材料的性质和可加工性会出现显著的降低。此外,在平行使用硅晶片和聚合物基材的半导体封装中,或者在其中用无机阻隔层来涂覆聚合物膜以赋予阻气性的涂层中,在加工和/或施用温度条件下发生改变时由于组成材料之间的具有高度的CTE不匹配,会产生产品缺陷(如在无机层中形成裂纹、基材翘曲、涂覆层剥落、基材失效等)。因为聚合物材料的高CTE和导致的聚合物材料的尺寸变化,可能限制诸如下一代半导体基材、印刷电路板(PCB)、封装、有机薄膜晶体管(OTFT)和柔性显示器基材等技术的发展。具体来说,目前在半导体和PCB领域中,由于聚合物材料具有比金属/陶瓷材料高得多的CTE,设计者在设计要求高度集成、小型化、灵活性、性能等的下一代部件时以及在确保部件的加工性和可靠性方面面临挑战。换言之,因为聚合物材料在加工温度下的高热膨胀性质,可能产生缺陷,可能限制可加工性,且部件的设计和确保零件的可加工性和可靠性可能成为关心的目标。因此,为了确保电子部件的可加工性和可靠性,必须改善聚合物材料的热膨胀性质,即尺寸稳定性。迄今为止,为了改善聚合物材料(如环氧树脂)的热膨胀性质,即获得低的CTE值,已使用了下述方法:(1)用无机颗粒(无机填料)和/或纤维制备环氧树脂的复合材料的方法,或者(2)设计并合成具有降低的CTE的新型环氧树脂的方法。当形成环氧化合物和作为填料的无机颗粒的复合材料来改善热膨胀性质时,需要大量直径约为2-30微米的无机二氧化硅颗粒以大幅降低CTE。但是,因为加入了大量无机颗粒,部件的可加工性和性能可能变差。即,大量无机颗粒的存在可降低流动性,且在填充窄空间时可能产生空穴。此外,因为添加了无机颗粒,材料的粘度可呈指数增长。此外,因为半导体结构的小型化,无机颗粒的尺寸趋于减小。当使用粒度小于或等于1微米的填料时,流动性(粘度增大)的降低可能更加严重。在使用具有大平均粒径的无机颗粒时,包含树脂和无机颗粒的组合物中填充不足的频率可能增加。虽然在使用包含有机树脂和用作填料的纤维的组合物时复合材料的CTE可大大降低,但纤维复合材料的CTE与硅芯片等的CTE相比可能仍然较高。如上所述,由于环氧树脂的复合材料技术的限制,制造用于下一代半导体基材、PCB等的高度集成和高性能的电子部件可能受到限制。因此,需要开发具有改善的耐热性(即低CTE和高玻璃化转变温度)以及改善的硬度的环氧复合材料,从而克服常规热固性聚合物复合材料因高CTE和较差的可加工性而造成的差的热学性质的缺陷。
技术实现要素:
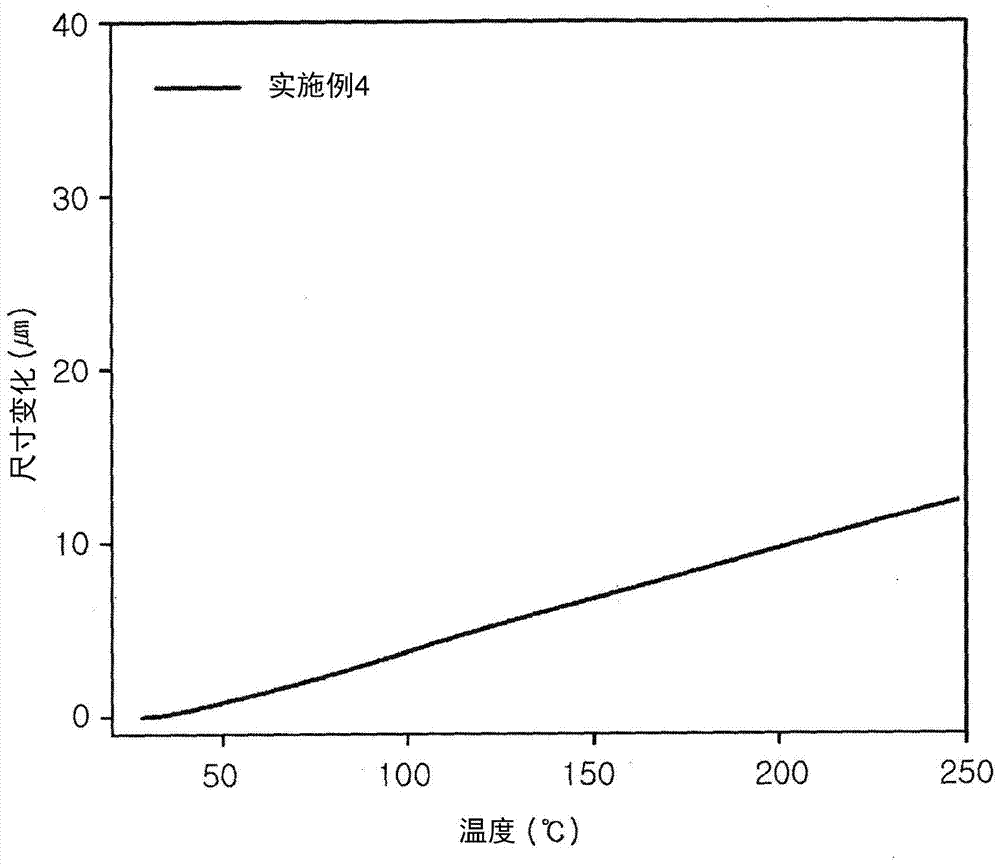
技术问题本发明的一个方面提供了具有烷氧基甲硅烷基的环氧化合物,具有改善的耐热性(特别是低CTE和高Tg)的所述化合物的复合材料和/或具有高良好的阻燃性的所述化合物的固化产物。本发明的另一个方面提供了制备以下物质的方法:具有烷氧基甲硅烷基的环氧化合物,具有改善的耐热性(特别是低CTE和高Tg)的所述化合物的复合材料和/或具有高良好的阻燃性的所述化合物的固化产物。本发明的另一个方面提供了包含以下物质的环氧组合物:具有烷氧基甲硅烷基的环氧化合物,具有改善的耐热性(特别是低CTE和高Tg)的所述化合物的复合材料和/或具有高良好的阻燃性的所述化合物的固化产物。本发明的另一个方面提供了本发明的一个实施方式所述的环氧组合物的固化产物,具有改善的耐热性(特别是低CTE和Tg)的所述组合物的复合材料和/或具有高良好的阻燃性的所述组合物的固化产物。此外,本发明的另一个方面还提供了本发明的一个实施方式所述的环氧组合物的应用。本发明的另一个方面提供了具有羟基和烯丙基的环氧化合物。技术方案根据本发明的一个方面,提供了具有烷氧基甲硅烷基的环氧化合物,其包含至少一个烷氧基甲硅烷基,所述烷氧基甲硅烷基为(1)独立地选自式S11至S16,(2)独立地选自式S21至S26,(3)独立地选自式S11至S16和式S31至S38,或(4)独立地选自式S21至S26和式S31至S38;以及核结构中的至少两个环氧基。[式S1]在式S11至S16中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S2]在式S21至S26中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S3]在式S31至S38中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第二方面,提供了具有烷氧基甲硅烷基的环氧化合物,其中,环氧基独立地选自第一方面所述的式S51至S58。[式S5(3)]在式S56至S58中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第三方面,所述环氧化合物可包括以下物质作为第一或第二方面所述的核结构:双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族或酚醛清漆单元。根据本发明的第四方面,所述核结构可选自第三方面所述的式A’至N’。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是并且在式K’至N’中,n是大于或等于1的整数且优选是1至1000的整数。根据本发明的第五方面,如果选自式A’至J’的核结构的数目大于或等于2,则式A’至I’的核结构可通过独立地选自式LG1至LG14的连接基团相连,且上述式J’的核结构可通过独立地选自第四方面所述的式LG2和LG9的连接基团相连。[式6(2)]在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的,且在式LG4、LG5、LG7、LG11、LG12和LG14中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第六方面,具有烷氧基甲硅烷基的环氧化合物还可包含式S4的取代基,所述式S4的取代基选自第一至第五方面中任一方面所述的式S41至S45。[式S4]根据本发明的第七方面,提供了具有羟基和烷基、包含选自式(A’)至(N’)的核结构以及至少一个选自式S41至S45的S4取代基和至少两个选自S51至S55的S5(1)环氧基的环氧化合物,其中,如果式(A’)至(J’)的核结构数目大于或等于2,则式(A’)至(J’)的核结构可通过选自式LG1至LG7的连接基团连接且上述式(J’)的核结构可通过式LG2的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数,[式S4][式S5(1)][式S6(1)]在式LG4、LG5和LG7中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第八方面,提供了一种制备环氧化合物的方法,包括第一步:使具有至少三个环氧基的环氧化合物的起始材料与烯丙醇在碱和可选的溶剂存在的情况下进行反应以制备中间体;以及第二步:使该中间体与下文式B1的化合物在碱和可选的溶剂存在的情况下进行反应以制备在独立地选自S11至S16的具有至少一个烷氧基甲硅烷基并具有至少两个环氧基的环氧化合物。[式B1]OCN(CH2)3SiR1R2R3在式B1中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S1]在式S11至S16中,R1至R3的中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第九方面,还可包括可选的第三步,在该步骤中使第二步中制备的环氧化合物与下文式B2的化合物在金属催化剂和可选的溶剂的存在下进行反应,以制备在独立地选自式S11至S16和式S31至S38的具有至少一个烷氧基甲硅烷基并在第八方面所述的核结构中具有至少两个环氧基的环氧化合物。[式B2]HSiR1R2R3在式B2中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S3]在式S31至S38中,R1至R3的中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第十方面,提供了一种制备包含烷氧基甲硅烷基的环氧化合物的方法,包括第一步:使具有至少三个环氧基的环氧化合物的起始材料与烯丙醇在碱和可选的溶剂存在的情况下进行反应以制备中间体;以及第二步:使该中间体与下式B2的化合物在金属催化剂和可选的溶剂存在的情况下进行反应以制备在独立地选自S21至S26的具有至少一个烷氧基甲硅烷基并具有至少两个环氧基的环氧化合物。[式B2]HSiR1R2R3在式B2中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S2]在式S21至S26中,R1至R3的中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第十一方面,还可包括可选的第三步:使第二步中制备的环氧化合物与下文式B1的化合物在碱和可选的溶剂的存在下进行反应的,以制备在独立地选自式S21至S26和式S31至S38的核结构中具有至少一个烷氧基甲硅烷基并在第十方面所述的核结构中具有至少两个环氧基的环氧化合物。[式B1]OCN(CH2)3SiR1R2R3在式B1中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S3]在式S31至S38中,R1至R3的中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。根据本发明的第十二方面,具有至少三个环氧基的环氧化合物的起始材料可包含选自式(A’)至(N’)的核结构和至少三个选自式S51至S54的环氧基,并且如果式(A’)至(J’)的核结构的数目大于或等于2,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且式(J’)的核结构可通过第八或第十方面所述的LG2的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。[式S5(0)][式S6(1)]在式LG4、LG5和LG7中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第十三方面,第一步中制备的中间体可包含选自式(A’)至(N’)的核结构、至少一个选自式S41至S45的取代基和至少两个选自式S51至S55的环氧基,并且当式(A’)至(N’)的核结构的数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且式(J’)的核结构可通过第八或第十方面所述的LG2的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数,[式S4][式S5(1)][式6(1)]在式LG4、LG5和LG7中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第十四方面,第二步中制备的具有烷氧基甲硅烷基的环氧化合物可包含选自式(A’)至(N’)的核结构、至少一个独立地选自式S11至S16的烷氧基甲硅烷基、至少两个选自式S51至S55和S57的环氧基和可选的选自式S41至S45的取代基,并且当式(A’)至(J’)的核结构的数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过第八方面所述的LG2和LG9的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。[式S4][式S5(2-1)]在式S57中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式6(2)]在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的,且在式LG4、LG5、LG7、LG11、LG12和LG14中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,且如上文所定义,且是根据核结构的数目重复的。根据本发明的第十五方面,第三步中制备的具有烷氧基甲硅烷基的环氧化合物可包含选自式(A’)至(N’)的核结构、至少一个选自式S11至S16以及式S31至S38的烷氧基甲硅烷基、至少两个选自式S51至S58的环氧基和可选的选自式S41至S45的取代基,并且当式(A’)至(J’)的核结构的数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过第九方面所述的LG2和LG9的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。[式S4][式S5(3-1)]在式S56至S58中,R1至R3的中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式6(2)]在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的,且在式LG4、LG5、LG7、LG11、LG12和LG14中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第十六方面,第二步中制备的具有烷氧基甲硅烷基的环氧化合物包含选自式(A’)至(N’)的核结构、至少一个选自式S21至S26的烷氧基甲硅烷基、至少两个选自式S51至S56的环氧基和可选的选自式S41至S45的取代基,并且当式(A’)至(J’)的核结构的数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且式(J’)的核结构可通过第十方面所述的LG2的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。[式S4][式S5(2-2)]在式S56中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式S6(1)]在式LG4、LG5和LG7中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第十七方面,第三步中制备的具有烷氧基甲硅烷基的环氧化合物可包含选自式(A’)至(N’)的核结构、至少一个选自S2取代基(其独立地选自式S21至S26)和S3取代基(其独立地选自式S31至S38)的烷氧基甲硅烷基、至少两个选自式S51至S58的环氧基和可选的选自式S41至S45的取代基,并且当式(A’)至(J’)的核结构的数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过第十一方面所述的LG2和LG9的连接基团连接。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。[式S4][式S5(3-2)]在式S56至S58中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的。[式6(2)]在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的,且在式LG4、LG5、LG7、LG11、LG12和LG14中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。根据本发明的第十八方面,在第八或第十方面所述的第一步中,0.1至10当量的烯丙醇相对于1当量的起始材料的环氧基进行反应。根据本发明的第十九方面,第八或第十方面所述的第一步可以在室温至120℃的温度下进行1至120小时。根据本发明的第二十方面,在第八方面所述的第二步中,0.1至5当量的上述式B1的烷氧基硅烷相对于1当量的中间体的羟基进行反应。根据本发明的第二十一方面,在第十方面所述的第二步中,0.1至5当量的上述式B2的烷氧基甲硅烷基相对于1当量的中间体的烯丙基进行反应。根据本发明的第二十二方面,第八或第十方面所述的第二步可以在室温至120℃的温度下进行1至72小时。根据本发明的第二十三方面,在第九方面所述的可选的第三步中,0.1至5当量的上述式B2的烷氧基硅烷相对于1当量的第二步制得的具有烷氧基甲硅烷基的环氧化合物的烯丙基进行反应。根据本发明的第二十四方面,在第十一方面所述的可选的第三步中,0.1至5当量的上述式B1的烷氧基硅烷相对于1当量的第二步中获得的具有烷氧基甲硅烷基的环氧化合物的羟基进行反应。根据本发明的第二十五方面,第九或第十一方面所述的可选的第三步可以在室温至120℃的温度下进行1至72小时。根据本发明的第二十六方面,提供了一种包含环氧化合物的环氧组合物,所述环氧化合物是第一至第六方面中任一方面所述的具有烷氧基甲硅烷基的环氧化合物。根据本发明的第二十七方面,在第二十六方面所述的环氧组合物中还可包括选自下组的至少一种环氧化合物:缩水甘油醚基环氧化合物、缩水甘油基环氧化合物、缩水甘油胺基环氧化合物、缩水甘油酯基环氧化合物、橡胶改性环氧化合物、脂肪族聚缩水甘油基环氧化合物和脂肪族缩水甘油胺基环氧化合物。根据本发明的第二十八方面,第二十七方面所述的环氧组合物可包括以下物质作为核结构:双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族化合物或酚醛清漆单元。根据本发明的第二十九方面,在第二十七方面所述的环氧组合物中,以环氧化合物的总重量为基准计,所述环氧组合物可包括10重量%-100重量%的具有烷氧基甲硅烷基的环氧化合物和0重量%-90重量%的选自下组的至少一种环氧化合物:缩水甘油醚基环氧化合物、缩水甘油基环氧化合物、缩水甘油胺基环氧化合物、缩水甘油酯基环氧化合物、橡胶改性环氧化合物、脂族聚缩水甘油基环氧化合物和脂族缩水甘油胺基环氧化合物。根据本发明的第三十方面,在第二十九方面所述的环氧组合物中,以环氧化合物的总重量为基准计,所述环氧组合物可包括30重量%-100重量%的具有烷氧基甲硅烷基的环氧化合物和0重量%-70重量%的选自下组的至少一种环氧化合物:缩水甘油醚基环氧化合物、缩水甘油基环氧化合物、缩水甘油胺基环氧化合物、缩水甘油酯基环氧化合物、橡胶改性环氧化合物、脂族聚缩水甘油基环氧化合物和脂族缩水甘油胺基环氧化合物。根据本发明的第三十一方面,在第二十六至三十方面中任一方面所述的环氧组合物中还可包含选自无机颗粒或纤维的至少一种填料。根据本发明的第三十二方面,在第三十一方面所述的环氧组合物中,所述无机颗粒可以是选自下组中的至少一种:选自二氧化硅、氧化锆、氧化钛、氧化铝、氮化硅和氮化铝的金属氧化物、以及T-10型硅倍半氧烷、梯型硅倍半氧烷和笼型硅倍半氧烷。根据本发明的第三十三方面,在第三十三方面所述的环氧组合物中,所述无机颗粒的含量可以占所述环氧组合物的固体总重量的5重量%-95重量%。根据本发明的第三十四方面,在第三十三方面所述的环氧组合物中,所述无机颗粒的含量可以占所述环氧组合物的固体总重量的30重量%-95重量%。根据本发明的第三十五方面,在第三十三方面所述的环氧组合物中,所述无机颗粒的含量可以占所述环氧组合物的固体总重量的5重量%-60重量%。根据本发明的第三十六方面,在第三十一方面所述的环氧组合物中,所述纤维可以是选自下组的至少一种玻璃纤维:E玻璃纤维、T玻璃纤维、S玻璃纤维、NE玻璃纤维、D玻璃纤维、H玻璃纤维、和石英;以及选自下组的有机纤维:液晶聚酯纤维、聚对苯二甲酸乙二醇酯纤维、全芳族纤维、多氧苯扎索尔(polyoxybenzasol)纤维、尼龙纤维、聚萘二甲酸乙二醇酯纤维、聚丙烯纤维、聚醚砜纤维、聚偏二氟乙烯纤维、硫化聚乙烯(polyethylenesulfide)纤维和聚醚醚酮纤维。根据本发明的第三十七方面,在第三十六方面所述的环氧组合物中,所述纤维可以是E玻璃纤维。根据本发明的第三十八方面,在第三十六方面所述的环氧组合物中,所述纤维可以是T玻璃纤维。根据本发明的第三十九方面,在第三十一方面所述的环氧组合物中,所述纤维的含量可以占所述环氧组合物的固体总重量的10重量%-90重量%。根据本发明的第四十方面,当第三十一方面所述的环氧组合物包含纤维时,其还可包含无机颗粒。根据本发明的第四十一方面,在第二十六至四十方面中的任一方面所述的环氧组合物中还可包含固化剂。根据本发明的第四十二方面,在第二十六至四十方面中的任一方面所述的环氧组合物中还可包含用于烷氧基甲硅烷基的反应催化剂。根据本发明的第四十三方面,在第四十二方面所述的环氧组合物中,所述用于烷氧基甲硅烷基的反应催化剂可以是选自下组的至少一种:选自硝酸、硫酸、盐酸、乙酸和磷酸的无机酸中的至少一种,氨,KOH,NH4OH,胺,过渡金属烷氧化物和锡化合物。根据本发明的第四十四方面,在第四十二方面所述的环氧组合物中,所用的反应催化剂可以占所述具有烷氧基甲硅烷基的环氧化物的0.01phr-10phr。根据本发明的第四十五方面,在第四十二方面所述的环氧组合物中还可包含水。根据本发明的第四十六方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的电子材料。根据本发明的第四十七方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的基材。根据本发明的第四十八方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的膜(film)。根据本发明的第四十九方面,提供了一种包含在基底层上放置金属层的层叠体,所述基底层使用第二十六至第四十五方面中任一方面所述的环氧组合物形成。根据本发明的第五十方面,提供了一种包含第四十九方面所述的层叠体的印刷电路板。根据本发明的第五十一方面,提供了一种包含第五十方面所述的印刷电路板的半导体装置。根据本发明的第五十二方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的半导体包装材料。根据本发明的第五十三方面,提供了一种包含第五十二方面所述的半导体包装材料的半导体装置。根据本发明的第五十四方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的粘合剂。根据本发明的第五十五方面,提供了一种包含第二十六至第四十五方面中的任一方面所述的环氧组合物的油漆。根据本发明的第五十六方面,提供了一种包含第二十六至第四十五方面中的任一方面所述的环氧组合物的复合材料。根据本发明的第五十七方面,提供了一种包含第二十六至第四十五方面中的任一方面所述的环氧组合物的预浸渍体。根据本发明的第五十八方面,提供了一种层叠体,所述层叠体包含置于第五十七方面所述的预浸渍体上的金属层。根据本发明的第五十九方面,提供了一种包含第二十六至第四十五方面中任一方面所述的环氧组合物的固化产物。根据本发明的第六十方面,第五十九方面所述的固化产物可具有小于或等于60ppm/℃的CTE。根据本发明的第六十一方面,第五十九方面所述的固化产物可具有等于或高于100℃的玻璃化转变温度,或者可不具有玻璃化转变温度。[有益效果]根据本发明的实施方式,由于环氧化合物的烷氧基甲硅烷基和填料(纤维和/或无机颗粒)之间以及环氧组合物中的烷氧基甲硅烷基之间的化学反应形成的化学键,包含具有烷氧基甲硅烷基的新颖环氧化合物的的组合物、复合材料和/或固化产物可改进耐热性。即,可降低环氧复合材料的CTE,并且可升高玻璃化转变温度或者可能不会出现玻璃化转变温度(无Tg)。此外,本发明的环氧化合物的固化产物还可具有良好的阻燃性。此外,当本发明的环氧组合物的组合物施用于金属膜时,由于金属膜的表面处的官能团和烷氧基甲硅烷基之间的化学键合,其可表现出良好的粘附性。此外,由于在包含具有烷氧基甲硅烷基的环氧化合物的组合物中化学键合效率增加,在包含具有烷氧基甲硅烷基的环氧化合物的组合物中可能无需使用常规环氧组合物中使用的硅烷偶联剂。包含所述环氧化合物的环氧组合物可具有良好的固化效率,通过固化所述环氧组合物形成的复合材料可具有良好的热膨胀性质,如低CTE和高玻璃化转变温度或无Tg。[附图说明]结合附图,通过以下详述能够更清楚地理解本发明的上述和其他方面、特性和其他优势,其中:图1显示了根据实施例2的复合材料随温度变化而产生的尺寸变化;图2显示了根据实施例4的复合材料随温度变化而产生的尺寸变化;且图3是显示实施例1和比较例1的复合材料的条带的燃烧状态的照片图。具体实施方式本发明提供了一种新颖的具有烷氧基甲硅烷基的环氧化合物、通过固化环氧组合物得到的复合材料,其具有改善的耐热性(具体而言是具有低CTE和高Tg,包括无Tg),和/或使用所述组合物形成的固化产物,其具有良好阻燃性、用于制备上述物质的方法、包含上述物质的环氧组合物、由该组合物制得的固化产物和该组合物的应用。在本发明中,“复合材料”指的是通过使用包含环氧化物和填料(纤维和/或无机颗粒)的组合物而形成的固化产物。在本发明中,“固化产物”通常指的是由包含环氧化合物——具体来说是环氧化合物和固化剂——的组合物形成的固化产物,例如,由包含环氧化合物、固化剂、以及至少一种选自下组的物质的组合物形成的固化产物:填料、可选和额外的固化剂、可选的固化催化剂和其它添加剂。此外,术语“固化产物”还用来指“部分固化的产物”。通常,只有使用无机颗粒和/或纤维强化的固化产物被称为复合材料,且固化产物的含义比复合材料宽。然而,可以认为用无机颗粒和/或纤维加强的术语固化产物与复合材料具有相同的含义。当通过使本发明的包含具有烷氧基甲硅烷基的环氧化合物的环氧组合物固化而形成复合材料时,环氧基可以与固化剂反应以进行交联反应,所述烷氧基甲硅烷基可以与填料(纤维和/或无机颗粒)的表面基团形成界面键合,和/或在烷氧基甲硅烷基之间形成化学键合。因此,在环氧复合材料系统中可获得相当高的化学键合效率,并因此取得低CTE和高玻璃化转变温度增加效果或无Tg。因此,可改善尺寸稳定性。此外,单独的硅烷偶联剂不是必须的。此外,包含本发明的环氧化合物的固化产物可具有良好的阻燃性。此外,在通过热固化和/或光固化本发明的包含具有烷氧基甲硅烷基的环氧化合物的环氧组合物时,可实现良好的固化性质。具体而言,对于包含–O-CH2-CHOHCH2O(CH2)3SiR1R2R3、-CH2-CHOHCH2O(CH2)3SiR1R2R3或–COO-CH2-CHOHCH2O(CH2)3SiR1R2R3(其中R1至R3中的至少一个是具有1-5个碳原子的烷氧基,且余者是具有1-10个碳原子的烷基)的烷氧基甲硅烷基化环氧化合物,与丙烯酰化合物相比,可避免由于氧气对光固化反应的干扰,并且由于烷氧基甲硅烷基化环氧化合物的固化收缩较小,可维持良好的粘附性,并可避免形成裂缝。此外,当把本发明的环氧组合物施用至化学处理的金属膜如铜膜时,可与通过金属表面处理而得到的金属的表面上的羟基等形成化学键合,从而具有与所述金属膜的良好粘附性。1.环氧化合物根据本发明的一个实施方式,提供了在核结构中具有至少一个烷氧基甲硅烷基和至少两个环氧基的环氧化合物。所述烷氧基甲硅烷基(1)可独立地选自式S11至S16,(2)可独立地选自式S21至S26,(3)可独立地选自式S11至S16和式S31至S38,或者(4)可独立地选自式S21至S26和式S31至S38。根据本发明的一个实施方式,在具有烷氧基甲硅烷基的环氧化合物中,环氧基:烷氧基甲硅烷基的当量比范围可以是1:10至10:1。[式S1]在式S11至S16中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,优选是具有2-3个碳原子的烷氧基,更优选是乙氧基,且余者是具有1-10个碳原子的烷基。所述烷氧基和烷基是直链或支链的。[式S2]在式S21至S26中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,优选是具有2-3个碳原子的烷氧基,更优选是乙氧基,且余者是具有1-10个碳原子的烷基。所述烷氧基和烷基是直链或支链的。[式S3]在式S31至S38中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,优选是具有2-3个碳原子的烷氧基,更优选是乙氧基,且余者是具有1-10个碳原子的烷基。所述烷氧基和烷基是直链或支链的。本发明的一个实施方式中所述的具有烷氧基甲硅烷基的环氧化合物中的环氧基独立地选自以下式S5(3)中的式S51至S58。[式S5(3)]在式S56至S58中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,优选是具有2-3个碳原子的烷氧基,更优选是乙氧基,且余者是具有1-10个碳原子的烷基。所述烷氧基和烷基是直链或支链的。所述烷氧基甲硅烷基化环氧化合物可是包括以下物质作为核结构的化合物:双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族或酚醛清漆单元。此外,在烷氧基甲硅烷基化环氧化合物中,所述核结构可以是芳族核结构且可以选自式A’至N’。在本发明中,核结构被理解为包括式A’至J’的结构和包含重复单元的结构(如式K’至N’)。在式A’中,-q-是–CH2-或直接连接,在式D’中,-r-是–C(CH3)2-、-CH2-、-C(CF3)2-、-SO2-、-S-、在式K’中,s是在式N’中,t是且在式K’至N’中,n是大于或等于1的整数。此外,当烷氧基甲硅烷基化环氧化合物中(A’)至(J’)的核结构数目大于或等于2时(其中所述核结构是相同类型,且也可适用于包含至少两种核结构的情况,如下文所述),所述核结构可通过连接基团LG连接。在需要的情况下,还可连接1-1000个核结构。具体而言,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团LG连接,且式(J’)的核结构可通过独立地选自式LG2至LG9的连接基团连接。通过连接基团,还可连接1-1000个核结构。对于至少三个核结构,可使用至少两个连接基团,且在这种情况下,各连接基团可以是相同或不同的。当具有多个连接基团时,所述连接基团可独立地选自多种连接基团。[式6(2)]在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,余者是具有1-10个碳原子的烷基,且所述烷氧基和烷基是直链或支链的,且在式LG4、LG5、LG7、LG11、LG12和LG14中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。在式LG8至LG14中,R1至R3中的至少一个是具有1-5个碳原子的烷氧基,优选是具有2-3个碳原子的烷氧基,且更优选是乙氧基,且余者是具有1-10个碳原子的烷基。所述烷氧基和烷基是直链或支链的。此外,本发明的具有烷氧基甲硅烷基的环氧化合物还可包括选自式S41至S45的取代基S4。[式S4]具体而言,例如,当所述核结构是上文式(F’)的萘核结构时,本发明的实施方式所述的环氧化合物可表示为下式(FI),且当所述核结构是上文式(G’)的联苯核结构时,本发明的实施方式所述的环氧化合物可表示为下式(GI)。在本发明的具有烷氧基甲硅烷基的环氧化合物和本申请所述的任何化合物中,基于本领域已知技术,本领域技术人员可清楚地理解取代基(如烷氧基甲硅烷基和环氧基)对核结构的键合(取代)和核结构通过连接基团的连接。后文中,将详细解释萘核结构和联苯核结构。此外,从解释中,本领域技术人员易于理解本发明中通过连接基团连接且具有环氧基、烷氧基甲硅烷基等的烷氧基甲硅烷基化环氧化合物的结构。此外,本发明的环氧化合物还可包括除环氧化合物单聚体以外的二聚体、三聚体等的混合物,且这些特征是本领域中显而易见的。在式FI和GI中,多个W中的至少一个可以是(1)独立地选自式S11至S16的S1取代基、(2)独立地选自式S21至S26的S2取代基、(3)独立地选自S1取代基(其独立地选自式S11至S16)和S3取代基(其独立地选自式S31至S38)的烷氧基甲硅烷基,或(4)独立地选自S2取代基(其独立地选自式S21至S26)和S3取代基(其独立地选自式S31至S38)的烷氧基甲硅烷基,且其中至少两个是独立地选自式S5(3)中式S51至S54和S56至S58的环氧取代基,除S1、S2、S3和S5(3)以外的其余基团可以独立地选自H和S4取代基(其独立地选自式S41至S45),LG选自式LG1至LG14,且m是0-1000的整数。在本发明的实施方式所述具有烷氧基甲硅烷基的环氧化合物中,考虑到固化反应期间与填料的反应稳定性和/或反应性,优选R1至R3中的至少一个烷氧基是乙氧基。在本说明书中,“烷氧基”指的是一价基团–OR(R是烷基),其可以是直链或支链的。在本说明书中,“烷基”指的是单价烃基,其可以是直链或支链的。此外,在通过使根据本发明实施方式的包含具有烷氧基甲硅烷基的环氧化合物的组合物固化而得到的复合材料时,可观察到低CTE,高玻璃化转变温度或无Tg。根据本发明的另一个方面,提供了具有羟基和烷基、包含选自式(A’)至(N’)的核结构以及至少一个选自式S41至S45的取代基和至少两个独立地选自S51至S55的环氧基的环氧化合物。其中,如果式(A’)至(J’)的核结构数目大于或等于2,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且式(J’)的核结构可通过式LG2的连接基团连接。通过连接基团,还可连接1-1000个核结构。[式S5(1)][式S6(1)]在式LG4、LG5和LG7中,是氢、缩水甘油基或其中是与另一核结构相连的连接部分,如上文所定义,且是根据核结构的数目重复的。2.制备环氧化合物的方法本发明的实施方式所述的烷氧基甲硅烷基化环氧化合物可通过环氧开环反应和烷氧基甲硅烷基化制备,所述环氧开环反应是通过环氧基与烯丙基化合物反应来进行。因此,根据本发明的实施方式,提供了制备烷氧基甲硅烷基化环氧化合物的方法,所述方法包括通过使具有至少三个环氧基的环氧化合物的起始材料与烯丙基化合物反应进行的环氧开环反应(第一步)、烷氧基甲硅烷基化(第二步)和可选的额外的烷氧基甲硅烷基化(第三步)。所述烷氧基甲硅烷基化可包括两种类型的异氰酸酯甲硅烷化和氢化甲硅烷化反应,并且根据异氰酸酯甲硅烷化和氢化甲硅烷化的反应顺序,主要可通过两种方法合成本发明的烷氧基甲硅烷基化环氧化合物。后文中将给出详细的描述,其关于通过起始材料的环氧开环反应(第一步)、异氰酸酯甲硅烷化(第二步)和可选的氢化甲硅烷化(第三步)来制备烷氧基甲硅烷基化环氧化合物的方法(称为制备方法1);以及通过起始材料的环氧开环反应(第一步)、氢化甲硅烷化(第二步)和可选和额外的异氰酸酯甲硅烷化(第三步)来制备烷氧基甲硅烷基化环氧化合物的方法(称为制备方法2)。(1)制备方法1在第一步中,通过使具有至少三个环氧基的环氧化合物的起始材料与烯丙醇(CH2CHCH2OH)反应来对环氧化合物的环氧基进行开环以获得中间体。在第一步中,具有至少三个环氧基的环氧化合物的起始材料与CH2CHCH2OH的反应是在碱和可选的溶剂存在的情况下进行的。在该情况下,对每1当量的起始材料的环氧基使用0.1-10当量的CH2CHCH2OH来进行反应。此外,可在需要的情况下使用可选的卤化铵以提高碱在反应溶剂中的溶解度。在使用卤化铵的情况下,每1当量的起始材料的环氧基使用0.05-5当量的卤化铵以提高碱在反应溶剂中的溶解度,从而缩短第一步的反应时间。所述起始材料可以是通常已知具有至少三个环氧基的材料的任何环氧化合物。例如,可包括具有至少三个环氧基的缩水甘油醚类环氧化合物、缩水甘油基类环氧化合物、缩水甘油胺类环氧化合物或缩水甘油酯类环氧化合物。更具体地,可包括具有至少三个环氧基并具有以下物质作为核结构的环氧化合物:双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族或酚醛清漆单元。更具体地,作为起始材料的具有至少三个环氧基的环氧化合物包含选自式(A’)至(N’)的一类核结构和至少三个选自式S51至S54的环氧基。此外,当式(A’)至(J’)的核结构数目大于或等于2时,式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且上述式(J’)的核结构可通过式LG2的连接基团连接。通过连接基团,还可连接1-1000个核结构。可根据反应物的种类改变第一步反应的反应温度和反应时间,并且可通过在例如室温(例如15C°至25C°)至120℃下进行1-120小时的反应来对环氧化合物起始材料的环氧基进行开环,从而生成中间体。所述中间体可以是包含至少一个独立地选自式S41至S45的取代基和至少两个独立地选自式S51至S55的环氧基的环氧化合物,且具体而言是缩水甘油醚型、缩水甘油基型、缩水甘油胺型或缩水甘油酯型环氧化合物。更具体地,所述中间体可以是一种环氧化合物,该环氧化合物具有以下物质作为核结构:包含至少一个独立地选自式S41至S45的取代基,至少两个独立地选自式S51至S55的环氧基,以及双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族或酚醛清漆单元。更具体地,所述中间体可包含选自式(A’)至(N’)的核结构、至少一个独立地选自式S41至S45的取代基和至少两个独立地选自式S51至S55的环氧基。当式(A’)至(J’)的核结构数目大于或等于2时,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且式(J’)的核结构可通过式LG2的连接基团连接。使用的碱可包括但不限于,例如KOH、NaOH、K2CO3、Na2CO3、KHCO3、NaHCO3、NaH、三乙胺和二异丙基乙基胺。这些碱可单独使用,或以两种或更多种碱的组合使用。考虑到反应效率,对于起始材料中每1当量的环氧基团,可使用0.1-5当量的碱。使用的卤化铵可包括但不限于,例如碘化四丁基铵(Bu4NI)、溴化四丁基铵(Bu4NBr)、溴化四乙基铵(Et4NBr)、四丁基硝酸铵(Bu4N+NO3-)和氯化铵(NH4Cl)。这些卤化铵可单独使用,或以两种或更多种卤化铵的组合使用。考虑到反应效率,对于起始材料中每1当量的环氧基,可使用0.05-5当量的卤化铵。此外,还可由本领域技术人员来确定卤化铵的使用。第一步反应过程中可以任选地使用溶剂。例如,在第一步反应中如果在反应温度下反应物的粘度适合于进行反应而不需要额外的溶剂,则可以不使用溶剂。也就是说,在反应物的粘度足够低,对反应物的混合和搅拌可以在不存在溶剂的条件下容易进行,则单独的溶剂不是必需的。本领域普通技术人员很容易对此作出决定。在使用溶剂的情况下,可以使用任意有机溶剂,前提是该溶剂能够适当地溶解反应物,不会引起对反应的不利影响,以及反应后能够易于除去。例如,可使用乙腈、四氢呋喃(THF)、甲基乙基酮(MEK)、丙酮、二甲基甲酰胺(DMF)、二甲亚砜(DMSO)、二氯甲烷、乙腈等,但不限于此。这些溶剂可单独使用,或作为两种或更多种溶剂的混合物使用。溶剂的量可不限定到具体范围内,且在充分溶解反应物并且不会对反应产生不利影响的所需范围内可使用合适量的溶剂。考虑上述要点,本领域普通技术人员可选定合适量的溶剂。在第二步中,通过使第一步中获得的中间体与下文式(B1)的烷氧基硅烷反应来对该中间体进行异氰酸酯甲硅烷化,以生成在核结构中具有至少一个独立地选自式S11至S16的烷氧基甲硅烷基和至少两个环氧基的环氧化合物。第二步中获得的产物还对应于烷氧基甲硅烷基化环氧化合物,一种本发明的实施方式所述的终产物。具体而言,通过使中间体与下文式B1的异氰酸酯烷氧基硅烷反应,第一步中形成的中间体的仲醇(羟基)可被甲硅烷化以形成独立地选自式S11至S16的烷氧基甲硅烷基。在第二步的反应中,第一步的中间体与下文式B1的烷氧基硅烷的反应可按化学计量进行。此外,如上文所述,式B1的烷氧基硅烷可与中间体的仲醇基团(羟基)反应。因此,考虑到上文所述各点,式B1的烷氧基硅烷与中间体的反应可以以每1当量的中间体醇基团对应0.1-5当量的烷氧基硅烷的比例来进行。[式B1]OCN(CH2)3SiR1R2R3在式B1中,R1至R3中的至少一个是C1-C5烷氧基,优选是C2-C3烷氧基,更优选是乙氧基,且余者是C1-C10烷基。所述烷氧基和烷基可以是直链或支链的。根据反应物,第二步反应的反应温度和反应时间可以是不同的,且可以是例如在室温(如15℃-25℃)至120℃的温度范围下进行1-72小时。第二步的反应是在碱存在的情况下进行的。使用的碱可包括但不限于,例如K2CO3、Na2CO3、KHCO3、NaHCO3、三乙胺和二异丙基乙基胺。这些碱可单独使用,或以两种或更多种碱的组合使用。考虑到反应效率,对于起始材料中每1当量的羟基,可使用0.1-5当量的碱。用于第二步反应的溶剂可以任选地使用溶剂。例如,在第二步反应中,如果在反应温度下反应物的粘度适合于进行反应而不需要额外的溶剂,则可以不使用溶剂。也就是说,在反应物的粘度足够低,对反应物的混合和搅拌可以在不存在溶剂的条件下容易进行,则额外的溶剂不是必需的。本领域普通技术人员很容易对此作出决定。在使用溶剂的情况下,可以使用任意非质子性溶剂,只要该溶剂能够适当地溶解反应物,不会引起对反应的不利影响,以及反应后能够易于除去即可。例如,可使用甲苯、乙腈、THF、MEK、DMF、DMSO、二氯甲烷、乙腈等,且不限于此。这些溶剂可单独使用,或作为两种或更多种溶剂的混合物使用。溶剂的量可不限定到具体范围内,且在充分溶解反应物并且不会对反应产生不利影响的所需范围内可使用合适量的溶剂。考虑上述要点,本领域普通技术人员可选定合适量的溶剂。如上文所述,第二步中获得的环氧化合物还具有环氧基和烷氧基甲硅烷基且对应于具有烷氧基甲硅烷基的环氧化合物(本发明的目标产物)。具体而言,第二步中获得的具有烷氧基甲硅烷基的环氧化合物具有选自式(A’)至(N’)的核结构、至少一个选自式S11至S16的烷氧基甲硅烷基、至少两个选自式S51至S55和S57的环氧基和可选的选自式S41至S45的上述式S4的取代基。当式(A’)至(J’)的核结构数目大于或等于2时,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过独立地选自LG2和LG9的连接基团连接。通过连接基团,还可连接1-1000个核结构。第二步后,还可根据需要进行可选的第三步氢化硅烷化。在第三步中,通过使第二步反应产物与上文式B2的烷氧基硅烷反应来对第二步的产物的烯丙基进行氢化甲硅烷化,以生成具有至少一个独立地选自式S11至S16和式S31至S38的烷氧基甲硅烷和至少两个环氧基的本发明的实施方式所述的烷氧基甲硅烷基化环氧化合物。在第三步的反应中,第二步的反应产物和下文式B2的烷氧基硅烷可通过化学计量当量摩尔比进行反应。考虑到上述各点,第二步的反应产物与下文式B2的烷氧基硅烷可以每1当量第二步的反应产物的烯丙基对应0.1-5当量式B2的烷氧基硅烷来进行反应。[式B2]HSiR1R2R3在式B2中,R1至R3中的至少一个是C1-C5烷氧基,优选是C2-C3烷氧基,更优选是乙氧基,且余者是C1-C10烷基。所述烷氧基和烷基是直链或支链的。根据反应物,第三步反应的反应温度和反应时间可以是不同的,且可以通过例如在室温(如15℃-25℃)至120℃的温度范围下反应1-72小时来完成反应。第三步的反应是在金属催化剂存在的情况下进行的。例如,铂催化剂PtO2或氯铂酸(H2PtCl6)可用作金属催化剂,但不限于此。考虑到反应效率,优选每1当量的第二步反应产物的烯丙基使用1x10-4-0.05当量的铂催化剂。可根据需要任选地在第三步中使用溶剂且所述溶剂的类型、量和用法与第二步中相同。具体而言,第三步中获得的具有烷氧基甲硅烷基的环氧化合物具有选自式(A’)至(N’)的核结构、至少一个独立地选自式S11至S16和式S31至S38的烷氧基甲硅烷基、至少两个选自式S51至S58的环氧基和可选的选自式S41至S45的取代基。当式(A’)至(J’)的核结构数目大于或等于2时,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过独立地选自LG2和LG9的连接基团连接。通过连接基团,还可连接1-1000个核结构。用于包含四苯基乙烷芳族核结构的环氧化合物的制备方法1的反应机制如下文所述。[制备方法1-1]其中,m是0-1000的整数。[制备方法1-2]其中,m是0-1000的整数。(2)制备方法2在制备方法2中,本发明的烷氧基甲硅烷基化环氧化合物通过起始材料的开环反应(第一步)、氢化甲硅烷化(第二步)和可选的异氰酸酯甲硅烷化(第三步)来制备。第一步起始材料的开环反应与制备方法1相同。随后,在第二步中,通过使第一步中获得的中间体与上文式(B2)的烷氧基硅烷反应来对该中间体进行氢化甲硅烷化,以生成具有至少一个独立地选自式S21至S26的烷氧基甲硅烷基和至少两个环氧基的环氧化合物。第二步中获得的产物还对应于烷氧基甲硅烷基化环氧化合物,一种本发明的实施方式所述的最终目标产物。具体而言,通过中间体与上文式(B2)的烷氧基硅烷反应,中间体的烯丙基被甲硅烷化以形成独立地选自式S21至S26的烷氧基甲硅烷基。在第二步的反应中,第一步的中间体与下文式B2的烷氧基硅烷的反应可按化学计量进行。此外,如上文所述,式B2的烷氧基硅烷可与中间体的烯丙基反应。因此,考虑到上文所述各点,式B2的烷氧基硅烷与中间体的反应可以以每1当量的中间体的烯丙基对应0.1-5当量的烷氧基硅烷的比例来进行。根据反应物,第二步反应的反应温度和反应时间可以是不同的,且可以是例如在室温(如15℃-25℃)至120℃的温度范围下进行1-72小时。第二步的反应是在金属催化剂存在的情况下进行的。例如,铂催化剂PtO2或H2PtCl6(氯铂酸)可用作金属催化剂,但不限于此。考虑到反应效率,优选每1当量的中间体的烯丙基使用1x10-4-0.05当量的铂催化剂。可根据需要任选地在第二步中使用溶剂且所述溶剂的类型、用法和量与制备方法1的第二步中相同。如上文所述,第二步中获得的环氧化合物还具有环氧基和烷氧基甲硅烷基且还对应于具有烷氧基甲硅烷基的环氧化合物(本发明的目标材料)。具体而言,第二步中获得的具有烷氧基甲硅烷基的环氧化合物具有至少一个选自式(A’)至(N’)的核结构、至少一个独立地选自式S21至S26的烷氧基甲硅烷基、至少两个选自式S51至S56的环氧基和可选的选自式S41至S45的取代基。当式(A’)至(J’)的核结构数目大于或等于2时,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG7的连接基团连接且上述式(J’)的核结构可通过式LG2的连接基团连接。通过连接基团,还可连接1-1000个核结构。第二步后,还可根据需要进行可选的第三步异氰酸酯甲硅烷化。在第三步中,通过使第二步反应产物与上文式B1的烷氧基硅烷反应来对第二步的产物的羟基进行异氰酸酯甲硅烷化,以生成在核结构中具有至少一个独立地选自式21至式26以及式S31至S38的烷氧基甲硅烷和至少两个环氧基的本发明的实施方式所述的烷氧基甲硅烷基化环氧化合物。在第三步的反应中,第二步的反应产物和下文式B1的烷氧基硅烷可通过化学计量当量摩尔比进行反应。考虑到上述各点,第二步的反应产物与下文式B1的烷氧基硅烷可以每1当量第二步的反应产物的羟基使用0.1-5当量式B1的烷氧基硅烷来进行反应。根据反应物种类,可以改变第三步反应的反应温度和反应时间,且可以是例如在室温(如15℃-25℃)至120℃的温度范围下进行1-72小时。第三步的反应是在碱存在的情况下进行的。使用的碱的类型与制备方法1的第二步中使用的相同。考虑到反应效率,优选相对于1当量的第二步反应产物的羟基使用0.1-5当量的碱。在第三步中,可根据需要任选地使用溶剂且所述溶剂的类型、量和用法与第二步中相同。具体而言,第三步中获得的具有烷氧基甲硅烷基的环氧化合物具有选自式(A’)至(N’)的核结构、至少一个独立地选自式S21至S26和式S31至S38的烷氧基甲硅烷基、至少两个选自式S51至S58的环氧基和可选的选自式S41至S45的取代基。当如果式(A’)至(J’)的核结构数目大于或等于2时,则式(A’)至(I’)的核结构可通过独立地选自式LG1至LG14的连接基团连接且式(J’)的核结构可通过独立地选自LG2和LG9的连接基团连接。通过连接基团,还可连接1-1000个核结构。[制备方法2-1]其中,m是0-1000的整数。[制备方法2-2]其中,m是0-1000的整数。3.环氧组合物根据本发明的另一个实施方式,环氧组合物包含具有烷氧基甲硅烷基的环氧化合物,其包含至少一个烷氧基甲硅烷基,该烷氧基甲硅烷基是(1)独立地选自式S11至S16的式S1的取代基、(2)独立地选自式S21至S26的式S2的取代基、(3)独立地选自式S11至S16和式S31至S38的取代基,或(4)独立地选自式S21至S26和式S31至S38的取代基;以及至少两个环氧基。具体而言,提供了包含上述1.环氧化合物的类别中所述的本发明任何实施方式中提供的任何烷氧基甲硅烷基化环氧化合物(下文中称作“烷氧基甲硅烷基化环氧化合物”)的环氧组合物。本发明提供的任意组合物可用于各种用途,例如电子材料,如电子部件,诸如半导体基材如IC基材,累积膜(build-upfilm),包封材料(包装材料),印刷电路板,粘合剂,油漆,复合材料等,但不限于此。此外,本发明提供的任意组合物可以是可固化的组合物和/或包含无机材料的可固化组合物。仅当根据本发明的上述或下述任意实施方式的任意环氧组合物包含本发明的任意实施方式中提供的烷氧基甲硅烷基化环氧化合物时,该环氧组合物可以包括本领域已知的任意类型和/或任意混合比。在这种情况下,不限制固化剂、固化促进剂(催化剂)、无机材料(填料)(例如,无机颗粒和/或纤维)、其它常规环氧化合物和其它添加剂的种类和混合比。此外,根据环氧组合物、固化产物和/或复合材料的应用和/或用途,考虑到控制物理性质的特点,所述环氧组合物、固化产物和/或复合材料可以与各种类型的常规环氧化合物一起使用。因此,在根据本发明的上述或下述任意实施方式的任意环氧组合物中,所述环氧化合物可包括任意类型的本领域公知的环氧化合物(下文称作“常规环氧化合物”)以及本发明的任意实施方式中提供的烷氧基甲硅烷基化环氧化合物(下文称作“本发明的烷氧基甲硅烷基化环氧化合物”)。所述常规环氧化合物可以是不受限制的本领域公知的任意环氧化合物,可以是例如至少一种选自下组的环氧化合物:缩水甘油醚基环氧化合物、缩水甘油基环氧化合物、缩水甘油胺基环氧化合物、缩水甘油酯基环氧化合物、橡胶改性环氧化合物、脂族聚缩水甘油基环氧化合物和脂族缩水甘油胺基环氧化合物。例如,所述常规环氧化合物可以是至少一种选自下组的环氧化合物:缩水甘油醚基环氧化合物、缩水甘油基环氧化合物、缩水甘油胺基环氧化合物、缩水甘油酯基环氧化合物、橡胶改性环氧化合物、脂族聚缩水甘油基环氧化合物和脂族缩水甘油胺基环氧化合物,其包含双酚A、双酚F、双酚S、联苯、萘、苯、硫代二苯酚、芴、蒽、异氰脲酸酯、三苯基甲烷、1,1,2,2-四苯基乙烷、四苯基甲烷、4,4'-二氨基二苯甲烷、氨基苯酚、环脂族或酚醛清漆单元作为核结构。以环氧化合物的总重量为基准计,根据本发明实施方式的任意环氧组合物可包括,但不限于:1-100重量%的根据本发明的烷氧基甲硅烷基化环氧化合物和0-99重量%的常规环氧化合物;例如10-100重量%的本发明的烷氧基甲硅烷基化环氧化合物和0-90重量%的常规环氧化合物;例如30-100重量%的本发明的烷氧基甲硅烷基化环氧化合物和0-70重量%的常规环氧化合物;例如50-100重量%的本发明的烷氧基甲硅烷基化环氧化合物和0-50重量%的常规环氧化合物;例如10重量%至小于100重量%的本发明的烷氧基甲硅烷基化环氧化合物和大于0重量%至90重量%的常规环氧化合物;例如30重量%至小于100重量%的本发明的烷氧基甲硅烷基化环氧化合物和大于0重量%至70重量%的常规环氧化合物;例如50重量%至小于100重量%的本发明的烷氧基甲硅烷基化环氧化合物和大于0重量%至50重量%的常规环氧化合物。此外,根据本发明的实施方式,提供了一种环氧组合物,其包含烷氧基甲硅烷基化环氧化合物和无机材料(填料)(例如无机颗粒和/或纤维)(下文称作“复合物组合物”)。仅当复合物组合物包含本发明的烷氧基甲硅烷基化环氧化合物和填料时,认为该复合物组合物包括具有本领域公知的任意类型和/或任意混合比的环氧组合物。不限制组成环氧组合物的固化剂、固化促进剂(催化剂)、无机材料(填料)(例如,无机颗粒和/或纤维)、常规环氧化合物和其它添加剂的种类和混合比。上述复合物组合物和根据本发明的上述或下述的任意组合物可额外地包含无机颗粒和/或纤维。可使用已知用于加强常规有机树脂的物理性质的任意无机颗粒。无机颗粒的示例可包括但不限于至少一种选自下组的材料:至少一种选自二氧化硅(包括如熔融二氧化硅和晶态二氧化硅)、氧化锆、氧化钛、氧化铝、氮化硅和氮化铝的金属氧化物、和T-10型硅倍半氧烷、梯型硅倍半氧烷和笼型硅倍半氧烷。这些无机颗粒可单独使用,或以两种或更多种无机颗粒的混合物形式使用。在混合特别大量二氧化硅的情况下,优选使用熔融二氧化硅。熔融二氧化硅可具有介于碎裂形状和球形之间的任意形状。但是,为了增加熔融二氧化硅的填充因子和限制复合材料的熔融粘度的增加,球形是优选的。考虑到复合材料的用途,特别是无机颗粒的可分散性等,可使用粒径为0.5nm至几十μm(例如50μm-100μm)的无机颗粒。因为无机颗粒分散在环氧化合物中且分散能力可随粒径而不同,所以可优选地将具有上述范围内的不同粒径尺寸的无机颗粒一起使用。此外,优选地,增加待混合的无机颗粒的粒径分布以增大无机颗粒的填充因子。在根据本发明的实施方式的环氧组合物中,考虑到环氧复合材料的CTE减少和施用时所需的适当粘度,可适当控制无机颗粒相对于环氧化合物的填充因子。例如,所述无机颗粒的量可为5-95重量%,例如5-90重量%,例如10-90重量%,例如30-95重量%,例如30-90重量%,例如5-60重量%,例如10-50重量%,该含量基于环氧组合物的固体含量的总重量(对于环氧固化产物而言,基于环氧固化产物的总重量)。更具体而言,在一个示例性实施方式中,当所述环氧组合物用作半导体包装剂等时,考虑到CTE值和材料可加工性,无机颗粒的含量可以是例如30-95重量%,例如30-90重量%,但不限于此,该含量基于环氧组合物的总固体重量(对于环氧固化产物而言,基于环氧固化产物的总重量)。在其它示例性实施方式中,当所述环氧组合物用于半导体基材时,考虑到CTE值和基材的强度,无机颗粒的含量可以是5-60重量%,例如10-50重量%,该含量基于环氧组合物的总固体重量(对于环氧固化产物而言,基于环氧固化产物的总量)。同时,当纤维用作无机材料时,复合材料可主要通过在环氧组合物内浸渍纤维来获得。因此,可不具体限定纤维的尺寸。可使用本技术领域常用的任意种类的纤维,且不限定纤维的尺寸。可使用用来改善常规已固化有机树脂的物理性质的任意常规纤维,但不限于此。具体来说,可使用玻璃纤维、有机纤维或它们的混合物。此外,本申请中所用术语“玻璃纤维”可包括玻璃纤维织物、玻璃纤维无纺产品等,以及玻璃纤维。玻璃纤维的示例可包括但不限于:E玻璃纤维、T玻璃纤维、S玻璃纤维、NE玻璃纤维、D玻璃纤维、石英玻璃纤维等。例如可包括E或T玻璃纤维。有机纤维可包括至少一种选自下组的材料:液晶聚酯纤维、聚对苯二甲酸乙二醇酯纤维、全芳族纤维、多氧苯扎索尔纤维、尼龙纤维、聚萘二甲酸乙二醇酯纤维、聚丙烯纤维、聚醚砜纤维、聚偏二氟乙烯纤维、聚硫化乙烯纤维和聚醚醚酮纤维。这些纤维可以单独地使用,或者以两种或更多种组合的形式使用。基于所述环氧组合物的固体总重量,本发明的环氧组合物中(例如在环氧组合物的玻璃纤维复合材料中)的纤维含量可以是10-90重量%,例如30-70重量%,此外例如35-65重量%。此外,基于所述固化产物的总重量,在所述环氧组合物的固化产物中(例如在玻璃纤维复合材料中)的纤维含量可以是10-90重量%,例如30-70重量%,此外例如35-65重量%。因此,树脂添加量的两可以是10-90重量%,例如30-70重量%,此外例如35-65重量%。考虑到耐热性和可加工性的提高,上述范围内的纤维含量可以是优选的。同时,在包含纤维的环氧组合物、固化产物等中,从固体总含量中排除纤维之后的固体份数称作树脂。在包含纤维的环氧组合物中,除纤维之外的剩余量是树脂含量。此外,在包含纤维的环氧组合物中可根据需要额外地包含无机颗粒。在这种情况下,考虑到物理性质和可加工性的改善,以树脂的总重量为基准计,可包含1-70重量%的无机颗粒。在这种情况中,无机颗粒的种类没有具体限制,可使用本领域已知的任意无机颗粒。例如,可使用上述无机颗粒。根据本发明的另一个实施方式,提供了烷氧基甲硅烷基化环氧组合物(后文中称作“含固化剂的组合物”),其包含本发明任意实施方式所述的环氧化合物和固化剂。任何含固化剂组合物只有在包含本发明的烷氧基甲硅烷基化环氧化合物和固化剂时,才可包含具有本领域中已知的任何类型和/或任何混合比例的环氧组合物。但是,组成环氧组合物的固化剂、固化促进剂(催化剂)、无机材料(填料)(例如,无机颗粒和/或纤维)、其它常规环氧化合物和其它添加剂的种类和混合比不受限制。根据本发明的另一个实施方式,提供了烷氧基甲硅烷基化环氧组合物(后文中称作“含反应催化剂组合物”),其包含本发明任意实施方式所述的环氧化合物和用于烷氧基甲硅烷基的反应催化剂(后文中称作“反应催化剂”)。任何含反应催化剂组合物只有在包含本发明的烷氧基甲硅烷基化环氧化合物和反应催化剂时,才可包含具有本领域中已知的任何类型和/或任何混合比例的环氧组合物。但是,组成环氧组合物的固化剂、固化促进剂(催化剂)、无机材料(填料)(例如,无机颗粒和/或纤维)、其它常规环氧化合物和其它添加剂的种类和混合比不受限制。在包含用于烷氧基甲硅烷基的反应催化剂的情况中,可预期改进的可加工性(例如快速的固化速率和/或低固化温度)。所述含固化剂组合物和含反应催化剂组合物还可包含常规环氧化合物作为环氧化合物。在这种情况下,常规环氧化合物的种类、烷氧基甲硅烷基化的环氧化合物和常规环氧化合物的混合比与上文所述相同。当含固化剂组合物和根据本发明实施方式的组合物中包含固化剂时,可使用公知作为环氧化合物的固化剂的任意固化剂。例如,可使用胺基树脂、苯酚基树脂、酸酐化合物等,但不限于此。具体来说,可使用脂族胺、脂环族胺、芳族胺、其它胺和改性的胺作为胺基固化剂,但不限于此。此外,可使用包括两个或更多个伯胺基的胺化合物。胺固化剂的具体示例可包括至少一种选自下组的芳族胺:4,4'-二甲基苯胺(二氨基二苯基甲烷、DAM或DDM)、二氨基二苯基砜(DDS)和间苯二胺,至少一种选自下组的脂族胺:二亚乙基三胺(DETA)、二亚乙基四胺、三亚乙基四胺(TETA)、间二甲苯二胺(MXTA)、甲烷二胺(MDA)、N,N'-二亚乙基二胺(N,N'-DEDA)、四亚乙基五胺(TEPA)和六亚甲基二胺,至少一种选自下组的脂环族胺:异佛尔酮二胺(IPDI)、N-氨基乙基哌嗪(AEP),和双(4-氨基3-甲基环己基)甲烷(larominc260),以及其它胺如二氰二胺(DICY)等,以及改性的胺如聚酰胺基化合物、环氧化物基化合物等。苯酚基固化剂的示例可包括但不限于:酚醛清漆树脂、三官能化苯酚酚醛清漆树脂、甲酚酚醛清漆树脂、双酚A酚醛清漆树脂、二甲苯酚醛清漆树脂、三苯基酚醛清漆树脂、联苯酚醛清漆树脂、苯酚对二甲苯树脂、苯酚4,4’-二甲基亚联苯树脂、苯酚二环戊二烯酚醛清漆树脂、二环戊二烯-苯酚(DCPD-苯酚)酚醛清漆树脂、新酚醛树脂(xylok)(对二甲苯改性的)树脂、联苯基苯酚树脂、萘基苯酚酚醛清漆树脂、三嗪基化合物、二羟基萘、二羟基苯等。酸酐固化剂的示例可包括但不限于:脂族酸酐如十二碳烯基琥珀酸酐(DDSA)、聚壬二酸聚酐等,脂环族酸酐如六氢邻苯二甲酸酐(HHPA)、甲基四氢邻苯二甲酸酐(MeTHPA)、甲基纳迪克(methylnadic)酸酐(MNA)等,芳族酸酐如偏苯三酸酐(TMA)、均苯四酸二酐(PMDA)、二苯甲酮四羧酸二酐(BTDA)等,含卤素酸酐化合物如四溴邻苯二甲酸酐(TBPA)、氯菌酸酐等。一般地,可通过固化剂与环氧基的反应程度来控制环氧复合材料的固化度。根据目标固化度的范围,可基于环氧化合物的环氧基的浓度来控制固化剂的量。例如,当在胺固化剂与环氧基的当量反应中使用胺固化剂时,环氧当量/胺当量的比例可优选地控制到0.5-2.0,例如0.8-1.5。尽管解释了胺基固化剂的固化剂的混合比,然而可根据所需的固化度范围,以环氧组合物中的环氧基的总浓度为基准,根据环氧官能团和固化剂的反应性官能团的化学反应,通过适当地混合化学计量量来使用未在该申请中单独说明但将其用于固化的苯酚基固化剂、酸酐基固化剂和用于固化环氧化合物的任意固化剂。上面描述的要素是本领域所公知的。对于阳离子光固化剂(也称作光引发剂),可使用该领域中公知的光固化剂,例如芳族磷鎓盐、芳族碘鎓盐、芳族锍盐等,但不限于此。具体来说,可使用二苯基碘鎓四(五氟苯基)硼酸盐、二苯基碘鎓六氟磷酸盐、二苯基碘鎓六氟锑酸盐、二(4-壬基苯基)碘鎓六氟磷酸盐、三苯基锍六氟磷酸盐、三苯基锍六氟锑酸盐、三苯基锍四(五氟苯基)硼酸盐、4,4'-双[二苯基锍]二苯硫醚双六氟磷酸盐、4,4'-双[二(β-羟基乙氧基)苯基锍]二苯硫醚双六氟磷酸盐等。所述光固化机可以以0.5-20份数/百份(phr)环氧化合物(以每100重量份的环氧化合物为基准计的重量份)的比例使用,可优选至少1phr,可优选最多15phr。根据需要,可额外地包含任选的固化促进剂(催化剂)来促进本发明提供的任意环氧组合物中的固化反应。可使用本技术领域中通常用于固化环氧组合物的任意固化促进剂(催化剂),例如可使用咪唑、叔胺,季铵、有机酸盐、磷化合物固化促进剂,但不限于此。具体来说,可列举例如咪唑固化促进剂如二甲基苄基胺、2-甲基咪唑(2MZ),2-十一烷基咪唑、2-乙基-4-甲基咪唑(2E4M)、2-苯基咪唑、1-(2-氰乙基)-2-烷基咪唑和2-十七烷基咪唑(2HDI);叔胺固化促进剂,如苄基二甲基胺(BDMA)、三(二甲基氨基甲基)苯酚(DMP-30)和三亚乙基二胺;季铵固化促进剂如溴化四丁铵等;二氮杂双环十一碳烯(DBU),或DBU的有机酸盐;磷化合物固化促进剂如三苯基膦、磷酸酯等,以及路易斯酸如BF3-单乙胺(BF3-MEA)等,但不限于此。还可使用通过固化促进剂盐的微胶囊化和络合形成的潜伏性固化促进剂。根据固化条件,这些化合物可单独使用,或以两种或更多种的混合物形式使用。固化促进剂的混合的量可以是本领域通常采用的混合量,不作限定。例如,以环氧化合物为基准计,可使用0.1-10phr(份数/百份树脂,以每100重量份的环氧化合物为基准计的重量份),例如0.2-5phr的固化促进剂。考虑到固化反应促进效果和对固化反应速率的控制,可优选地使用上述固化促进剂的范围。通过使用上述范围的固化促进剂,但可实现快速固化,并可预期改善加工生产量。当含反应催化剂的组合物和根据本发明任意实施方式的组合物中包含用于烷氧基甲硅烷基的反应催化剂时,用于烷氧基甲硅烷基的反应催化剂可以是至少一种选自例如硝酸、硫酸、盐酸、乙酸和磷酸的无机酸、包含羧基的有机酸、氨、KOH、NH4OH、胺、过渡金属烷氧化物、金属氧化物、有机酸盐和金属卤化物、和锡化合物(例如二月桂酸二丁基锡、辛酸锡、2-乙基己酸锡(II)等)。这些化合物可单独使用,或以两种或更多种化合物的混合物的形式使用。用于烷氧基甲硅烷基的反应催化剂的混合比不受具体限制;但是考虑到反应性,对于本发明的环氧化合物,可使用0.01-10phr的用于烷氧基甲硅烷基的反应催化剂。在包含用于烷氧基甲硅烷基的反应催化剂的组合物中,可额外地包含水以提高反应催化剂的效率。所述混合比不受具体限制;但是考虑到作为催化剂的效率和反应活性,相对于1当量的烷氧基甲硅烷基,可包含0.01-20当量的水。在环氧组合物中,根据需要,在不破坏环氧组合物的物理性质的范围内,可混合其它添加剂如脱模剂,表面处理剂,阻燃剂,增塑剂,杀细菌剂,流平剂,消泡剂,着色剂,稳定剂,耦联剂,粘度控制剂,稀释剂、橡胶、热塑性树脂等,以控制环氧组合物的物理性质。例如,当使用本发明的任意组合物形成薄膜时,以及当使用挠性不足的组合物形成薄层时,由此形成的薄层可能是脆性的,可能很容易形成裂纹。例如当本发明的组合物包括大量的无机颗粒时,可能会出现这种现象。因此,为了通过赋予组合物以溶解性来改进薄膜的可加工性,可向本发明的环氧组合物中加入橡胶和/或热塑性树脂。对于热塑性树脂和橡胶改性的环氧树脂,可使用本领域公知的树脂。对于橡胶,可使用本领域已知的任意橡胶,前提是该橡胶不会溶解在组合物中使用的溶剂中并且在组合物中保持分散的状态。这类橡胶可包括,例如丙烯腈丁二烯橡胶、丁二烯橡胶、丙烯酰基橡胶、芯-壳类型的橡胶颗粒、交联的丙烯腈丁二烯橡胶颗粒、交联的苯乙烯丁二烯橡胶颗粒、丙烯酰基橡胶颗粒等,但不限于此。这些材料可以单独使用,或者同时使用其中至少两种材料。当使用颗粒形状的橡胶时,考虑到改进物理性质,平均粒径可优选为0.005-1微米,更优选0.2-0.6微米。考虑到物理性质,以环氧组合物的固体重量为基准计,可以以0.5-10重量%的量比例来混合橡胶颗粒。对于热塑性树脂,可使用苯氧基树脂、聚乙烯基缩醛树脂、聚酰亚胺树脂、聚酰胺酰亚胺树脂、聚醚砜树脂、聚砜树脂等,但不限于此。这些材料可以单独使用,或者同时使用其中至少两种材料。考虑到物理性质,以环氧组合物的固体重量为基准计,可以以0.5-60重量%,优选3-50重量%的比例来混合热塑性树脂。如上所述,在本申请中术语“环氧组合物”应理解成包括本发明的环氧化合物和组成该环氧组合物的其它成分,例如在本领域中根据需要混合的任选的固化剂、固化促进剂(催化剂)、无机材料(填料)(例如无机颗粒和/或纤维)、其它常规环氧化合物、溶剂和其它添加剂。一般地,考虑到环氧组合物的可加工性等,任选地可使用溶剂来控制环氧组合物的固体物料的量和/或粘度。同时,本发明中使用的“环氧组合物的固体总含量”涉及除液体组分(例如组成环氧组合物的溶剂)之外的固体组分的总量。根据本发明的一个示例性实施方式提供的环氧组合物可用于电子材料。所述电子材料可包括例如电子部件,例如用于半导体的基材、膜、预浸渍体、通过在使用本发明的组合物形成的基底层上放置金属层获得的层叠体、基材、包封材料(包装材料)、累积膜(基材)、印刷电路板等。此外,所述环氧组合物可用于各种应用,如粘合剂、油漆和复合材料。根据本发明的其它示例性实施方式,提供了包含含有本发明的烷氧基甲硅烷基化环氧化合物的组合物的电子材料,或者使用含有本发明的烷氧基甲硅烷基化环氧化合物的组合物制造的电子材料。此外,提供了包括该电子材料的半导体设备,或基本上使用或使用该电子材料制造的半导体设备。具体来说,半导体设备可以是包括印刷电路板(例如用于安装半导体器件)的半导体设备和/或包含半导体包装材料的半导体设备,所述半导体包装材料包括含有本发明的烷氧基甲硅烷基化环氧化合物的组合物或通过基本上使用或使用本发明的烷氧基甲硅烷基化环氧化合物的组合物制得。此外,提供了包含含有本发明的任意实施方式所提供的任意环氧组合物的固化剂、粘合剂、油漆或复合材料或通过基本上使用或使用本发明的任意实施方式所提供的任意环氧组合物制得的固化剂、粘合剂、油漆或复合材料。根据本发明的其它示例性实施方式,可提供包含本发明的示例性实施方式所提供的环氧组合物的固化产物,或者通过基本上使用或使用本发明的示例性实施方式所提供的环氧组合物制备的固化产物。当实际应用采用本发明的示例性实施方式提供的环氧组合物时,例如把环氧组合物用作电子材料等时,可使用由环氧组合物形成的固化产物。在本领域中,通常把包括环氧化合物和无机组分填料的组合物形成的固化产物称为复合材料。上述本发明的示例性实施方式所提供的烷氧基甲硅烷基化环氧化合物可在复合材料中显示良好的耐热性和/或在固化产物中显示良好的阻燃性。具体来说,所述复合材料可显示低CTE,例如其CTE小于或等于15ppm/℃,例如小于或等于12ppm/℃、例如小于或等于10ppm/℃、例如小于或等于8ppm/℃、例如小于或等于6ppm/℃、例如小于或等于4ppm/℃。CTE值越小则复合材料的物理性质越好,且CTE的下限不受具体限制。例如,包括根据本发明的示例性实施方式的任意烷氧基甲硅烷基化环氧化合物作为环氧化合物、玻璃纤维(如E玻璃纤维和/或T玻璃纤维)作为无机材料、且具有30-60重量%的树脂含量(树脂含量可以包括或不包括无机颗粒)的复合材料,可具有小于或等于10ppm/℃、例如小于或等于8ppm/℃、例如小于或等于6ppm/℃、例如小于或等于4ppm/℃的CTE。此外,例如包括根据本发明的示例性实施方式的任意烷氧基甲硅烷基化环氧化合物作为环氧化合物、以及无机颗粒作为无机材料(如60-80重量%、如70-80重量%的二氧化硅颗粒)的复合材料,可具有小于或等于20ppm/℃、例如小于或等于15ppm/℃、例如小于或等于10ppm/℃、例如小于或等于8ppm/℃、例如小于或等于6ppm/℃、例如小于或等于4ppm/℃的CTE。此外,根据本发明的复合材料(含无机材料的固化产物)的Tg可高于100℃,例如高于或等于130℃,此外例如高于或等于250℃。或者,所述复合材料可以是无Tg的。当Tg值较高时,复合材料的物理性质是良好的,且没有具体限定Tg的上限值。同时,由根据本发明的烷氧基甲硅烷基化环氧化合物形成的固化产物(不含无机材料的固化产物)可具有50ppm/℃-150ppm/℃的CTE。在本发明中,除非另有特定说明,通过范围限定的值包括下限、上限、该范围中的任意子范围、和包括在该范围中的所有数值。例如,C1-C10应理解成包括全部的C1、C2、C3、C4、C5、C6、C7、C8、C9和C10。此外,在没有定义数值范围的下限或上限时,发现更小或更大的值可提供更好的性质。此外,在没有定义极限时,可包括任意值。例如,小于或等于4ppm/℃的CTE应理解成包括在该范围的每一个值,如4、3.5、3、2.7、2、1.4、1、0.5ppm/℃等的CTE。下文将参考优选实施方式详细地描述本发明。但是,以下实施方式用于说明,本发明不限于此。合成实施例1:具有烷氧基甲硅烷基的联萘环氧化合物(结构A-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的双(2,7-双(环氧乙烷-2-基甲氧基)萘-1-基)甲烷(DIC公司,EXA-4700,式A环氧)并分别加入THF和乙腈以溶解环氧化合物。随后向其中加入0.93g的NaOH、1.13g的Et4NBr和26.09g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体A-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.70-2.88(m,4H),3.26-3.37(m,2H),3.69-4.17(m,18H),4.90(s,2H),5.20-5.28(m,4H),5.86-5.98(m,2H),6.88-7.18(m,4H),7.42-7.62(m,6H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体A-0和乙腈并搅拌。随后向其中加入9.41g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.92g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂A-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=4Hz,4H),1.06-1.28(m,18H),1.42-1.72(m,4H),2.71-2.88(m,4H),3.15(t,J=8Hz,4H),3.26-3.37(m,2H),3.69-3.83(m,30H),4.90(s,2H),5.13-5.29(m,6H),5.86-5.98(m,2H),6.88-7.18(m,4H),7.42-7.62(m,6H)。上述合成实施例1的合成方案如下。上述合成实施例1的主要反应示于上述合成实施例1的合成方案中。然而,生成的化合物可具有1:1的环氧基:烯丙基比例,根据情况也可以是4:0、3:1、1:3和0:4。上述合成实施例中的环氧基:烯丙基比例指具有不同环氧基:烯丙基比例的全部混合的环氧化合物中的环氧基:烯丙基平均比例。此外,在合成方案中,取代在核结构的特定位置上进行以方便进行说明;然而,在能够发生取代的实际反应中,取代可在核结构的任意位置上进行。本发明应理解为包括上述所有情况。类似地,这些要点适用于下述合成实施例。此外,上述合成方案中还显示了烯丙基和缩水甘油醚官能团之间的反应;但烯丙基可与缩水甘油基反应。合成实施例2具有烷氧基甲硅烷基的联萘环氧化合物(结构A-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例1的第一步中获得的中间体A-0、135mg的PtO2、5.37g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧A-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.22(t,J=8Hz18H),1.56-1.61(m,4H),2.70-2.88(m,4H),3.24-4.17(m,32H),4.92(s,2H),6.88-7.18(m,4H),7.42-7.62(m,6H)。上述合成实施例2的合成反应如下。合成实施例3具有烷氧基甲硅烷基的四苯基乙烷环氧化合物(结构B-1(1))的合成(方法1)(1)第一步向双颈烧瓶中加入10g的四苯基乙烷的四缩水甘油醚(日本化药有限公司(NipponKayaku),GTR1800,式B-环氧),并分别加入20ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.84g的NaOH、1.01g的Et4NBr和23.32g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体B-0(1)。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.68(dd,J=4.8Hz,2.8Hz,2H),2.84(t,J=4.8Hz,2H),3.22-3.27(m,2H),3.80-3.84(m,4H),3.87-3.91(m,4H),4.00-4.01(m,4H),4.05-4.13(m,6H),4.47(s,2H),5.20(m,4H),5.87(m,2H),6.65(d,J=8.4Hz,8H),7.00(d,J=8.0Hz,8H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体B-0(1)和乙腈并搅拌。随后向其中加入8.57g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.48g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂B-1(1)。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,4H),1.22(t,J=8Hz,18H),1.60(t,J=8Hz,4H),2.70(dd,J=4.8Hz,2.8Hz,2H),2.86(t,J=4.8Hz,2H),3.15(t,J=8Hz,4H),3.50-3.60(m,4H),3.78-3.91(m,20H),3.97-4.13(m,8H),4.55(s,2H),5.13-5.26(m,6H),5.85(m,2H),6.67(d,J=8.8Hz,8H),7.00(d,J=8.4Hz,8H)。上述合成实施例3的合成方案如下。合成实施例4和5:具有烷氧基甲硅烷基的四苯基乙烷环氧化合物的合成(方法1)通过与上文合成实施例3的第一步和第二步相同的方法合成式B-1(2)和B-1(3)的化合物,不同之处在于如下文表B1和B2中所述改变用量。实施例4和实施例5中合成的式B-1的环氧基:烷氧基甲硅烷基的比例分别是2:1和3:1。[表B1]用于合成化合物B-1的第一步中使用的反应物用量[表B2]用于合成化合物B-1的第二步中使用的反应物量B-1(2)第一步NMR(环氧基:烯丙基2:1)1HNMR(400MHz,CDCl3):δ=2.68(dd,J=4.8Hz,2.8Hz,2.67H),2.84(t,J=4.8Hz,2.67H),3.22-3.27(m,2.67H),3.80-3.84(m,2.67,1.33H),3.87-3.91(m,2.66H),4.00-4.01(m,2.67,1.33H),4.05-4.13(m,2.67,1.33H),4.47(s,2H),5.20(m,2.66H),5.87(m,1.33H),6.65(d,J=8.4Hz,8H),7.00(d,J=8.0Hz,8H)。B-1(2)第二步NMR(环氧基:烷氧基甲硅烷基=2:1)1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,4H),1.22(t,J=8Hz,18H),1.60(t,J=8Hz,4H),2.70(dd,J=4.8Hz,2.8Hz,2H),2.86(t,J=4.8Hz,2H),3.15(t,J=8Hz,4H),3.50-3.60(m,4H),3.77-3.91(m,20H),3.97-4.13(m,8H),4.55(s,2H),5.13-5.26(m,6H),5.85(m,2H),6.67(d,J=8.8Hz,8H),7.00(d,J=8.4Hz,8H)。B-1(3)第一步NMR(环氧基:烯丙基3:1)1HNMR(400MHz,CDCl3):δ=2.68(dd,J=4.8Hz,2.8Hz,3H),2.84(t,J=4.8Hz,3H),3.22-3.27(m,3H),3.80-3.84(m,4H),3.87-3.91(m,2H),4.00-4.01(m,4H),4.05-4.13(m,3H),4.47(s,2H),5.20(m,2H),5.87(m,1H),6.65(d,J=8.4Hz,8H),7.00(d,J=8.0Hz,8H)。B-1(3)第二步NMR(环氧基:烷氧基甲硅烷基=3:1)1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,2H),1.22(t,J=8Hz,9H),1.60(t,J=8Hz,2H),2.68(dd,J=4.8Hz,2.8Hz,3H),2.84(t,J=4.8Hz,3H),3.15(t,J=8Hz,2H),3.22-3.27(m,3H),3.78-3.84(m,10H),3.87-3.91(m,2H),4.00-4.13(m,7H),4.47(s,2H),5.13-5.26(m,3H),5.87(m,1H),6.65(d,J=8.4Hz,8H),7.00(d,J=8.0Hz,8H)。合成实施例6具有烷氧基甲硅烷基的四苯基乙烷环氧化合物(结构B-2)的合成(方法2)向双颈烧瓶中加入10g的上述合成实施例3的第一步中获得的中间体B-0(1)、123mg的PtO2、4.89g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成最终的目标材料具有烷氧基甲硅烷基的环氧B-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.21(t,J=8Hz18H),1.57-1.61(m,4H),2.65(dd,J=4.8Hz,2.8Hz,2H),2.82(t,J=4.8Hz,2H),3.24-3.28(m,4H2H),3.51-3.55(m,2H),3.71-3.74(m,2H),3.80(q,J=8Hz,12H),3.82-3.88(m,2H),3.96-4.01(m,4H),4.04-4.08(m,4H),4.53(s,2H),6.64(d,J=8.4Hz,8H),7.00(d,J=8.0Hz,8H)。上述合成实施例6的合成方案如下。合成实施例7具有烷氧基甲硅烷基的四苯基乙烷环氧化合物(结构B-3)的合成(方法3)向双颈烧瓶中加入10g的上述合成实施例3中获得的具有烷氧基甲硅烷基的中间体B-1(1)、74mg的PtO2、2.93g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成最终的目标材料具有烷氧基甲硅烷基的环氧B-3,其环氧基:烷氧基甲硅烷基比例为1:2。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,8H),1.22(t,J=8Hz,36H),1.60(t,J=8Hz,8H),2.70(dd,J=4.8Hz,2.8Hz,2H),2.86(t,J=4.8Hz,2H),3.15(t,J=8Hz,8H),3.50-3.60(m,4H),3.81-3.91(m,4H),3.78-3.83(m,28H),3.97-4.13(m,4H),4.55(s,2H),5.13-5.15(m,2H),6.67(d,J=8.8Hz,8H),7.00(d,J=8.4Hz,8H)。上述合成实施例7的合成方案如下。合成实施例8:具有烷氧基甲硅烷基的四苯基乙烷环氧化合物(结构B-3)的合成(方法4)向双颈烧瓶中加入10g的合成实施例6中合成的具有烷氧基甲硅烷基的环氧B-2和乙腈并搅拌。随后向其中加入5.13g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和2.68g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:2的环氧树脂B-3。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,8H),1.22(t,J=8Hz,36H),1.60(t,J=8Hz,8H),2.70(dd,J=4.8Hz,2.8Hz,2H),2.86(t,J=4.8Hz,2H),3.15(t,J=8Hz,8H),3.50-3.60(m,4H),3.81-3.91(m,4H),3.78-3.83(m,28H),3.97-4.13(m,4H),4.55(s,2H),5.13-5.15(m,2H),6.67(d,J=8.8Hz,8H),7.00(d,J=8.4Hz,8H)。上述合成实施例8的合成方案如下。合成实施例9具有烷氧基甲硅烷基的三苯基甲烷环氧化合物(结构C-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的三苯基甲烷的三缩水甘油醚(阿德瑞奇公司(Aldrich),式C-环氧),并分别加入20ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.85g的NaOH、1.03g的Et4NBr和23.65g的烯丙醇,随后在室温下搅拌3小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体C-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.72-2.75(m,2H),2.88-2.90(m,2H),3.31-3.36(m,2H),3.94(dd,2H,J=11.9Hz,5.6Hz),3.80-4.14(m,7H),4.17(dd,2H,J=12.0Hz,3.6Hz),5.20-5.28(m,2H),5.41(s,1H),5.86-5.98(m,1H),6.73(d,2H,J=8.8Hz),6.82(d,4H,J=8.8Hz),6.95(d,2H,J=8.8Hz),6.99(d,4H,J=8.8Hz)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体C-0和乙腈并搅拌。随后向其中加入6.11g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和3.19g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为2:1的环氧树脂C-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,2H),1.22(t,J=8Hz,9H),1.60(t,J=8Hz,2H),2.72-2.75(m,2H),2.88-2.90(m,2H),3.15(t,J=8Hz,2H),3.31-3.36(m,2H),3.78-4.14(m,15H),4.17(dd,2H,J=12.0Hz,3.6Hz),5.13-5.28(m,3H),5.41(s,1H),5.86-5.98(m,1H),6.73(d,2H,J=8.8Hz),6.82(d,4H,J=8.8Hz),6.95(d,2H,J=8.8Hz),6.99(d,4H,J=8.8Hz)。上述合成实施例9的合成方案如下。合成实施例10具有烷氧基甲硅烷基的三苯基甲烷环氧化合物(结构C-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例9的第一步中获得的中间体C-0、88mg的PtO2、3.48g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧C-2,其环氧基:烷氧基甲硅烷基比例为2:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,2H),1.23(t,J=8Hz9H),1.55-1.61(m,2H),2.72-2.75(m,2H),2.88-2.90(m,2H),3.24-3.28(m,2H),3.31-3.36(m,2H),3.80-4.17(m,15H),5.41(s,1H),6.73(d,2H,J=8.8Hz),6.82(d,4H,J=8.8Hz),6.95(d,2H,J=8.8Hz),6.99(d,4H,J=8.8Hz)。上述合成实施例10的合成方案如下。合成实施例11:具有烷氧基甲硅烷基的双酚环氧化合物(结构D-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的四缩水甘油基-4,4’-二氨基二苯基甲烷(阿德瑞奇公司,式D-环氧),并分别加入29.6ml的THF和乙腈以溶解环氧化合物。随后向其中加入1.23g的NaOH、1.49g的Et4NBr和34.37g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基团:烯丙基比例为1:1的中间体D-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.58-2.60(m,2H),2.78-2.81(m,2H),3.16-3.20(m,2H),3.490-3.49(m,2H),3.73-4.14(m,18H),5.21-5.28(m,4H),5.84-5.97(m,2H),6.74-6.77(m,4H),7.08-7.12(m,4H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体D-0和乙腈并搅拌。随后向其中加入11.76g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和6.14g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂D-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,4H),1.25(t,J=8Hz,18H),1.62(t,J=8Hz,4H),2.58-2.60(m,2H),2.78-2.81(m,2H),3.15-3.20(m,6H),3.49-3.40(m,2H),3.78-4.14(m,30H),5.13-5.29(m,6H),5.84-5.97(m,2H),6.74-6.77(m,4H),7.08-7.12(m,4H)。上述合成实施例11的合成方案如下。合成实施例12具有烷氧基甲硅烷基的双酚环氧化合物(结构D-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例11的第一步中获得的中间体D-0、189mg的PtO2、6.71g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧D-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.21(t,J=8Hz18H),1.57-1.63(m,4H),2.58-2.60(m,2H),2.78-2.81(m,2H),3.16-3.28(m,6H),3.40-3.49(m,2H),3.73-4.14(m,26H),6.74-6.77(m,4H),7.08-7.13(m,4H)。上述合成实施例12的合成方案如下。合成实施例13具有烷氧基甲硅烷基的氨基苯酚环氧化合物(结构E-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的N,N-二缩水甘油基-4-缩水甘油氧基苯胺(N,N-diglycidyl-4-glycidyloxyaniline)(希巴葛杰公司(CibaGeigy),MY-0510,式E-环氧),并分别加入34ml的THF和乙腈以溶解环氧化合物。随后向其中加入1.41g的NaOH、1.71g的Et4NBr和39.27g的烯丙醇,随后在室温下搅拌3小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为2:1的中间体E-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.58-2.60(m,1.3H),2.73-2.90(m,2H),3.16-3.20(m,0.7H),3.31-3.35(m,1.3H),3.40-3.49(m,2H),3.76-4.14(m,9H),4.16-4.20(m,0.7H),5.20-5.28(m,2H),5.86-5.98(m,1H),6.62-6.69(m,2H),6.80-6.83(m,2H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体E-0和乙腈并搅拌。随后向其中加入9.44g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.93g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为2:1的环氧树脂E-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,2H),1.22(t,J=8Hz,9H),1.60(t,J=8Hz,2H),2.58-2.60(m,1.3H),2.73-2.90(m,2H),3.15-3.21(m,2.7H),3.31-3.35(m,1.3H),3.40-4.20(m,17.7H),5.13-5.28(m,3H),5.86-5.98(m,1H),6.62-6.69(m,2H),6.80-6.83(m,2H)。上述合成实施例13的合成方案如下。合成实施例14具有烷氧基甲硅烷基的氨基苯酚环氧化合物(结构E-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例13的第一步中获得的中间体E-0、135mg的PtO2、5.39g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧E-2,其环氧基:烷氧基甲硅烷基比例为2:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=4Hz,2H),1.21(t,J=8Hz9H),1.56-1.61(m,2H),2.58-2.61(m,1.3H),2.74-2.90(m,2H),3.16-3.28(m,2.7H),3.31-3.49(m,3.3H),3.80(q,J=8Hz,6H),3.76-4.20(m,7.7H),6.62-6.69(m,2H),6.80-6.83(m,2H)。上述合成实施例14的合成方案如下。合成实施例15具有烷氧基甲硅烷基的萘环氧化合物(结构F-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的1,5-缩水甘油氧基-2,6-二缩水甘油基萘(1,5-diglycidyloxy-2,6-diglycidylnaphthalene)(非市售可得,但可以通过以下参考实施例中韩国工业技术研究院的韩国注册专利第10-1252063号公开的合成方法来合成,F-环氧),并分别加入33ml的THF和乙腈以溶解环氧化合物。随后向其中加入1.35g的NaOH、1.64g的Et4NBr和37.77g的烯丙醇,随后在室温下搅拌3小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体F-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.52-2.57(m,1H),2.61-2.66(m,2H)2.73-2.81(m,3H),2.90-2.93(m,2H),3.16-3.18(m,1H),3.35-3.37(m,1H),3.80-4.13(m,13H),4.22-4.25(m,1H),5.20-5.27(m,4H),5.87-6.00(m,2H),7.28(d,J=8.5Hz,2H),7.75(d,J=8.5Hz,2H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体F-0和乙腈并搅拌。随后向其中加入12.65g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和6.61g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂F-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=8Hz,4H),1.25(t,J=8Hz,18H),1.65(t,J=8Hz,4H),2.52-2.57(m,1H),2.61-2.66(m,2H)2.73-2.81(m,3H),2.90-2.93(m,2H),3.16-3.20(m,5H),3.35-3.37(m,1H),3.76-4.13(m,25H),4.22-4.25(m,1H),5.12-5.28(m,6H),5.87-6.00(m,2H),7.28(d,J=8.5Hz,2H),7.75(d,J=8.5Hz,2H)。上述合成实施例15的合成方案如下。合成实施例16具有烷氧基甲硅烷基的萘环氧化合物(结构F-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例15的第一步中获得的中间体F-0、181mg的PtO2、7.22g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧F-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.21(t,J=8Hz18H),1.57-1.61(m,4H),2.52-2.57(m,1H),2.61-2.66(m,2H)2.73-2.81(m,3H),2.90-2.93(m,2H),3.16-3.18(m,1H),3.24-3.28(m,4H),3.35-3.37(m,1H),3.78-4.13(m,21H),4.22-4.25(m,1H),7.28(d,J=8.5Hz,2H),7.75(d,J=8.5Hz,2H)。上述合成实施例16的合成方案如下。合成实施例17具有烷氧基甲硅烷基的联苯环氧化合物(结构G)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的3,3’-二缩水甘油基-4,4’-二缩水甘油氧基联苯(3,3’-diglycidyl-4,4’-diglycidyloxybiphenyl)(非市售可得,但可以通过以下参考实施例2中韩国工业技术研究院的韩国注册专利第10-1252063号公开的合成方法来合成,G-环氧),并分别加入31ml的THF和乙腈以溶解环氧化合物。随后向其中加入1.27g的NaOH、1.54g的Et4NBr和35.37g的烯丙醇,随后在室温下搅拌3小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体G-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.53-2.57(m,1H),2.61-2.65(m,2H),2.73-2.81(m,3H),2.88-2.92(m,2H),3.16-3.18(m,1H),3.35-3.37(m,1H),3.80-4.04(m,13H),4.22-4.25(m,1H),5.20-5.26(m,4H),5.86-5.99(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体G-0和乙腈并搅拌。随后向其中加入12.02g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和6.28g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机相。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂G-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.62(t,J=8Hz,4H),1.24(t,J=8Hz,18H),1.62(t,J=8Hz,4H),2.53-2.57(m,1H),2.61-2.65(m,2H),2.73-2.81(m,3H),2.88-2.92(m,2H),3.15-3.18(m,5H),3.35-3.37(m,1H),3.78-4.04(m,25H),4.22-4.25(m,1H),5.12-5.27(m,6H),5.86-5.99(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H)。上述合成实施例17的合成方案如下。合成实施例18具有烷氧基甲硅烷基的联苯环氧化合物(结构G-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例17的第一步中获得的中间体G-0、172mg的PtO2、6.86g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧G-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.23(t,J=8Hz18H),1.57-1.62(m,4H),2.53-2.57(m,1H),2.61-2.65(m,2H),2.73-2.81(m,3H),2.88-2.92(m,2H),3.16-3.18(m,1H),3.24-3.28(m,4H),3.35-3.37(m,1H),3.80-4.04(m,21H),4.22-4.25(m,1H),6.73-6.75(m,2H),7.03-7.05(m,4H)。上述合成实施例18的合成方案如下。合成实施例19具有烷氧基甲硅烷基的芴环氧化合物(结构H-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的9,9-二(3-缩水甘油基-4-二缩水甘油氧基苯基)芴(9,9-di(3-glycidyl-4-diglycidyloxyphenyl)fluorene)(非市售可得,但可以通过以下参考实施例3中韩国工业技术研究院的韩国注册专利第10-1252063号公开的合成方法来合成,H-环氧),并分别加入21ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.90g的NaOH、1.10g的Et4NBr和25.27g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体H-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.53-2.56(m,1H),2.59-2.66(m,2H),2.72-2.81(m,3H),2.89-2.93(m,2H),3.16-3.18(m,1H),3.35-3.37(m,1H),3.79-4.15(m,13H),4.22-4.25(m,1H),5.20-5.28(m,4H),5.86-5.98(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体H-0和乙腈并搅拌。随后向其中加入9.17g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.79g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂H-1。1HNMR(400MHz,CDCl3):δ=0.62(t,J=8Hz,4H),1.23(t,J=8Hz,18H),1.61(t,J=8Hz,4H),2.52-2.56(m,1H),2.60-2.66(m,2H),2.72-2.81(m,3H),2.89-2.93(m,2H),3.15-3.20(m,5H),3.35-3.37(m,1H),3.78-4.15(m,25H),4.22-4.25(m,1H),5.13-5.28(m,6H),5.86-5.98(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。上述合成实施例19的合成方案如下。合成实施例20具有烷氧基甲硅烷基的芴环氧化合物(结构H-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例19的第一步中获得的中间体H-0、131mg的PtO2、5.23g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧H-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,4H),1.22(t,J=8Hz18H),1.55-1.61(m,4H),2.53-2.66(m,3H),2.72-2.81(m,3H),2.89-2.93(m,2H),3.16-3.18(m,1H),3.24-3.28(m,4H),3.35-3.37(m,1H),3.79-4.25(m,22H),6.73-6.75(m,2H),7.03-7.05(m,4H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。上述合成实施例20的合成方案如下。合成实施例21具有烷氧基甲硅烷基的四苯基甲烷环氧化合物(结构I-1)的合成(方法1)(预期实施例1)(1)第一步向双颈烧瓶中加入10g的3,3’-二缩水甘油基-4,4’-二缩水甘油氧基四苯基甲烷(3,3’-diglycidyl-4,4’-diglycidyloxytetraphenylmethane)(I-环氧),并分别加入22ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.90g的NaOH、1.09g的Et4NBr和25.18g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体I-0。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体I-0和乙腈并搅拌。随后向其中加入9.14g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.78g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基团:烷氧基甲硅烷基比例为1:1的环氧树脂I-1。上述合成实施例21的合成方案如下。合成实施例22具有烷氧基甲硅烷基的四苯基甲烷环氧化合物(结构I-2)的合成(方法2)(预期实施例2)向烧瓶中加入10g的上述合成实施例21的第一步中获得的中间体I-0、131mg的PtO2、5.22g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧I-2,其环氧基:烷氧基甲硅烷基比例为1:1。上述合成实施例22的合成方案如下。合成实施例23具有烷氧基甲硅烷基的异氰脲酸酯环氧化合物(结构J-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的三(2,3-环氧丙基)异氰酸酯(阿德瑞奇公司,式J-环氧),并分别加入31.5ml的THF和乙腈以溶解环氧化合物。随后向其中加入1.31g的NaOH、1.59g的Et4NBr和36.63g的烯丙醇,随后在室温下搅拌3小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为2:1的中间体J-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.61-2.64(m,2H),2.71-2.75(m,2H),3.13-3.20(m,2H),3.75-4.14(m,11H),5.19-5.27(m,2H),5.86-5.97(m,1H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体J-0和乙腈并搅拌。随后向其中加入8.91g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.66g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为2:1的环氧树脂J-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.63(t,J=8Hz,2H),1.26(t,J=8Hz,9H),1.63(t,J=8Hz,2H),2.60-2.64(m,2H),2.71-2.75(m,2H),3.13-3.20(m,4H),3.75-4.14(m,17H),5.13-5.29(m,3H),5.86-5.97(m,1H)。上述合成实施例23的合成方案如下。合成实施例24具有烷氧基甲硅烷基的异氰脲酸酯环氧化合物(结构J-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例23的第一步中获得的中间体J-0、128mg的PtO2、5.09g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧J-2,其环氧基:烷氧基甲硅烷基比例为2:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.59(t,J=4Hz,2H),1.23(t,J=8Hz,9H),1.57-1.61(m,2H),2.61-2.64(m,2H),2.71-2.75(m,2H),3.13-3.28(m,4H),3.75-4.14(m,15H)。上述合成实施例24的合成方案如下。合成实施例25具有烷氧基甲硅烷基的苯酚酚醛清漆(phenolnovolac)环氧化合物(结构K-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的苯酚酚醛清漆环氧化合物(国都化工公司(KukdoChemicalCo.),YDPN,K-环氧),并分别加入19ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.79g的NaOH、0.96g的Et4NBr和22.00g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体K-0。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.60-2.73(m,6.89H),3.29-3.32(m,3.51H),3.78-4.17(m,35.59H),4.47-4.49(m,7.61H),5.20-5.41(m,6.91H),6.00-6.04(m,3.66H),6.70-7.14(m,21.68H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体K-0和乙腈并搅拌。随后向其中加入8.16g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.26g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基团:烷氧基甲硅烷基比例为1:1的环氧树脂K-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,7.33),1.22(t,J=8Hz,32.94H),1.60(t,J=8Hz,7.71H),2.60-2.73(m,6.89H),3.15-3.32(m,10.87H),3.78-4.18(m,57.74H),4.47-4.49(m,7.61H),5.13-5.41(m,10.62H),6.00-6.04(m,3.66H),6.70-7.14(m,21.68H)。上述合成实施例25的合成方案如下。对于上述合成方案的K-0结构,酚醛清漆环氧化合物的烯丙醇的反应位点被认为不具有交替结构(alternatingstructure),但具有沿主链随机分布的环氧官能团和烯丙基结构。基于相同的理由,K-1结构中环氧基和烷氧基甲硅烷基的浓度是1:1,但环氧基和烷氧基甲硅烷基的位置不具有交替结构但具有随机结构。显示了上述方案中所示交替结构以便于调节环氧基和修饰的官能团的比例,且其不同于真实化合物的结构。后文中,核结构适用于K’至N’的酚醛清漆环氧化合物的合成方案和结构。合成实施例26具有烷氧基甲硅烷基的苯酚酚醛清漆环氧化合物(结构K-2)的合成(方法2)向双颈烧瓶中加入10g的上述合成实施例25的第一步中获得的中间体K-0、117mg的PtO2、4.66g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成最终的目标材料具有烷氧基甲硅烷基的环氧K-2,其环氧基:烷氧基甲硅烷基烯丙基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.62(t,J=4Hz,7.41H),1.22(t,J=8Hz33.91H),1.55-1.61(m,7.33H),2.60-2.73(m,6.89H),3.24-3.32(m,11.01H),3.78-4.17(m,58.58H),6.70-7.14(m,21.68H)。上述合成实施例26的合成方案如下。合成实施例27具有烷氧基甲硅烷基的甲酚酚醛清漆环氧化合物(结构L-1(1))的合成(方法1)(1)第一步向双颈烧瓶中加入10g的邻甲酚醛清漆环氧化合物(日本化药有限公司,EOCN-1020,结构L-环氧),并分别加入22ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.91g的NaOH、1.10g的Et4NBr和25.39g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体L-0(1)。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.19-2.32(m,3.28H),2.64-2.91(m,1.21H),3.20-3.36(m,0.72H),3.62-4.13(m,12.15H),5.20-5.28(m,1.33H),5.86-5.98(m,0.62H),6.71-7.04(m,3.00H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体L-0(1)和乙腈并搅拌。随后向其中加入9.97g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和5.21g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:1的环氧树脂L-1(1)。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.63(t,J=8Hz,1.25H),1.26(t,J=8Hz,5.58H),1.62(t,J=8Hz,1.25H),2.17-2.32(m,3.28H),2.64-2.91(m,1.21H),3.15-3.36(m,1.97H),3.62-4.13(m,16.57H),5.13-5.30(m,1.95H),5.86-5.98(m,0.62H),6.71-7.04(m,3.00H)。上述合成实施例27的合成方案如下。合成实施例28和29:具有烷氧基甲硅烷基的苯甲酚酚醛清漆环氧化合物的合成(方法1)通过与上文合成实施例27的第一步和第二步相同的方法合成式L-1的化合物,不同之处在于如下文表L1和L2中所述改变细节。实施例28和实施例29中合成的式L-1的环氧基:烷氧基甲硅烷基的比例分别是2:1和3:1。[表L1]用于合成化合物L-1的第一步中使用的反应物用量[表L2]用于合成化合物L-1的第二步中使用的反应物用量L-1(2)第一步NMR(环氧基:烯丙基=2:1)1HNMR(400MHz,CDCl3):δ=2.19-2.32(m,3.28H),2.64-2.91(m,1.79H),3.20-3.36(m,0.90H),3.62-4.13(m,10.01H),5.20-5.28(m,0.95H),5.86-5.98(m,0.45H),6.71-7.04(m,3.00H)。L-1(2)第二步NMR(环氧基:烷氧基甲硅烷基=2:1)1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,1.00H),1.22(t,J=8Hz,4.21H),1.60(t,J=8Hz,0.92H),2.19-2.32(m,3.28H),2.64-2.91(m,1.79H),3.15(t,J=8Hz,0.98H),3.20-3.36(m,0.90H),3.78-3.83(m,2.70H),3.62-4.13(m,10.01H),5.12-5.28(m,1.42H),5.86-5.98(m,0.45H),6.71-7.04(m,3.00H)。L-1(3)第一步NMR(环氧基:烯丙基3:1)1HNMR(400MHz,CDCl3):δ=2.19-2.32(m,3.28H),2.64-2.91(m,2.11H),3.20-3.36(m,1.12H),3.62-4.13(m,9.6H),5.20-5.28(m,0.73H),5.86-5.98(m,0.35H),6.71-7.04(m,3.00H)。L-1(3)第二步NMR(环氧基:烷氧基甲硅烷基3:1)1HNMR(400MHz,CDCl3):δ=0.61(t,J=8Hz,0.71H),1.22(t,J=8Hz,3.89H),1.60(t,J=8Hz,0.73H),2.19-2.32(m,3.28H),2.64-2.91(m,2.11H),3.15(t,J=8Hz,0.70H),3.20-3.36(m,1.12H),3.78-3.83(m,2.21H),3.62-4.13(m,9.6H),5.13-5.28(m,1.14H),5.86-5.98(m,0.35H),6.71-7.04(m,3.00H)。合成实施例30具有烷氧基甲硅烷基的甲酚酚醛清漆环氧化合物(结构L-2)的合成(方法2)向双颈烧瓶中加入10g的上述合成实施例25的第一步中合成的中间体L-0、143mg的PtO2、5.69g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧L-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=4Hz,1.35H),1.21(t,J=8Hz6.39H),1.57-1.61(m,1.36H),2.19-2.32(m,3.28H),2.64-2.91(m,1.21H),3.20-3.36(m,2.16H),3.62-4.13(m,13.94H),6.71-7.04(m,3.00H)。上述合成实施例30的合成方案如下。合成实施例31具有烷氧基甲硅烷基的甲酚酚醛清漆环氧化合物(结构L-3)的合成(方法3)加入10g的上述合成实施例25中获得的具有烷氧基甲硅烷基的中间体L-1(1)、80mg的PtO2、3.20g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成最终的目标材料具有烷氧基甲硅烷基的环氧L-3,其环氧基:烷氧基甲硅烷基比例为1:2。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.63(t,J=8Hz,2.62H),1.26(t,J=8Hz,11.97H),1.60-1.63(m,2.62H),2.17-2.32(m,3.28H),2.64-2.91(m,2.58H),3.15-3.36(m,1.97H),3.62-4.13(m,18.72H),6.71-7.04(m,3.00H)。上述合成实施例31的合成方案如下。合成实施例32具有烷氧基甲硅烷基的甲酚酚醛清漆环氧化合物(结构L-3)的合成(方法4)加入10g的合成实施例30中合成的中间体L-2和乙腈并搅拌。随后向其中加入6.57g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和3.43g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基:烷氧基甲硅烷基比例为1:2的环氧树脂L-3。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.63(t,J=8Hz,2.62H),1.26(t,J=8Hz,11.97H),1.60-1.63(m,2.62H),2.17-2.32(m,3.28H),2.64-2.91(m,2.58H),3.15-3.36(m,1.97H),3.62-4.13(m,18.72H),6.71-7.04(m,3.00H)。上述合成实施例32的合成方案如下。合成实施例33具有烷氧基甲硅烷基的甲酚酚醛清漆环氧化合物(结构M-1)的合成(方法1)(1)第一步向双颈烧瓶中加入10g的双酚A酚醛清漆环氧化合物(DIC,LF,结构M-环氧),并分别加入18ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.74g的NaOH、0.90g的Et4NBr和20.63g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基团:烯丙基比例为1:1的中间体M-0。1HNMR(400MHz,CDCl3):δ=1.62(m,31.92H),2.58-2.77(m,10.99H),3.28-3.33(m,4.67H),3.67-4.18(m,49.42H),4.47-4.51(m,11.90H),5.22-5.44(m,10.74H),6.06-6.15(m,5.12H),6.65-6.72(m,20.21H),7.26-7.31(m,12.68H)。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体M-0和乙腈并搅拌。随后向其中加入7.72g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和4.03g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基团:烷氧基甲硅烷基比例为1:1的环氧树脂M-1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.60(t,J=8Hz,10.62H),1.22(t,J=8Hz,47.08H),1.60-1.63(m,43.54H),2.58-2.77(m,10.99H),3.15(t,J=8Hz,10.82H),3.28-3.33(m,4.67H),3.67-4.18(m,80.13H),4.47-4.51(m,11.90H),5.13-5.42(m,15.88H),6.06-6.15(m,5.12H),6.65-6.72(m,20.21H),7.26-7.31(m,12.68H)。上述合成实施例33的合成方案如下。合成实施例34具有烷氧基甲硅烷基的双酚酚醛清漆环氧化合物(结构M-2)的合成(方法2)向烧瓶中加入10g的上述合成实施例33的第一步中获得的中间体M-0、111mg的PtO2、4.41g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧M-2,其环氧基:烷氧基甲硅烷基比例为1:1。NMR数据如下。1HNMR(400MHz,CDCl3):δ=0.62(t,J=4Hz,10.66H),1.23(t,J=8Hz47.05H),1.55-1.63(m,42.46H),2.58-2.76(m,10.99H),3.24-3.33(m,15.67H),3.67-4.18(m,71.78H),4.46-4.51(m,11.90H),6.66-6.72(m,20.21H),7.25-7.31(m,12.68H)。上述合成实施例34的合成方案如下。合成实施例35具有烷氧基甲硅烷基的萘酚醛清漆环氧化合物(结构N-1)的合成(方法1)(预期实施例1)(1)第一步向双颈烧瓶中加入10g的萘酚醛清漆环氧化物(结构N-环氧),并分别向其中加入16ml的THF和乙腈以溶解环氧化合物。随后向其中加入0.66g的NaOH、0.80g的Et4NBr和18.52g的烯丙醇,随后在室温下搅拌6小时。向其中加入10ml的饱和NH4Cl溶液以终止反应。此后,使用旋转蒸发仪部分除去溶剂,并使用150ml的乙酸乙酯和50ml的自来水处理反应残留物三次。分离有机层,使用MgSO4除去残留的H2O并使用旋转蒸发仪和真空泵对有机层进行完全干燥以生成环氧基:烯丙基比例为1:1的中间体N-0。(2)第二步向双颈烧瓶中加入10g的第一步中合成的中间体N-0和乙腈并搅拌。随后向其中加入7.03g的3-(三乙氧基甲硅烷基)丙基异氰酸酯和3.68g的二异丙基乙胺,随后在65℃下反应30小时。反应完成后,将反应混合物冷却至室温,使用100ml乙酸乙酯和40ml的饱和NH4Cl溶液对反应混合物进行处理。分离有机层,并向其中加入MgSO4以除去残留的H2O。使用蒸发仪除去溶剂并使用己烷浆料纯化有机层。除去上清己烷层并使用真空泵完全干燥后,生成了环氧基团:烷氧基甲硅烷基比例为1:1的环氧树脂N-1。上述合成实施例35的合成方案如下。合成实施例36具有烷氧基甲硅烷基的萘酚醛清漆环氧化合物(结构N-2)的合成(方法2)(预期实施例4)向烧瓶中加入10g的上述预期实施例3的第一步中获得的中间体N-0、101mg的PtO2、4.01g的三乙氧基硅烷和150ml的甲苯并在室温下搅拌5分钟。升温至80℃并将反应混合物加热并搅拌24小时。反应完成后,将温度下降至室温,并用硅藻土过滤器除去无机材料。通过蒸发除去甲苯并使用真空泵完全干燥产物以生成具有烷氧基甲硅烷基的环氧N-2,其环氧基:烷氧基甲硅烷基比例为1:1。上述合成实施例36的合成方案如下。参照实施例1合成实施例15的起始材料的合成(1)第一步:合成1,5-双(烯丙氧基)萘把20.0g的1,5-二羟基萘(西格玛奥德里奇公司(Sigma-Aldrich))、27.0ml的烯丙基溴(西格玛奥德里奇公司)、103.61g的K2CO3和500ml的丙酮加入安装了回流冷凝管的1,000ml双颈烧瓶中,然后在室温下混合。使用80℃的回流温度将均匀混合的溶液回流过夜。完成反应后,使反应混合物冷却至室温,使用硅藻土过滤并蒸发以获得粗产物。使用乙酸乙酯萃取粗产物中的目标组分,用水清洗三次并用MgSO4干燥。通过过滤除去MgSO4并使用蒸发仪除去溶剂以生成作为中间体11的1,5-双(烯丙氧基)萘。NMR数据如下。1HNMR(400MHz,CDCl3):δ=4.70(dt,J=5.2Hz,1.6Hz,4H),5.32-5.34(m,2H),5.49-5.54(m,2H),6.12-6.21(m,2H),6.84(d,J=8.0Hz,2H),7.35(dd,J=7.6,0.8Hz,2H),7.89(d,J=8.8Hz,2H)。(2)第二步:合成2,6-二烯丙基萘-1,5-二醇把在第一步中获得的20.0g的中间体11和100ml的1,2-二氯苯(西格玛奥德里奇公司)添加到安装了回流冷凝管的1,000ml双颈烧瓶中,随后在室温下混合均匀。在190℃的回流温度下使均匀的溶液回流8小时。完成反应后,使反应混合物冷却至室温并通过真空烘箱除去溶剂以生成作为中间体12的2,6-二烯丙基萘-1,5-二醇。如此获得的中间体12的NMR数据如下。1HNMR(400MHz,CDCl3):δ=3.57(dt,J=6.4Hz,1.6Hz,4H),5.21-5.27(m,4H),5.50(s,2H),6.02-6.12(m,2H),7.21(d,J=8.4Hz,2H),7.70(d,J=8.4Hz,2H)。(3)第三步:合成2,2’-(2,6-二烯丙基萘-1,5-二基)双(氧基)双(亚甲基)二环氧乙烷把在第二步中获得的20.0g的中间产物12、65.07ml的表氯醇(西格玛奥德里奇公司)、74.15gofK2CO3,和300ml的乙腈添加到安装了回流冷凝管的1,000ml双颈烧瓶中,随后在室温下混合。随后升高温度并在80℃下反应过夜。完成反应后,使反应混合物冷却至室温并使用硅藻土过滤并蒸发有机溶剂以生成中间体13。中间体13的NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.77(dd,J=2.6Hz,2H),2.93(dd,J=4.4Hz,2H),3.44-3.48(m,2H),3.61(d,J=6.4Hz,4H),3.91(dd,J=6.0Hz,2H),4.24(dd,J=2.8Hz,2H),5.07-5.12(m,4H),5.98-6.08(m,2H),7.34(d,J=8.4Hz,2H),7.88(d,J=8.4Hz,2H)。(4)第四步:合成2,2’-(1,5-双(环氧乙烷-2-基甲氧基)萘-2,6-二基)双(亚甲基)二环氧乙烷向500ml烧瓶中加入10.0g的2,2’-(2,6-二烯丙基萘-1,5-二基)双(氧基)双(亚甲基)二环氧乙烷、19.08g的77mol%3-氯过氧苯甲酸和200ml的二氯甲烷并在室温下搅拌2天。随后,使用硫代硫酸钠水溶液处理反应残留物并使用乙酸乙酯萃取。然后,反应产物用1N氢氧化钠水溶液和盐水洗涤,用MgSO4干燥并用滤器过滤。除去溶剂并通过柱色谱纯化以生成2,2’-(1,5-双(环氧乙烷-2-基甲氧基)萘-2,6-二基)双(亚甲基)二环氧乙烷。产物的NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.53-2.57(m,2H),2.73-2.81(m,6H),2.89-2.92(m,4H),3.16-3.18(m,2H),3.35-3.37(m,2H),3.90-3.97(m,2H),4.22-4.25(m,2H),7.28(d,J=8.5Hz,2H),7.75(d,J=8.5Hz,2H)。参考实施例1的合成机制如下。参照实施例2合成实施例17的起始材料的合成(1)第一步:合成4,4’-双(烯丙氧基)联苯把10.0g的联苯-4,4’-二醇(西格玛奥德里奇公司)、11.61ml的烯丙基溴(西格玛奥德里奇公司)、44.56g的K2CO3和500ml的丙酮加入安装了回流冷凝管的1,000ml双颈烧瓶中,并在室温下混合。使用80℃的回流温度将均匀混合的溶液回流过夜。完成反应后,使反应混合物冷却至室温,使用硅藻土过滤并蒸发以获得粗产物。使用乙酸乙酯从粗产物中萃取目标组分,用水清洗三次并用MgSO4干燥。通过过滤除去MgSO4并使用蒸发仪除去溶剂以生成作为中间体21的4,4-双(烯丙氧基)联苯。NMR数据如下。1HNMR(400MHz,CDCl3):δ=4.56(dt,J=5.2Hz,1.6Hz,4H),5.30-5.33(m,2H),5.41-5.44(m,2H),6.03-6.12(m,2H),6.96(td,J=3.0,2.2,8.8Hz,4H),7.46(td,J=3.0,2.2,8.8Hz,4H)。(2)第二步:合成3,3’-二烯丙基联苯-4,4’-二醇把在第一步中获得的10.0g的中间体21和100ml的1,2-二氯苯(西格玛奥德里奇公司)添加到安装了回流冷凝管的1,000ml双颈烧瓶中,随后在室温下混合均匀。在190℃的回流温度下使均匀的溶液回流72小时。完成反应后,使反应混合物冷却至室温并通过真空烘箱除去溶剂以生成作为中间体22的3,3-二烯丙基联苯-4,4-二醇。NMR数据如下。1HNMR(400MHz,CDCl3):δ=3.35(d,J=6.4Hz,4H),5.14-5.25(m,6H),6.00-6.10(m,2H),6.84(dd,J=2.0Hz,7.2Hz,2H),7.29(dd,J=10.6Hz,4H)。(3)第三步:合成2,2’-(3,3’-二烯丙基联苯-4,4’-二基)双(氧基)双(亚甲基)二环氧乙烷把在第二步中获得的10.0g的中间产物22、30.38ml的表氯醇(西格玛奥德里奇公司)、35.13gofK2CO3,和300ml的乙腈添加到安装了回流冷凝管的1,000ml双颈烧瓶中,并在室温下混合。随后升高温度并在80℃下反应过夜。完成反应后,使反应混合物冷却至室温并使用硅藻土过滤并蒸发有机溶剂以生成中间体23。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.75(dd,J=2.6Hz,2H),2.87(dd,J=4.2Hz,2H),3.11-3.35(m,6H),3.96(dd,J=5.4Hz,2H),4.25(dd,J=3.2Hz,2H),5.03-5.13(m,4H),5.93-6.03(m,2H),6.81(d,J=7.2Hz,2H),7.34-7.42(m,4H)。(4)第四步:合成2,2’-(4,4-双(环氧乙烷-2-基甲氧基)联苯-3,3’-二基)双(亚甲基)二环氧乙烷向500ml烧瓶中加入10.0g的2,2’-(3,3’-二烯丙基联苯-4,4’-二基)双(氧基)双(亚甲基)二环氧乙烷、17.77g的77mol%3-氯过氧苯甲酸和200ml的二氯甲烷并在室温下搅拌2天。随后,使用硫代硫酸钠水溶液处理反应混合物并使用乙酸乙酯萃取。然后,对反应产物用1N氢氧化钠水溶液和盐水洗涤,用MgSO4干燥并用滤器过滤。除去溶剂并通过柱色谱纯化以生成2,2’-(4,4’-双(环氧乙烷-2-基甲氧基)联苯-3,3’-二基)双(亚甲基)二环氧乙烷。终产物的NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.53-2.57(m,2H),2.73-2.81(m,6H),2.89-2.92(m,4H),3.16-3.18(m,2H),3.35-3.37(m,2H),3.90-3.97(m,2H),4.22-4.25(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H)。参考实施例2的合成机制如下。参照实施例3合成实施例19的起始材料的合成(1)第一步:合成9,9-双(4-烯丙氧基)苯基-9H-芴把10.0g的4,4’-(9H-芴-9,9-二基)二苯酚(西格玛奥德里奇公司)、6.17ml的烯丙基溴(西格玛奥德里奇公司)、23.68g的K2CO3和500ml的丙酮加入安装了回流冷凝管的1,000ml双颈烧瓶中,并在室温下混合。使用80℃的回流温度将均匀混合的溶液回流过夜。完成反应后,使反应混合物冷却至室温,使用硅藻土过滤并蒸发以获得粗产物。使用乙酸乙酯从粗产物中萃取目标组分,用水清洗三次并用MgSO4干燥。通过过滤除去MgSO4并使用蒸发仪除去溶剂以生成作为中间体31的9,9-双(4-烯丙氧基)苯基-9H-芴。NMR数据如下。1HNMR(400MHz,CDCl3):δ=4.46(td,J=1.4,2.4Hz,4H),5.25(qd,J=1.6,1.2,10.4Hz,2H),5.35-5.38(m,2H),5.97-6.06(m,2H),6.75(td,J=3.2,2.0,8.8Hz,4H),7.10(td,J=3.2,2.0,8.8Hz,4H),7.23-7.39(m,6H),7.70-7.79(m,2H)。(2)第二步:合成4,4’-(9H-芴-9,9-二基)双(2-烯丙基苯酚)把在第一步中获得的10.0g的中间体31和100ml的1,2-二氯苯(西格玛奥德里奇公司)添加到安装了回流冷凝管的1,000ml双颈烧瓶中,随后在室温下混合均匀。在190℃的回流温度下使均匀的溶液回流96小时。完成反应后,使反应混合物冷却至室温并通过真空烘箱除去溶剂以生成作为中间体32的4,4’-(9H-芴-9,9-二基)双(2-烯丙基苯酚)。NMR数据如下。1HNMR(400MHz,CDCl3):δ=3.28(d,J=6.0Hz,4H),5.04-5.09(m,4H),5.21(s,2H),5.87-5.97(m,2H),6.62(d,J=8.4Hz,2H),6.88(dd,J=2.4,6.0Hz,2H),6.96(d,J=2.4Hz,2H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。(3)第三步:合成2,2’-(4,4’-(9H-芴-9,9-二基)双(2-烯丙基-4,1-亚苯基))双(氧基)双(亚甲基)二环氧乙烷把在第二步中获得的10.0g的中间产物32、18.16ml的表氯醇(西格玛奥德里奇公司)、21.00gofK2CO3和300ml的乙腈添加到安装了回流冷凝管的1,000ml双颈烧瓶中,并在室温下混合。随后升高温度并在80℃下反应过夜。完成反应后,使反应混合物冷却至室温并使用硅藻土过滤,并蒸发有机溶剂以生成中间体33。NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.75(dd,J=2.6Hz,2H),2.87(dd,J=4.2Hz,2H),3.11-3.35(m,6H),3.96(dd,J=5.4Hz,2H),4.12(dd,J=3.2Hz,2H),4.97-5.03(m,4H),5.93-6.03(m,2H),6.69(d,J=8.4Hz,2H),6.80-6.83(m,2H),7.05(s,2H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。(4)第四步:合成2,2’-(5,5’-(9H-芴-9,9-二基)双(2-(环氧乙烷-2-基甲氧基)-5,1-亚苯基))双(亚甲基)-二环氧乙烷向500ml烧瓶中加入10.0g的2,2’-(4,4’-(9H-芴-9,9-二基)双(2-烯丙基-4,1-亚苯基))双(氧基)双(亚甲基)二环氧乙烷、12.39g的77mol%3-氯过氧苯甲酸和200ml的二氯甲烷并在室温下搅拌2天。随后,使用硫代硫酸钠水溶液处理反应混合物并使用乙酸乙酯萃取。然后,反应产物用1N氢氧化钠水溶液和盐水洗涤,用MgSO4干燥并用滤器过滤。除去溶剂并通过柱色谱纯化以生成2,2’-(5,5’-(9H-芴-9,9-二基)双(2-(环氧乙烷-2-基甲氧基)-5,1-亚苯基))双(亚甲基)-二环氧乙烷。终产物的NMR数据如下。1HNMR(400MHz,CDCl3):δ=2.53-2.57(m,2H),2.73-2.81(m,6H),2.89-2.92(m,4H),3.16-3.18(m,2H),3.35-3.37(m,2H),3.90-3.97(m,2H),4.22-4.25(m,2H),6.73-6.75(m,2H),7.03-7.05(m,4H),7.22-7.36(m,6H),7.74(d,J=7.2Hz,2H)。参考实施例3的合成机制如下。评估物理性质:制造固化产物和评估耐热性环氧复合材料的生产环氧玻璃纤维复合材料(固化产物)的生产根据表1所示的配方,将环氧化合物、固化剂、固化催化剂和反应催化剂溶解在甲基乙基酮中,使得固体含量为40总量%,并进行混合以获得均匀的溶液。把玻璃纤维(日东纺公司(NittoboCo.)的玻璃纤维织物,E-玻璃2116或T-玻璃2116)用由此得到的混合物浸渍,以生产包括环氧化合物的玻璃纤维复合材料。然后,将复合材料放置在真空烘箱中,加热至100℃以除去溶剂,然后在预热至120℃的热压机中在120℃下固化2小时,在180℃下固化2小时,在高于200℃的温度下固化2小时,以生产玻璃纤维复合膜(4mmx16mmx0.1mm)。在制造复合材料膜的同时,根据压机的压力和树脂的粘度控制复合材料膜的树脂含量。复合材料膜中的树脂含量示于下表1中。此外,当用于玻璃纤维复合材料的组合物包含二氧化硅时,根据下表1所示的配方将环氧化合物和二氧化硅浆料(70重量%的固含量,2-甲氧基乙醇溶剂,1微米的二氧化硅平均尺寸)溶解在甲基乙基酮中,使得固体含量为40%。以1,500rpm的速率将由此获得的混合物混合1小时,加入固化剂,然后再混合50分钟。最后,加入固化催化剂和反应催化剂,再混合10分钟,以获得环氧混合物。用环氧混合物来浸泡玻璃纤维(日东纺公司的玻璃纤维织物,E-玻璃2116)来生产玻璃纤维复合材料。然后,在与上述相同的条件下进行相同的固化处理,以生产复合材料膜。(2)环氧填料复合材料(固化产物)的生产根据下表2所示的配方,将除固化剂、固化催化剂和反应催化剂以外的所有的环氧化合物、二氧化硅浆料(70重量%的固含量,2-甲氧基乙醇溶剂,1微米的二氧化硅平均尺寸)和可选的热塑性聚合物组分溶解在甲基乙基酮中,使得固体含量为40重量%。以1,500rpm的速率将由此获得的混合物混合1小时,加入固化剂,然后再混合50分钟。最后,加入固化催化剂和反应催化剂,再混合10分钟,以获得环氧混合物。然后,将混合物放置在加热至100℃的真空烘箱中,以除去溶剂,然后在预热至120℃的热压机中,在120℃下固化2小时,在180℃下固化2小时,在高于200℃的温度下固化2小时,以生产环氧填料(无机颗粒)复合材料(5mmx5mmx3mm)。2.耐热物理性质的评估使用热机械分析仪(Thermo-mechanicalanalyzer)来评价下表1和2中所述的本发明实施例和比较例的固化产物随温度变化而发生的尺寸变化,结果如下表1和2中所示。生产环氧玻璃纤维复合材料膜的试样使其具有4mmx16mmx0.1mm的尺寸,并生产填料复合材料的试样使其具有5mmx5mmx3mm的尺寸。[表1-1]环氧玻璃纤维复合材料[表1-2]环氧玻璃纤维复合材料[表1-3]环氧玻璃纤维复合材料[表1-4]环氧玻璃纤维复合材料[表1-5]环氧玻璃纤维复合材料[表1-6]环氧玻璃纤维复合材料[表1-7]环氧玻璃纤维复合材料[表2-1]环氧填料复合材料[表2-2]环氧填料复合材料[表2-3]环氧填料复合材料[表2-4]环氧填料复合材料注:表1和2中的化合物如下。(1)EXA-4700:联萘环氧化合物(日本的DIC公司)(2)GTR1800:联萘环氧化合物(日本化药有限公司)(3)TMTE:三苯基甲烷环氧化合物(阿德瑞奇公司)(4)AP:氨基苯酚环氧化合物(希巴葛杰公司,MY-0510)(5)DGEBA:双酚A的二缩水甘油醚(阿德瑞奇公司)(6)3615:橡胶改性的DGEBA环氧树脂(Strruktol公司(StrruktolCo.))(7)EOCN:邻甲酚酚醛清漆的环氧树脂(日本化药有限公司)(8)HF-1M:苯酚酚醛清漆固化剂(明和塑料工业公司(MeiwaPlasticIndustries))(9)TPP:三苯基膦(阿德瑞奇公司)(10)Tin-OC:乙基己酸锡(II)(阿德瑞奇公司)(11)YX-4000H:联苯环氧化合物(优卡壳环氧化物公司(YukaShellEpoxyCo.))(12)HR5TM:热塑性聚合物(积水公司(Sekisui))*TgL:无Tg且未表现出玻璃化转变如上表1所示,使用本发明的烷氧基甲硅烷基修饰的酚醛清漆环氧化合物的玻璃纤维复合材料的耐热性得到极大改进,并表现出低CTE和高玻璃化转变温度或者无Tg。具体而言,实施例1-43所述的玻璃纤维复合材料具有1.18-8.9ppm/℃的CTE,小于不具有烷氧基甲硅烷基的环氧树脂复合材料(比较例1-6)的12.8-17.0ppm/℃的CTE值,并且显示无Tg。此外,实施例44-83的无机颗粒(填料)的CTE值为6.45-10.51ppm/℃,并可实现非常良好的CTE且无Tg。此外,如图1和2所示,实施例2-4的具有烷氧基甲硅烷基的环氧复合材料表现出无Tg转变。通过本发明观察到的具有烷氧基甲硅烷基的环氧化合物的良好CTE和玻璃化转变温度性质可被认为是由于烷氧基甲硅烷基与玻璃纤维和/或无机颗粒(填料)之间有效地形成界面键合而获得的。3.阻燃性的评价点燃上文表1中实施例1和比较例1所述的复合材料带,燃烧后的带的光学照片见图3。如图3所示,本发明实施例1所述环氧化合物的复合材料带在1-2秒内自发熄灭(图3中左侧照片)。但是,比较例1的不含烷氧基甲硅烷基的复合材料带完全烧掉(图3中右侧照片,照片中未燃烧的部分对应于镊子夹持的部分),因此,可理解本发明所述烷氧基甲硅烷基化环氧化合物具有良好的阻燃性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1