含氟高支化聚合物和包含该聚合物的树脂组合物的制作方法
技术领域
本发明涉及含氟高支化聚合物,详细而言,涉及可以作为树脂的表面改性剂使用的具有氟烷基的高支化聚合物和包含该聚合物的树脂组合物。
背景技术:
聚合物(高分子)材料近年来在多个领域中越来越多被利用。与此相伴,与各个领域相对应,作为基体的聚合物的性状以及其表面、界面的特性变得重要。通过使用例如表面能低的氟系化合物作为表面改性剂,可期待防水防油性、防污性、非粘合性、剥离性、脱模性、滑动性、耐磨耗性、防反射特性、耐化学性等与界面控制有关的特性的提高,提出了各种方案。
作为与使用了氟系聚合物的热塑性树脂的表面改性有关的公开内容,提出了例如,通过四氟乙烯-乙烯共聚物(ETFE)的配合而提高了脱模性的聚4-甲基-1-戊烯(PMP)树脂膜(专利文献1)、防水防油性优异的含氟聚烯烃(专利文献2)。
此外,作为与使用了氟系聚合物的光固化性和热固性透明树脂的表面改性有关的公开内容,可列举例如,使用了具有含氟聚醚的氟系聚合物的热固性环氧树脂的表面处理剂(专利文献3),此外公开了,包含含氟表面活性剂和/或具有环状结构的氟系聚合物的光固化性丙烯酸类树脂的脱模性优异并进行了表面改性(专利文献4)。
另一方面,作为用于合成高支化聚合物(超支化聚合物)的某方法,提出了使具有2个以上自由基聚合性双键的单体在自由基聚合引发剂的存在下进行聚合的方法。提出了例如,通过使用了具有2个乙烯基的苯乙烯化合物(专利文献5,非专利文献1、2、3、4)、或具有2个甲基丙烯酰基的甲基丙烯酰基化合物(专利文献5,非专利文献5、6)、或具有2个二烯丙基的烯丙基化合物(非专利文献7)、具有2个乙烯基的其它乙烯基化合物(专利文献5,非专利文献8、9)和偶氮系聚合引发剂的所谓引发剂片段插入型(共)聚合来制造高支化聚合物的方法。
然而,到现在为止,还未提出使用高支化聚合物进行光固化性和热固性透明树脂的表面改性的方法,特别是对于上述引发剂片段插入型高支化聚合物而言,还没有关于含有氟烷基的表面能低的高支化聚合物的报告。
专利文献
专利文献1:日本特开2005-307059号公报
专利文献2:日本特开平1-289817号公报
专利文献3:日本特开2009-7488号公报
专利文献4:国际公开第2006/114958号小册子
专利文献5:日本专利第4009700号公报
非专利文献
非专利文献1:Tsuneyuki Sato,Nobuyuki Sato,Makiko Seno,Tomohiro Hirano,J.Polymm.Sci.Part A,41,3038-3047(2003)
非专利文献2:Tsuneyuki Sato,Naoki Higashida,Tomohiro Hirano,Makiko Seno,J.Polymm.Sci.Part A,42,1609-1617(2004)
非专利文献3:Tomohiro Hirano,Naoki Higashida,Hongwai Wang,Makiko Seno,Tsuneyuki Sato,J.Appl.Polym.Sci.,100,664-670(2006)
非专利文献4:Tsuneyuki Sato,Hiroki Nobutane,Tomohiro Hirano,Makiko Seno,Macromol.Mater.Eng.,291,162-172(2006)
非专利文献5:Tsuneyuki Sato,Takashi Miyagi,Tomohiro Hirano,Makiko Seno,Polym.Int.,53,1503-1511(2004)
非专利文献6:Tomohiro Hirano,Hiroshi Ihara,Takashi Miyagi,Hongwei Wang,Makiko Seno,Tsuneyuki Sato,Macromol.Chem.Phys.,206,860-868(2005)
非专利文献7:Tsuneyuki Sato,Kazuki Nomura,Tomohiro Hirano,Makiko Seno,J.Appl.Polym.Sci.,102,408-415(2006)
非专利文献8:Tsuneyuki Sato,Atushi Ono,Tomohiro Hirano,Makiko Seno,J.Polymm.Sci.Part A,44,2328-2337(2006)
非专利文献9:Tsuneyuki Sato,Yukiko Arima,Makiko Seno,Tomohiro Hirano,Polym.Int.,53,1138-1144(2004)
技术实现要素:
上述的线状氟系聚合物可以对一部分的热塑性树脂赋予一定的表面改性效果,但一般与树脂的混合、分散性差,特别是分散在以聚甲基丙烯酸甲酯(PMMA)为代表的热塑性透明树脂中的情况下,可能会发生相分离,损坏透明树脂的透明性。
此外,这些氟系聚合物对有机溶剂的溶解性低,即使使用这些聚合物进行光固化性和热固性树脂的表面改性,也难以适用于使用有机溶剂进行成膜的工艺。
即,要求保持充分的透明性、对有机溶剂具有高溶解性、还具有表面改性效果的新的化合物。
本发明人为了达成上述目而进行了反复深入研究,结果发现,通过采用以往未研究过的在高支化聚合物中导入氟烷基而得的含氟高支化聚合物作为树脂的表面改性剂,从而不仅对有机溶剂的溶解性优异,而且对基体树脂的混合、分散性优异、在基体树脂中不发生凝聚、表面改性性优异,可获得具有高透明性的成型体和皮膜,从而完成了本发明。
即,在本发明中,作为第1观点,涉及一种含氟高支化聚合物,是通过将分子内具有2个以上自由基聚合性双键的单体A在相对于该单体A 1摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合而获得的含氟高支化聚合物,其特征在于,所述单体的至少一部分或所述聚合引发剂的任意一者是分子内具有氟烷基的物质。
作为第2观点,涉及一种含氟高支化聚合物,是通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合而获得的。
作为第3观点,涉及一种含氟高支化聚合物,是通过使分子内具有2个以上自由基聚合性双键的单体A在相对于该单体A 1摩尔为5摩尔%~200摩尔%的分子内具有氟烷基的聚合引发剂C的存在下进行聚合而获得的。
作为第4观点,涉及一种含氟高支化聚合物,是通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的分子内具有氟烷基的聚合引发剂C的存在下进行聚合而获得的。
作为第5观点,涉及第2观点~第4观点的任一项所述的含氟高支化聚合物,所述单体A是具有乙烯基或(甲基)丙烯酰基的任意一者或两者的化合物。
作为第6观点,涉及第5观点所述的含氟高支化聚合物,所述单体A是二乙烯基化合物或二(甲基)丙烯酸酯化合物。
作为第7观点,涉及第6观点所述的含氟高支化聚合物,所述单体A是乙二醇二(甲基)丙烯酸酯。
作为第8观点,涉及第2观点或第4观点所述的含氟高支化聚合物,在使用所述单体B的情况下,是相对于所述单体A 1摩尔,以0.05摩尔~3摩尔的比例使用所述单体B而获得的。
作为第9观点,涉及第8观点所述的含氟高支化聚合物,所述单体B是具有至少1个乙烯基或(甲基)丙烯酰基中的任意一者的化合物。
作为第10观点,涉及第9观点所述的含氟高支化聚合物,所述单体B是下述式[1]所示的化合物,
式[1]中,R1表示氢原子或甲基,并且,R2表示碳原子数2~12的可以被羟基取代的氟烷基。
作为第11观点,涉及第10观点所述的含氟高支化聚合物,所述单体B是下述式[2]所示的化合物,
式[2]中,
R1表示与所述式[1]中的定义相同的含义,
X表示氢原子或氟原子,
m表示1或2,并且
n表示0~5的整数。
作为第12观点,涉及第1观点~第11观点的任一项所述的含氟高支化聚合物,所述聚合引发剂C是偶氮系聚合引发剂。
作为第13观点,涉及第12观点所述的含氟高支化聚合物,所述聚合引发剂C是2,2’-偶氮二异丁酸二甲酯。
作为第14观点,涉及第12观点所述的含氟高支化聚合物,所述聚合引发剂C是2,2’-偶氮二(2,4,4-三甲基戊烷)。
作为第15观点,涉及一种清漆,其含有第1观点~第14观点的任一项所述的含氟高支化聚合物。
作为第16观点,涉及一种薄膜,其包含第1观点~第14观点的任一项所述的含氟高支化聚合物。
作为第17观点,涉及一种光聚合性组合物,其含有:(a)第1观点~第14观点的任一项所述的含氟高支化聚合物、(b)光聚合性化合物和(c)光聚合引发剂。
作为第18观点,涉及第17观点所述的光聚合性组合物,所述(b)光聚合性化合物是多官能(甲基)丙烯酸酯化合物。
作为第19观点,涉及第18观点所述的光聚合性组合物,所述(b)光聚合性化合物是三环癸烷二甲醇二(甲基)丙烯酸酯。
作为第20观点,涉及第17观点~第19观点的任一项所述的光聚合性组合物,所述(a)含氟高支化聚合物的含量相对于(b)光聚合性化合物的总质量为0.01质量%~20质量%。
作为第21观点,涉及一种树脂成型品,是使第17观点~第20观点的任一项所述的光聚合性组合物光聚合而制作的。
作为第22观点,涉及一种树脂组合物,其含有:(a)第1观点~第14观点的任一项所述的含氟高支化聚合物、和(d)热塑性树脂或热固性树脂。
作为第23观点,涉及第22观点所述的树脂组合物,所述(d)热塑性树脂是聚甲基丙烯酸甲酯树脂。
作为第24观点,涉及第22观点所述的树脂组合物,所述(d)热塑性树脂是聚乳酸树脂。
作为第25观点,涉及第22观点~第24观点的任一项所述的树脂组合物,所述(a)含氟高支化聚合物的含量相对于(d)热塑性树脂或热固性树脂的总质量为0.01质量%~20质量%。
作为第26观点,涉及一种树脂成型品,是由第22观点~第25观点的任一项所述的树脂组合物制作的。
作为第27观点,涉及一种制造含氟高支化聚合物的方法,其中,通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合,来制造含氟高支化聚合物。
作为第28观点,涉及一种制造含氟高支化聚合物的方法,其中,通过使分子内具有2个以上自由基聚合性双键的单体A在相对于该单体A 1摩尔为5摩尔%~200摩尔%的分子内具有氟烷基的聚合引发剂C的存在下进行聚合,来制造含氟高支化聚合物。
作为第29观点,涉及一种制造含氟高支化聚合物的方法,其中,通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的分子内具有氟烷基的聚合引发剂C的存在下进行聚合,来制造含氟高支化聚合物。
作为第30观点,涉及一种制造含氟高支化聚合物的方法,其包括下述工序:
使分子内具有2个以上自由基聚合性双键的单体A在相对于该单体A1摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合来制造高支化聚合物的工序,
使含有氟烷基的醇D与所得的高支化聚合物反应的工序。
作为第31观点,涉及一种压印用脱模剂,其包含第1观点~第14观点的任一项所述的含氟高支化聚合物。
作为第32观点,涉及一种光压印成型体,其是通过将具有微细图案的模具按压在第17观点~第20观点的任一项所述的光聚合性组合物的层上,通过光照射使该层固化,脱模,从而获得的转印有所述微细图案的光压印成型体。
作为第33观点,涉及一种热压印成型体,其是通过将具有微细图案的模具按压在第22观点~第25观点的任一项所述的树脂组合物的层上,将该层加热到该树脂的玻璃化转变温度以上,冷却后脱模,从而获得的转印有所述微细图案的热压印成型体。
以往的线状高分子一般为绳状形状,与此相对,本发明的含氟高支化聚合物由于主动地导入有分支结构,因而与线状高分子相比分子间的缠绕少而显示微粒的行为。即在作为基体的树脂中的移动变得容易。
因此,将本发明的含氟高支化聚合物配合在光聚合性组合物、含有热塑性树脂或热固性树脂的树脂组合物中制成树脂成型品的情况下,可以容易移动到界面(成型品表面)而有助于界面控制,使得树脂的表面改性提高。
此外,本发明的含氟高支化聚合物与作为基体的树脂的混合、分散性高,可以在树脂中不发生凝聚等地混合、分散,并可以制造透明性优异的树脂成型品。
此外,本发明的含氟高支化聚合物发挥高分子化合物这样的特性,可以以简单的涂布、干燥操作直接形成薄膜状的结构体。而且,本发明的含氟高支化聚合物不仅可以溶于N,N’-二甲基甲酰胺(DMF)、四氢呋喃(THF),而且也可溶于丙酮、甲苯等,因此可以不限定溶剂地形成清漆的形态,也可以形成薄膜。
此外,本发明的树脂成型品不仅可制成上述那样的透明性优异的成型品,而且可以制成表面改性了的树脂成型品,例如可以制成对混合、成型机械等各种机械、模具的脱模性、或对膜等其它树脂成型品的剥离性、以及防水防油性、防污性也优异的成型品。
附图说明
图1是显示实施例1中制造的高支化聚合物1的1H NMR光谱的图。
图2是显示实施例1中制造的高支化聚合物1的13C NMR光谱的图。
图3是显示实施例2中制造的高支化聚合物2的1H NMR光谱的图。
图4是显示实施例2中制造的高支化聚合物2的13C NMR光谱的图。
图5是显示实施例3中制造的高支化聚合物3的1H NMR光谱的图。
图6是显示实施例3中制造的高支化聚合物3的13C NMR光谱的图。
图7是显示实施例4中制造的高支化聚合物4的1H NMR光谱的图。
图8是显示实施例4中制造的高支化聚合物4的13C NMR光谱的图。
图9是显示实施例5中制造的高支化聚合物5的1H NMR光谱的图。
图10是显示实施例5中制造的高支化聚合物5的13C NMR光谱的图。
图11是显示实施例6中制造的高支化聚合物6的1H NMR光谱的图。
图12是显示实施例6中制造的高支化聚合物6的13C NMR光谱的图。
图13是显示实施例7中制造的高支化聚合物7的1H NMR光谱的图。
图14是显示实施例7中制造的高支化聚合物7的13C NMR光谱的图。
图15是显示实施例8中制造的高支化聚合物8的1H NMR光谱的图。
图16是显示实施例8中制造的高支化聚合物8的13C NMR光谱的图。
图17是显示实施例9中制造的高支化聚合物9的1H NMR光谱的图。
图18是显示实施例9中制造的高支化聚合物9的13C NMR光谱的图。
图19是显示实施例10中制造的高支化聚合物10的1H NMR光谱的图。
图20是显示实施例10中制造的高支化聚合物10的13C NMR光谱的图。
图21是显示实施例11中制造的高支化聚合物11的1H NMR光谱的图。
图22是显示实施例11中制造的高支化聚合物11的13C NMR光谱的图。
图23是显示比较例1中制造的高支化聚合物12的1H NMR光谱的图。
图24是显示比较例1中制造的高支化聚合物12的13C NMR光谱的图。
图25是显示实施例17中获得的高支化聚合物3/PMMA块体膜(bulk film)或PMMA单独膜的剥离强度试验结果的图。
图26是显示实施例18中制造的高支化聚合物13的1H NMR光谱的图。
图27是显示实施例18中制造的高支化聚合物13的13C NMR光谱的图。
图28是显示实施例19中制造的高支化聚合物14的1H NMR光谱的图。
图29是显示实施例19中制造的高支化聚合物14的13C NMR光谱的图。
图30是显示实施例20中制造的高支化聚合物15的1H NMR光谱的图。
图31是显示实施例20中制造的高支化聚合物15的13C NMR光谱的图。
图32是显示实施例21中制造的高支化聚合物16的1H NMR光谱的图。
图33是显示实施例21中制造的高支化聚合物16的13C NMR光谱的图。
图34是显示实施例22中制造的高支化聚合物17的1H NMR光谱的图。
图35是显示实施例22中制造的高支化聚合物17的13C NMR光谱的图。
图36是显示实施例24中制造的高支化聚合物19的1H NMR光谱的图。
图37是显示实施例24中制造的高支化聚合物19的13C NMR光谱的图。
图38是显示实施例25中制造的高支化聚合物20的1H NMR光谱的图。
图39是显示实施例25中制造的高支化聚合物20的13C NMR光谱的图。
图40是显示实施例26中制造的高支化聚合物21的1H NMR光谱的图。
图41是显示实施例26中制造的高支化聚合物21的13C NMR光谱的图。
图42是显示实施例27中制造的高支化聚合物22的1H NMR光谱的图。
图43是显示实施例27中制造的高支化聚合物22的13C NMR光谱的图。
图44是显示实施例28中制造的高支化聚合物23的1H NMR光谱的图。
图45是显示实施例28中制造的高支化聚合物23的13C NMR光谱的图。
图46是显示实施例29中制造的高支化聚合物24的1H NMR光谱的图。
图47是显示实施例29中制造的高支化聚合物24的13C NMR光谱的图。
图48是显示实施例30中制造的高支化聚合物25的1H NMR光谱的图。
图49是显示实施例30中制造的高支化聚合物25的13C NMR光谱的图。
图50是显示实施例31中制造的高支化聚合物26的1H NMR光谱的图。
图51是显示实施例31中制造的高支化聚合物26的13C NMR光谱的图。
图52是显示实施例32中制造的高支化聚合物27的1H NMR光谱的图。
图53是显示实施例32中制造的高支化聚合物27的13C NMR光谱的图。
图54是显示实施例33中制造的高支化聚合物28的1H NMR光谱的图。
图55是显示实施例33中制造的高支化聚合物28的13C NMR光谱的图。
图56是显示实施例34中制造的高支化聚合物29的1H NMR光谱的图。
图57是显示实施例34中制造的高支化聚合物29的13C NMR光谱的图。
图58是显示实施例35中制造的高支化聚合物30的1H NMR光谱的图。
图59是显示实施例35中制造的高支化聚合物30的13C NMR光谱的图。
图60是显示实施例36中制造的高支化聚合物31的1H NMR光谱的图。
图61是显示实施例36中制造的高支化聚合物31的13C NMR光谱的图。
图62是显示比较例2中制造的高支化聚合物32的1H NMR光谱的图。
图63是显示比较例2中制造的高支化聚合物32的13C NMR光谱的图。
图64是显示比较例3中制造的直链状聚合物1的1H NMR光谱的图。
图65是显示比较例3中制造的直链状聚合物1的13C NMR光谱的图。
图66是显示比较例4中制造的直链聚合物2的13C NMR光谱的图。
图67是显示比较例5中制造的直链聚合物3的13C NMR光谱的图。
图68是显示实施例43中获得的微细图案的光学显微镜的照片的图。
图69是显示比较例6中获得的微细图案的光学显微镜的照片的图。
图70是显示实施例44中获得的微细图案的光学显微镜的照片的图。
图71是显示实施例44中获得的微细图案的光学显微镜的照片的图。
图72是显示比较例7中获得的微细图案的光学显微镜的照片的图。
图73是显示比较例7中获得的微细图案的光学显微镜的照片的图。
图74是显示实施例43和实施例44、以及比较例6和比较例7中使用的硅制模件的微细图案的光学显微镜的照片的图。
图75是显示实施例45中制造的高支化聚合物33的1H NMR光谱的图。
图76是显示实施例45中制造的高支化聚合物33的13C NMR光谱的图。
图77是显示实施例46中制造的高支化聚合物34的1H NMR光谱的图。
图78是显示实施例46中制造的高支化聚合物34的13C NMR光谱的图。
图79是显示实施例47中制造的高支化聚合物35的1H NMR光谱的图。
图80是显示实施例47中制造的高支化聚合物35的13C NMR光谱的图。
图81是显示实施例48中制造的高支化聚合物36的1H NMR光谱的图。
图82是显示实施例48中制造的高支化聚合物36的13C NMR光谱的图。
图83是显示实施例49中制造的高支化聚合物37的1H NMR光谱的图。
图84是显示实施例49中制造的高支化聚合物37的13C NMR光谱的图。
图85是显示实施例50中制造的高支化聚合物38的1H NMR光谱的图。
图86是显示实施例50中制造的高支化聚合物38的13C NMR光谱的图。
图87是显示实施例54中制造的高支化聚合物39的1H NMR光谱的图。
图88是显示实施例54中制造的高支化聚合物39的13C NMR光谱的图。
图89是显示实施例55中制造的高支化聚合物40的1H NMR光谱的图。
图90是显示实施例55中制造的高支化聚合物40的13C NMR光谱的图。
图91是显示实施例56中制造的高支化聚合物41的1H NMR光谱的图。
图92是显示实施例56中制造的高支化聚合物41的13C NMR光谱的图。
图93是显示实施例57中制造的高支化聚合物42的1H NMR光谱的图。
图94是显示实施例57中制造的高支化聚合物42的13C NMR光谱的图。
图95是显示实施例58中制造的高支化聚合物43的1H NMR光谱的图。
图96是显示实施例58中制造的高支化聚合物43的13C NMR光谱的图。
具体实施方式
<含氟高支化聚合物>
本发明的含氟高支化聚合物是通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合而获得的。
本发明的含氟高支化聚合物是通过使分子内具有2个以上自由基聚合性双键的单体A在相对于该单体A 1摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合而获得的。此时,上述单体的至少一部分或上述聚合引发剂的任意一者在分子内具有氟烷基。
优选地,本发明的含氟高支化聚合物是通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的聚合引发剂C的存在下进行聚合而获得的。
或者,是通过使分子内具有2个以上自由基聚合性双键的单体A与相对于该单体A 1摩尔为5摩尔%~200摩尔%的分子内具有氟烷基的聚合引发剂C的存在下进行聚合而获得的。
或者,是通过使分子内具有2个以上自由基聚合性双键的单体A与分子内具有氟烷基和至少1个自由基聚合性双键的单体B在相对于该单体A和该单体B的总摩尔为5摩尔%~200摩尔%的分子内具有氟烷基聚合引发剂C的存在下进行聚合而获得的。
在本发明中,分子内具有2个以上自由基聚合性双键的单体A优选具有乙烯基或(甲基)丙烯酰基的任意一者或两者,特别优选为二乙烯基化合物或二(甲基)丙烯酸酯化合物。另外,在本发明中,(甲基)丙烯酸酯化合物是指丙烯酸酯化合物和甲基丙烯酸酯化合物两者。例如(甲基)丙烯酸是指丙烯酸和甲基丙烯酸。
作为这样的单体A,可例示例如,以下的(A1)~(A7)所示的有机化合物。
(A1)烯属烃:
(A1-1)脂肪族烯属烃类;异戊二烯、丁二烯、3-甲基-1,2-丁二烯、2,3-二甲基-1,3-丁二烯、1,2-聚丁二烯、戊二烯、己二烯、辛二烯等
(A1-2)脂环式烯属烃;环戊二烯、环己二烯、环辛二烯、降冰片二烯等
(A1-3)芳香族烯属烃;二乙烯基苯、二乙烯基甲苯、二乙烯基二甲苯、三乙烯基苯、二乙烯基联苯、二乙烯基萘、二乙烯基芴、二乙烯基咔唑、二乙烯基吡啶等
(A2)乙烯基酯、烯丙基酯、乙烯基醚、烯丙基醚、乙烯基酮:
(A2-1)乙烯基酯;己二酸二乙烯酯、马来酸二乙烯酯、邻苯二甲酸二乙烯酯、间苯二甲酸二乙烯酯、衣康酸二乙烯酯、(甲基)丙烯酸乙烯酯等
(A2-2)烯丙酯;马来酸二烯丙酯、邻苯二甲酸二烯丙酯、间苯二甲酸二烯丙酯、己二酸二烯丙酯、(甲基)丙烯酸烯丙酯等
(A2-3)乙烯基醚;二乙烯基醚、二甘醇二乙烯基醚、三乙二醇二乙烯基醚等
(A2-4)烯丙基醚;二烯丙基醚、二烯丙氧基乙烷、三烯丙氧基乙烷、四烯丙氧基乙烷、四烯丙氧基丙烷、四烯丙氧基丁烷、四甲基烯丙氧基乙烷等
(A2-5)乙烯基酮;二乙烯基酮、二烯丙基酮等
(A3)(甲基)丙烯酸酯:
乙二醇二(甲基)丙烯酸酯、三乙二醇二(甲基)丙烯酸酯、丙二醇二(甲基)丙烯酸酯、新戊二醇二(甲基)丙烯酸酯、三羟甲基丙烷三(甲基)丙烯酸酯、双三羟甲基丙烷四(甲基)丙烯酸酯、三(甲基)丙烯酸甘油酯、季戊四醇四(甲基)丙烯酸酯、烷氧基钛三(甲基)丙烯酸酯、1,6-己二醇二(甲基)丙烯酸酯、2-甲基-1,8-辛二醇二(甲基)丙烯酸酯、1,9-壬二醇二(甲基)丙烯酸酯、1,10-癸二醇二(甲基)丙烯酸酯、三环癸烷二甲醇二(甲基)丙烯酸酯、二氧杂环己烷二醇二(甲基)丙烯酸酯、2-羟基-1-丙烯酰氧基-3-甲基丙烯酰氧基丙烷、2-羟基-1,3-二(甲基)丙烯酰氧基丙烷、9,9-双[4-(2-(甲基)丙烯酰氧基乙氧基)苯基]芴、十一碳烯酰基乙二醇二(甲基)丙烯酸酯、双[4-(甲基)丙烯酰基硫苯基]硫化物、双[2-(甲基)丙烯酰基硫乙基]硫化物、1,3-金刚烷二醇二(甲基)丙烯酸酯、1,3-金刚烷二甲醇二(甲基)丙烯酸酯等
(A4)具有聚亚烷基二醇链的烯属化合物:
聚乙二醇(分子量300)二(甲基)丙烯酸酯、聚丙二醇(分子量500)二(甲基)丙烯酸酯等
(A5)含氮烯属化合物:
二烯丙基胺、异氰脲酸二烯丙酯、氰脲酸二烯丙酯、亚甲基双(甲基)丙烯酰胺、双马来酰亚胺等
(A6)含硅烯属化合物:
二甲基二乙烯基硅烷、二乙烯基甲基苯基硅烷、二苯基二乙烯基硅烷、1,3-二乙烯基-1,1,3,3-四甲基二硅氮烷、1,3-二乙烯基-1,1,3,3-四苯基二硅氮烷、二乙氧基二乙烯基硅烷等
(A7)含氟烯属化合物:
1,4-二乙烯基全氟丁烷、1,6-二乙烯基全氟己烷、1,8-二乙烯基全氟辛烷等
其中优选的是上述(A1-3)组的芳香族烯属烃化合物、(A2)组的乙烯基酯、烯丙酯、乙烯基醚、烯丙基醚和乙烯基酮、(A3)组的(甲基)丙烯酸酯、(A4)组的具有聚亚烷基二醇链的烯属化合物、以及(A5)组的含氮烯属化合物。
特别优选的是属于(A1-3)组的二乙烯基苯、属于(A2)组的邻苯二甲酸二烯丙酯、属于(A3)组的乙二醇二(甲基)丙烯酸酯、1,3-金刚烷二甲醇二(甲基)丙烯酸酯、三环癸烷二甲醇二(甲基)丙烯酸酯、以及属于(A5)组的亚甲基双(甲基)丙烯酰胺。其中,二乙烯基苯、乙二醇二(甲基)丙烯酸酯和三环癸烷二甲醇二(甲基)丙烯酸酯是优选的,特别地乙二醇二(甲基)丙烯酸酯是更优选的。
在本发明中,分子内具有氟烷基和至少1个自由基聚合性双键的单体B优选具有乙烯基或(甲基)丙烯酰基的任意一者中的至少1个,特别优选为上述式[1]所示的化合物,更优选为式[2]所示的化合物。
作为这样的单体B,可列举例如2,2,2-三氟乙基(甲基)丙烯酸酯、2,2,3,3,3-五氟丙基(甲基)丙烯酸酯、2-(全氟丁基)乙基(甲基)丙烯酸酯、2-(全氟己基)乙基(甲基)丙烯酸酯、2-(全氟辛基)乙基(甲基)丙烯酸酯、2-(全氟癸基)乙基(甲基)丙烯酸酯、2-(全氟-3-甲基丁基)乙基(甲基)丙烯酸酯、2-(全氟-5-甲基己基)乙基(甲基)丙烯酸酯、2-(全氟-7-甲基辛基)乙基(甲基)丙烯酸酯、1H,1H,3H-四氟丙基(甲基)丙烯酸酯、1H,1H,5H-八氟戊基(甲基)丙烯酸酯、1H,1H,7H-十二氟庚基(甲基)丙烯酸酯、1H,1H,9H-十六氟壬基(甲基)丙烯酸酯、1H-1-(三氟甲基)三氟乙基(甲基)丙烯酸酯、1H,1H,3H-六氟丁基(甲基)丙烯酸酯、3-全氟丁基-2-羟基丙基(甲基)丙烯酸酯、3-全氟己基-2-羟基丙基(甲基)丙烯酸酯、3-全氟辛基-2-羟基丙基(甲基)丙烯酸酯、3-(全氟-3-甲基丁基)-2-羟基丙基(甲基)丙烯酸酯、3-(全氟-5-甲基己基)-2-羟基丙基(甲基)丙烯酸酯、3-(全氟-7-甲基辛基)-2-羟基丙基(甲基)丙烯酸酯等。
在本发明中,关于使上述单体A与上述单体B共聚的比例,从反应性、表面改性效果的观点出发,优选相对于上述单体A 1摩尔,上述单体B为0.05摩尔~3.0摩尔,特别优选为0.1摩尔~1.5摩尔。
关于本发明的含氟高支化聚合物,也可以与上述单体A一起、或与上述单体A和上述单体B一起,使分子内具有至少1个自由基聚合性双键且不具有氟烷基的单体E共聚。作为这样的单体E,优选为具有至少1个乙烯基或(甲基)丙烯酰基的化合物、和马来酰亚胺化合物。
其中,优选为2-(2-乙烯氧基乙氧基)乙基丙烯酸酯等含有乙烯基醚基的(甲基)丙烯酸酯化合物;甲基丙烯酸缩水甘油酯等含有环氧基的(甲基)丙烯酸酯化合物;3-甲基丙烯酰氧基丙基三乙氧基硅烷等含有烷氧基甲硅烷基的(甲基)丙烯酸酯化合物;环己基马来酰亚胺、N-苄基马来酰亚胺等马来酰亚胺化合物等。
在本发明中,关于使上述单体E共聚的比例,从反应性、表面改性效果的观点出发,优选相对于上述单体A 1摩尔,上述单体E为0.05摩尔~3.0摩尔,特别优选为0.1摩尔~1.5摩尔。
作为本发明中的聚合引发剂C,优选使用偶氮系聚合引发剂。作为偶氮系聚合引发剂,可以列举例如以下的(1)~(6)所示的化合物。
(1)偶氮腈化合物:
2,2’-偶氮二异丁腈、2,2’-偶氮二(2-甲基丁腈)、2,2’-偶氮二(2,4-二甲基戊腈)、1,1’-偶氮二(1-环己烷甲腈)、2,2’-偶氮二(4-甲氧基-2,4-二甲基戊腈)、2-(氨基甲酰基偶氮)异丁腈等;
(2)偶氮酰胺化合物:
2,2’-偶氮二{2-甲基-N-[1,1-双(羟基甲基)-2-羟基乙基]丙酰胺}、2,2’-偶氮二{2-甲基-N-[2-(1-羟基丁基)]丙酰胺}、2,2’-偶氮二[2-甲基-N-(2-羟基乙基)丙酰胺]、2,2’-偶氮二[N-(2-丙烯基)-2-甲基丙酰胺]、2,2’-偶氮二(N-丁基-2-甲基丙酰胺)、2,2’-偶氮二(N-环己基-2-甲基丙酰胺)等;
(3)环状偶氮脒化合物:
2,2’-偶氮二[2-(2-咪唑啉-2-基)丙烷]二盐酸盐、2,2’-偶氮二[2-(2-咪唑啉-2-基)丙烷]二硫酸盐二水合物、2,2’-偶氮二[2-[1-(2-羟基乙基)-2-咪唑啉-2-基]丙烷]二盐酸盐、2,2’-偶氮二[2-(2-咪唑啉-2-基)丙烷]、2,2’-偶氮二(1-亚氨基-1-吡咯烷基-2-甲基丙烷)二盐酸盐等;
(4)偶氮脒化合物:
2,2’-偶氮二(2-甲基丙脒)二盐酸盐、2,2’-偶氮二[N-(2-羧基乙基)-2-甲基丙脒]四水合物等;
(5)其它:
2,2’-偶氮二异丁酸二甲酯、4,4’-偶氮二-4-氰基戊酸、2,2’-偶氮二(2,4,4-三甲基戊烷)、1,1’-偶氮二(1-乙酰氧基-1-苯基乙烷)、二甲基1,1’-偶氮二(1-环己烷甲酸酯)、4,4’-偶氮二(4-氰基戊酸)等。
(6)含有氟烷基的偶氮系聚合引发剂:
4,4’-偶氮二(4-氰基戊酸-2-(全氟甲基)乙酯)、4,4’-偶氮二(4-氰基戊酸-2-(全氟丁基)乙酯)、4,4’-偶氮二(4-氰基戊酸-2-(全氟己基)乙酯)等。
在上述偶氮系聚合引发剂中,从所得的高支化聚合物的表面能的观点出发,优选具有极性较低的取代基的引发剂,特别优选为2,2’-偶氮二异丁酸二甲酯或2,2’-偶氮二(2,4,4-三甲基戊烷)。
此外,作为含有氟烷基的偶氮系聚合引发剂,可以适合使用4,4’-偶氮二(4-氰基戊酸-2-(全氟甲基)乙酯)、4,4’-偶氮二(4-氰基戊酸-2-(全氟己基)乙酯)。
上述聚合引发剂C,相对于上述单体A 1摩尔、或相对于上述单体A和上述单体B的总摩尔数,以5摩尔%~200摩尔%的量使用,优选以15摩尔%~200摩尔%、更优选以15摩尔%~170摩尔%、进一步优选以50摩尔%~100摩尔%的量使用。
<含氟高支化聚合物的制造方法>
本发明的含氟高支化聚合物是通过在相对于上述单体A、或单体A和单体B为规定量的聚合引发剂C的存在下进行聚合而获得的,作为该聚合方法,可列举公知的方法,例如溶液聚合、分散聚合、沉淀聚合和本体聚合等,其中优选溶液聚合或沉淀聚合。从分子量控制方面出发,特别优选通过有机溶剂中的溶液聚合来实施反应。
作为此时使用的有机溶剂,可列举苯、甲苯、二甲苯、乙苯、四氢化萘等芳香族烃系溶剂;正己烷、正庚烷、矿物油精、环己烷等脂肪族或脂环式烃系溶剂;氯代甲烷、溴代甲烷、碘代甲烷、二氯甲烷、氯仿、四氯化碳、三氯乙烯、全氯乙烯、邻二氯苯等卤系溶剂;乙酸乙酯、乙酸丁酯、乙酸甲氧基丁酯、甲基溶纤剂乙酸酯、乙基溶纤剂乙酸酯、丙二醇单甲基醚乙酸酯等酯系或酯醚系溶剂;乙醚、四氢呋喃、1,4-二烷、甲基溶纤剂、乙基溶纤剂、丁基溶纤剂、丙二醇单甲基醚等醚系溶剂;丙酮、甲基乙基酮、甲基异丁基酮、二正丁基酮、环己酮等酮系溶剂;甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、2-乙基己醇、苄醇等醇系溶剂;N,N-二甲基甲酰胺、N,N-二甲基乙酰胺等酰胺系溶剂;二甲亚砜等亚砜系溶剂、N-甲基-2-吡咯烷酮等杂环式化合物系溶剂、以及它们的2种以上的混合溶剂。
其中优选的是芳香族烃系溶剂、卤系溶剂、酯系溶剂、醚系溶剂、酮系溶剂、醇系溶剂、酰胺系溶剂、亚砜系溶剂等,特别优选的是甲苯、二甲苯、邻二氯苯、乙酸丁酯、丙二醇单甲基醚乙酸酯、丙二醇单甲基醚、1,4-二烷、甲基溶纤剂、甲基异丁基酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺等。
在有机溶剂的存在下进行上述聚合反应的情况下,聚合反应物全体中的有机溶剂的含量相对于上述单体A的1质量份优选为1~100质量份,更优选为5~50质量份。
聚合反应在常压、加压密闭下、或减压下进行,从装置和操作的简便性出发,优选在常压下进行。此外,优选在N2等惰性气体气氛下进行。
聚合反应的温度优选为50~200℃,更优选为70~150℃。
更优选地,在聚合反应的温度比上述聚合引发剂C的10小时半衰期温度高20℃以上的温度下实施,更具体而言,优选通过将包含上述单体A、上述单体B、上述聚合引发剂C和有机溶剂的溶液向保持在比该聚合引发剂C的10小时半衰期温度高20℃以上的温度的该有机溶剂中滴加,从而进行聚合反应。
此外进一步更优选在反应压力下的上述有机溶剂的回流温度下实施聚合反应。
聚合反应结束后,用任意方法回收所得的含氟高支化聚合物,根据需要进行洗涤等后处理。作为从反应溶液中回收高分子的方法,可列举再沉淀等方法。
另外,也可以通过在相对于分子内不含氟烷基的单体A为规定量的分子内不含氟烷基的聚合引发剂C的存在下进行聚合,制造不含氟的高支化聚合物,然后使含有氟烷基的醇D反应,从而制造含氟高支化聚合物。另外,为了可以与含有氟烷基的醇D进行缩合反应,这里所用的聚合引发剂C优选末端包含羧酸。
作为这里所使用的含有氟烷基的醇D,可列举例如,2-(全氟丁基)-乙醇、2-(全氟己基)乙醇、1H,1H-三氟乙醇、1H,1H-五氟乙醇、2-(全氟丁基)乙醇、3-(全氟丁基)丙醇、2-(全氟己基)乙醇、3-(全氟己基)丙醇、1H,1H,3H-四氟丙醇、1H,1H,5H-八氟戊醇、1H,1H,7H-十二氟庚醇、2H-六氟-2-丙醇、1H,1H,3H-六氟丁醇等。
这里,上述含有氟烷基的醇D的使用量相对于上述单体A 1摩尔为0.05摩尔~3.0摩尔,特别优选为0.1摩尔~1.5摩尔。
实际的反应中,将不含氟的高支化聚合物和含有氟烷基的醇D溶解于可以溶解它们的适当溶剂(DMF、DMAc等)中,在4,4’-二甲基氨基吡啶(DMAP)、三乙胺等催化剂和N,N’-二异丙基碳二亚胺(DIC)、N,N’-二环己基碳二亚胺(DCC)等脱水缩合剂的存在下进行缩合反应。
缩合反应在常压、加压密闭下、或减压下进行,从装置和操作的简便性出发,优选在常压下进行。此外,优选在N2等惰性气体气氛下进行。
缩合的温度优选为室温(大约25℃)~200℃,更优选为50~150℃。
所得的含氟高支化聚合物的重均分子量(以下简写为Mw)为由凝胶渗透色谱(GPC)测得的以聚苯乙烯换算优选为1,000~200,000,更优选为2,000~100,000,最优选为5,000~60,000。
<清漆和薄膜的制造方法>
作为形成包含本发明的含氟高支化聚合物的薄膜的具体方法,首先,将含氟高支化聚合物溶解或分散在溶剂中制成清漆的形态(膜形成材料),通过铸涂法、旋涂法、刮涂法、浸涂法、辊涂法、棒涂法、模涂法、喷墨法、印刷法(凸版、凹版、平版、丝网印刷等)等将该清漆涂布在基板上,然后,用电热板或烘箱等干燥而成膜。
在这些涂布方法中,优选旋涂法。在使用旋涂法的情况下,由于可以在短时间内涂布,因此具有即使是挥发性高的溶液也可以利用,而且可以进行均匀性高的涂布这样的优势。
作为在上述清漆的形态中使用的溶剂,只要是可溶解含氟高支化聚合物的溶剂即可,可列举例如丙酮、四氢呋喃(THF)、甲苯、N,N’-二甲基甲酰胺(DMF)、环己酮、丙二醇单甲基醚(PGME)、丙二醇单甲基醚乙酸酯(PGMEA)、丙二醇单乙基醚、乳酸乙酯、二甘醇单乙基醚、丁基溶纤剂、γ-丁内酯、1,1,2,3,3,3-六氟丙基-3,3,4,5,5,5-六氟-2-戊基醚等。这些溶剂可以单独使用,也可以2种以上溶剂混合。
此外,溶解或分散于上述溶剂的浓度是任意的,但相对于含氟高支化聚合物和溶剂的总质量(合计质量),含氟高支化聚合物的浓度为0.001~90质量%,优选为0.002~80质量%,更优选为0.005~70质量%。
对所形成的包含含氟高支化聚合物的薄膜的厚度没有特别的限制,通常为0.01μm~50μm,优选为0.05μm~20μm。
<光聚合性组合物和由该组合物制作的成型品>
本发明还涉及含有(a)上述含氟高支化聚合物、(b)光聚合性化合物和(c)光聚合引发剂的光聚合性组合物。
作为上述(b)光聚合性化合物,只要是分子内具有1个以上、优选为1个~6个会由于光聚合引发剂的作用而聚合的聚合性的部位的化合物,就没有特别的限制。另外,对于本发明中的聚合性化合物的定义,并非是所谓的高分子物质的化合物,而是不仅包含狭义的单体化合物(单体),还包含二聚体、三聚体、低聚物、反应性高分子。
作为聚合性的部位,可列举作为自由基聚合性的部位的烯属不饱和键、或作为阳离子聚合性的部位的乙烯基醚结构、乙烯基硫醚结构、和环氧环、氧杂环丁烷环等环状醚结构等。因此,作为光聚合性化合物,可列举具有作为自由基聚合性的部位的烯属不饱和键的化合物、或具有作为阳离子聚合性的部位的乙烯基醚结构、环氧环或氧杂环丁烷环的化合物。
在上述光聚合性化合物中,优选具有2个以上具有烯属不饱和键的部位的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯化合物。
作为这样的聚合性化合物,可以列举例如,上述单体A中例示的(A3)(甲基)丙烯酸酯、以及(A4)具有聚亚烷基二醇链的烯属化合物。
其中,优选三环癸烷二甲醇二(甲基)丙烯酸酯、乙二醇二(甲基)丙烯酸酯、三羟甲基丙烷三(甲基)丙烯酸酯、双三羟甲基丙烷四(甲基)丙烯酸酯、季戊四醇四(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯、1,6-己二醇二(甲基)丙烯酸酯、二氧杂环己烷二醇二(甲基)丙烯酸酯、9,9-双[4-(2-(甲基)丙烯酰氧基乙氧基)苯基]芴、乙氧基化双酚A(甲基)丙烯酸酯等,特别优选三环癸烷二甲醇二(甲基)丙烯酸酯。
作为上述具有乙烯基醚结构的化合物,可以列举例如,2-氯乙基乙烯基醚、正丁基乙烯基醚、三乙二醇二乙烯基醚、1,4-环己烷二甲醇二乙烯基醚、三羟甲基乙烷三乙烯基醚、乙烯基缩水甘油醚等。
作为上述具有环氧环的化合物,可以列举例如,二甘油多缩水甘油醚、季戊四醇多缩水甘油醚、1,4-双(2,3-环氧丙氧基全氟异丙基)环己烷、山梨糖醇多缩水甘油醚、三羟甲基丙烷多缩水甘油醚、间苯二酚二缩水甘油醚、1,6-己二醇二缩水甘油醚、聚乙二醇二缩水甘油醚、苯基缩水甘油醚、对叔丁基苯基缩水甘油醚、己二酸二缩水甘油酯、邻苯二甲酸二缩水甘油酯、二溴苯基缩水甘油醚、1,2,7,8-二环氧辛烷、1,6-二羟甲基全氟己烷二缩水甘油醚、4,4’-双(2,3-环氧丙氧基全氟异丙基)二苯基醚、2,2-双(4-缩水甘油基氧基苯基)丙烷、3,4-环氧环己基甲基-3’,4’-环氧环己烷甲酸酯、3,4-环氧环己烯基甲基-3’,4’-环氧环己烯甲酸酯、3,4-环氧环己基环氧乙烷、2-(3,4-环氧环己基)-3’,4’-环氧-1,3-二烷-5-螺环己烷、1,2-亚乙基二氧双(3,4-环氧环己基甲烷)、4’,5’-环氧-2’-甲基环己基甲基-4,5-环氧-2-甲基环己烷甲酸酯、乙二醇双(3,4-环氧环己烷甲酸酯)、双(3,4-环氧环己基甲基)己二酸酯、二-2,3-环氧环戊基醚、ダイセル化学工业(株)制EHPE3150(制品名)、三菱化学(株)制157S70(制品名)等。
作为上述具有氧杂环丁烷环的化合物,可以列举例如,3-乙基-3-羟基甲基氧杂环丁烷、3-乙基-3-(苯氧基甲基)氧杂环丁烷、3,3-二乙基氧杂环丁烷、3-乙基-3-(2-乙基己氧基甲基)氧杂环丁烷等具有1个氧杂环丁烷环的化合物;1,4-双{[(3-乙基-3-氧杂环丁烷基)甲氧基]甲基}苯、二(3-乙基-3-氧杂环丁烷基甲基)醚、季戊四醇四(3-乙基-3-氧杂环丁烷基甲基)醚等具有2个以上氧杂环丁烷环的化合物。
作为上述(c)光聚合引发剂,也可以使用公知的光聚合引发剂,可列举例如,苯偶姻类;二苯甲酮类;苯偶酰缩酮类;α-羟基酮类;α-氨基酮类;酰基氧化膦类;噻吨酮类;碘盐;或锍盐等。具体而言,可列举例如チバ·ジャパン(株)制(以下为商品名)的イルガキュア(注册商标)184、イルガキュア369、イルガキュア500、イルガキュア651、イルガキュア784、イルガキュア819、イルガキュア907、イルガキュア1000、イルガキュア1300、イルガキュア1700、イルガキュア1800、イルガキュア1850、イルガキュア2959、チバ·ジャパン(株)制的ダロキュア(注册商标)1173、ADEKA(株)制アデカオプトマーSP-170、アデカオプトマーCP-77、ラムバーティ社制ESACURE(注册商标)1720等,但不限于这些。这样光聚合引发剂也可以多种组合使用。
此外,在使用具有作为阳离子聚合性的部位的乙烯基醚结构、环氧环或氧杂环丁烷环的化合物作为光聚合性化合物的情况下,作为光聚合引发剂,使用基本上在曝光时生成路易斯酸或布朗斯台德酸的光产酸剂。
作为光产酸剂,只要是在曝光时生成路易斯酸或布朗斯台德酸的化合物,就没有特别的限制,可以列举例如,二芳基碘盐化合物、三芳基锍盐化合物、重氮盐化合物等盐化合物、铁芳烃络合物化合物等。
作为二芳基碘盐化合物,可列举例如,二苯基碘、4,4’-二氯二苯基碘、4,4’-二甲氧基二苯基碘、4,4’-二叔丁基二苯基碘、(4-甲基苯基)[4-(2-甲基丙基)苯基]碘、3,3’-二硝基苯基碘等碘的、四氟硼酸盐、六氟磷酸盐、六氟砷酸盐、六氟锑酸盐等。
作为三芳基锍盐化合物,可以列举例如,三苯基锍、4-叔丁基三苯基锍、三(4-甲基苯基)锍、三(4-甲氧基苯基)锍、4-硫苯基三苯基锍等锍的、四氟硼酸盐、六氟磷酸盐、六氟砷酸盐、六氟锑酸盐等。
作为铁芳烃络合物化合物,可列举例如双环戊二烯基-(η6-异丙基苯)-铁(II)六氟磷酸盐等。
在上述光聚合性组合物中,(a)含氟高支化聚合物、(b)光聚合性化合物和(c)光聚合引发剂的配合量如下所述。
即,相对于(b)光聚合性化合物,(a)含氟高支化聚合物优选为0.01质量%~20质量%,特别优选为0.1质量%~20质量%。
此外,相对于(b)光聚合性化合物,(c)光聚合引发剂优选为0.1质量%~20质量%,优选为0.5质量%~10质量%。只要是在上述范围内,可以在不降低透射率的情况下使(b)光聚合性化合物聚合。
在上述光聚合性组合物中,只要不破坏本发明的效果,可以根据需要适当配合一般地添加的添加剂,例如,光敏化剂、阻聚剂、聚合引发剂、流平剂、表面活性剂、附着性赋予剂、增塑剂、紫外线吸收剂、抗氧化剂、储存稳定剂、抗静电剂、无机填充剂、颜料、染料等。
关于本发明的上述光聚合性组合物,可以通过涂布在基材上进行光聚合(固化),从而制成固化膜、叠层体等成型品。
作为上述基材,可以列举例如,塑料(聚碳酸酯、聚甲基丙烯酸酯、聚苯乙烯、聚酯、聚烯烃、环氧、三聚氰胺、三乙酰纤维素、ABS、AS、降冰片烯系树脂等)、金属、木材、纸、玻璃、板岩等。这些基材的形状可以为板状、膜状或三维成型体。
本发明的光聚合性组合物的涂布方法可以使用上文的<清漆和薄膜的制造方法>中描述的各种涂布方法等。另外,优选预先使用孔径为0.2μm左右的过滤器等将光聚合性组合物过滤,然后进行涂布。
涂布后,优选接着用电热板或烘箱等进行预干燥,然后照射紫外线等活性光线而进行光固化。作为活性光线,可列举紫外线、电子束、X射线等。作为紫外线照射所用的光源,可以使用太阳光线、化学灯、低压水银灯、高压水银灯、金属卤化物灯、氙灯等。
然后,通过进行后烘烤,具体是通过使用电热板、烘箱等进行加热,从而可以使聚合结束。
另外,涂布形成的膜的厚度在干燥、固化后通常为0.01μm~50μm,优选为0.05μm~20μm。
<树脂组合物和由该组合物制作的成型品>
本发明还涉及含有(a)上述含氟高支化聚合物、(d)热塑性树脂或热固性树脂的树脂组合物。
作为上述热塑性树脂,没有特别的限制,可列举例如PE(聚乙烯)、PP(聚丙烯)、EVA(乙烯-乙酸乙烯酯共聚物)、EEA(乙烯-丙烯酸乙酯共聚物)等聚烯烃系树脂;PS(聚苯乙烯)、HIPS(高抗冲聚苯乙烯)、AS(丙烯腈-苯乙烯共聚物)、ABS(丙烯腈-丁二烯-苯乙烯共聚物)、MS(甲基丙烯酸甲酯-苯乙烯共聚物)等聚苯乙烯系树脂;聚碳酸酯树脂;氯乙烯树脂;聚酰胺树脂;聚酰亚胺树脂;PMMA(聚甲基丙烯酸甲酯)等(甲基)丙烯酸类树脂;PET(聚对苯二甲酸乙二醇酯)、聚对苯二甲酸丁二醇酯、聚萘二甲酸乙二醇酯、聚萘二甲酸丁二醇酯、PLA(聚乳酸)、聚-3-羟基丁酸、聚己内酯、聚琥珀酸丁二醇酯、聚丁二酸乙二醇酯/聚己二酸乙二醇酯等聚酯树脂;聚苯醚树脂;改性聚苯醚树脂;聚缩醛树脂;聚砜树脂;聚苯硫醚树脂;聚乙烯醇树脂;聚乙醇酸;改性淀粉;乙酸纤维素、三乙酸纤维素;壳多糖、脱乙酰壳多糖;木质素;甲基系有机硅树脂、甲基苯基系有机硅树脂、改性有机硅树脂等有机硅树脂等。
其中优选聚酰亚胺树脂、(甲基)丙烯酸类树脂、聚酯树脂或有机硅树脂,更优选聚甲基丙烯酸甲酯树脂或聚乳酸树脂。
另外,在上述热塑性树脂为有机硅树脂的情况下,优选添加反应促进剂。
作为反应促进剂,没有特别的限制,可列举例如,包含铝、钛、锡等的有机金属化合物;硫酸、盐酸、乙酸、磷酸等无机或有机酸;氢氧化钠、氢氧化钾等碱;氨、二乙醇胺、三乙醇胺等胺化合物等。
此外,在上述热塑性树脂为有机硅树脂的情况下,从上述含氟高支化聚合物向上述热塑性树脂分散的分散性提高方面出发,作为上述含氟高支化聚合物,优选为其分子内具有烷氧基甲硅烷基结构的含氟高支化聚合物。
作为聚酰亚胺树脂,可列举例如由亚乙基四羧酸二酐、环丁烷四羧酸二酐、苯均四酸二酐、3,3’,4,4’-二苯甲酮四羧酸二酐、2,2’,3,3’-联苯四羧酸二酐等酸二酐成分与对苯二胺、间苯二胺、邻苯二胺、3,4’-二氨基二苯基醚、4,4’-二氨基二苯基醚、3,3’-二氨基二苯基硫化物、4,4’-二氨基二苯基砜、3,3’-二氨基二苯甲酮、4,4’-二氨基二苯甲酮、4,4’-二氨基二苯基甲烷等二胺成分制造的聚酰亚胺树脂。
此外,在上述热塑性树脂为聚酰亚胺树脂的情况下,从上述含氟高支化聚合物向上述热塑性树脂分散的分散性提高方面出发,作为上述含氟高支化聚合物,优选为其分子内具有酰亚胺结构的含氟高支化聚合物。
此外,作为上述热固性树脂,也没有特别的限制,可列举例如酚树脂、脲树脂、三聚氰胺树脂、不饱和聚酯树脂、聚氨酯树脂、环氧树脂等。
作为环氧树脂,可列举例如双酚A型环氧树脂、双酚F型环氧树脂、双酚S型环氧树脂、卤代双酚A型环氧树脂等双酚型环氧树脂;二聚酸缩水甘油酯型环氧树脂;聚亚烷基醚型环氧树脂;苯酚酚醛清漆型环氧树脂、邻甲酚酚醛清漆型环氧树脂等酚醛清漆型环氧树脂;联苯型环氧树脂;二环戊二烯型环氧树脂等脂环式环氧树脂;萘酚型环氧树脂;萘型环氧树脂;含有杂环的环氧树脂;二缩水甘油基环氧树脂;缩水甘油基胺型环氧树脂等。
其中,优选为脂环式环氧树脂或酚醛清漆型环氧树脂。
另外,在上述热固性树脂为环氧树脂的情况下,优选添加固化剂。
作为固化剂,没有特别的限制,可列举例如,有机酸酐、酚树脂、脂肪族胺类、芳香族胺类、双氰胺类等。其中优选为有机酸酐。
此外,在上述热固性树脂为环氧树脂的情况下,从上述含氟高支化聚合物向上述热固性树脂分散的分散性提高方面出发,作为上述含氟高支化聚合物,优选为其分子内具有金刚烷、三环癸烷、环己烷等脂环结构、环氧基和/或羟基的含氟高支化聚合物。
作为有机酸酐,可列举例如四氢邻苯二甲酸酐、甲基四氢邻苯二甲酸酐、六氢邻苯二甲酸酐、甲基六氢邻苯二甲酸酐、桥亚甲基四氢化邻苯二甲酸酐、甲基桥亚甲基四氢化邻苯二甲酸酐、三烷基四氢邻苯二甲酸酐、邻苯二甲酸酐、偏苯三甲酸酐、均苯四甲酸酐、二苯甲酮四甲酸酐、十二烯基琥珀酸酐、乙二醇偏苯三酸酐、甘油三偏苯三酸酐等。
在上述树脂组合物中,相对于(d)热塑性树脂或热固性树脂,(a)含氟高支化聚合物的配合量优选为0.01质量%~20质量%,特别优选为0.1质量%~20质量%。
在上述树脂组合物中,可以与热塑性树脂或热固性树脂一起合并使用一般添加的添加剂,例如,抗静电剂、润滑剂、热稳定剂、抗氧化剂、光稳定剂、荧光剂、加工助剂、交联剂、分散剂、发泡剂、阻燃剂、消泡剂、增强剂、颜料等。
关于本发明的上述树脂组合物,可以通过注射成型、挤出成型、加压成型、吹塑成型等任意成型方法来获得膜、片、或成型品等树脂成型品。
本发明的树脂成型品,如上所述,处于如下状态:与成型品内部(深部)相比,上述含氟高支化聚合物在成型品表面(界面)存在更多。因此,可以制成对成型品制作时使用的混合、成型机械等各种机械、模具的脱模性、对膜等其它树脂成型品的剥离性等、以及防水防油性、防污性优异的成型品。
实施例
以下,列举实施例更具体地说明本发明,但本发明不限于下述实施例。
另外,在实施例中,试料的调制和物性的分析中使用的装置和条件如下所述。
(1)凝胶渗透色谱(GPC)
装置:東ソー(株)制HLC-8220GPC
柱:Shodex KF-804L,KF-805L
柱温:40℃
溶剂:四氢呋喃
检测器:RI
(2)1H NMR光谱和13C NMR光谱
装置:日本电子データム(株)制JNM-ECA700
溶剂:CDCl3
内标:四甲基硅烷
(3)离子色谱(F定量分析)
装置:日本ダイオネクス(株)制ICS-1500
溶剂:2.7毫摩尔/L Na2CO3+0.3毫摩尔/L NaHCO3
检测器:电导率
(4)玻璃化转变温度(Tg)测定
装置:Perkin elmer社制Diamond DSC
测定条件:氮气气氛下
升温速度:5℃/分钟(25-160℃)
(5)5%重量减少时温度(Td5%)测定
装置:(株)リガク制TG8120
测定条件:空气气氛下
升温速度:10℃/分钟(25-500℃)
(6)椭圆光度法(折射率和膜厚测定)
装置:J.A.Woollam社制ESM-300
(7)接触角测定
装置:AST Products社制VCA Optima
测定溶剂:水和二碘甲烷
测定温度:20℃
(8)旋涂机
装置1:ミカサ(株)制MS-A100
装置2(实施例16):(株)共和理研制,旋涂机K-359SD2
(9)电热板
装置:アズワン(株)制MH-180CS,MH-3CS
(10)UV照射装置
装置:アイグラフィックス(株)制H02-L41
(11)高精度微细形状测定机(膜厚测定)
装置:(株)小坂研究所制ET-4000A
(12)雾度计(全光透射率和浊度测定)
装置:日本电色工业(株)制,NDH5000
(13)真空加热
装置:东京理化器械(株)制真空恒温干燥器VOS-201SD
(14)加压成型
装置:テスター产业(株)制台式テストプレスS型SA-303-II-S
(15)X射线光电子分光(XPS)测定
装置:Physical Electronics社制PHI ESCA 5800
X射线源:单色化AI Kα射线(2mmΦ)
X射线输出:200W,14mA
光电子放射角度:45度
(16)拉伸试验机
装置:(株)エー·アンド·デイ制テンシロン万能试验机
(17)刮刀(涂膜制作)
装置:ヨシミツ精机(株)制刮刀YD-1型(涂布厚1mil)
(18)棱镜耦合器(膜厚测定)
装置:Metricon社制2010型棱镜耦合器
(19)纳米压印装置
装置:明昌机工(株)制纳米压印机NM-0801HB
此外,简写符号表示以下意思。
EGDMA:乙二醇二甲基丙烯酸酯(新中村化学工业(株)制,制品名:1G)
DVB:二乙烯基苯(新日铁化学(株)制,制品名:DVB-960)
DCP:三环癸烷二甲醇二甲基丙烯酸酯(新中村化学工业(株)制,制品名:DCP)
PGHM:2-羟基-1,3-二甲基丙烯酰氧基丙烷(新中村化学工业(株)制,制品名:701)
ADM:1,3-金刚烷二甲醇二甲基丙烯酸酯(出光兴产(株)制)
MMA:甲基丙烯酸甲酯(纯正化学工业(株)制)
C4FHM:1H,1H,5H-八氟戊基甲基丙烯酸酯(大阪有机化学工业(株)制,制品名:V-8FM)
C4FHA:1H,1H,5H-八氟戊基丙烯酸酯(大阪有机化学工业(株)制,制品名:V-8F)
C6FHA:1H,1H,7H-十二氟庚基丙烯酸酯(ダイキン化成品贩卖(株)制,制品名:R-5610)
C4FA:2-(全氟丁基)乙基丙烯酸酯(ダイキン化成品贩卖(株)制,制品名:R-1420)
C6FM:2-(全氟己基)乙基甲基丙烯酸酯(ダイキン化成品贩卖(株)制,制品名:M-1620)
C6FA:2-(全氟己基)乙基丙烯酸酯(ダイキン化成品贩卖(株)制,制品名:R-1620)
VEEA:2-(2-乙烯氧基乙氧基)乙基丙烯酸酯(日本触媒(株)制,制品名:VEEA)
GMA:甲基丙烯酸缩水甘油酯(纯正化学(株)制)
TESMA:3-甲基丙烯酰氧基丙基三乙氧基硅烷(信越シリコーン(株)制,制品名:KBE-503)
CHMI:环己基马来酰亚胺(东京化成工业(株)制)
BMI:N-苄基马来酰亚胺(东京化成工业(株)制)
MAIB:2,2’-偶氮二异丁酸二甲酯(大塚化学(株)制,制品名:MAIB)
DCHC:二甲基1,1’-偶氮二(1-环己烷甲酸酯)(和光纯药工业(株)制,制品名:VE-073)
AMBN:2,2’-偶氮二(2-甲基丁腈)(和光纯药工业(株)制,制品名:V-59)
AF1:4,4’-偶氮二(4-氰基戊酸-2-(全氟甲基)乙酯)(和光纯药工业(株)制,AE-041)
AF6:4,4’-偶氮二(4-氰基戊酸-2-(全氟己基)乙酯)(和光纯药工业(株)制,ACVA-PFO)
AVCA:4,4’-偶氮二(4-氰基戊酸)(和光纯药工业(株)制,制品名:V-501)
C4FOH:2-(全氟丁基)乙醇(ダイキン化成品贩卖(株)制,制品名:A-1420)
C6FOH:2-(全氟己基)乙醇(ダイキン化成品贩卖(株)制,制品名:A-1620)
DMAP:4,4’-二甲基氨基吡啶(和光纯药工业(株)制)
DIC:N,N’-二异丙基碳二亚胺(东京化成工业(株)制)
A-DCP:三环癸烷二甲醇二丙烯酸酯(新中村化学工业(株)制,制品名:A-DCP)
TPTA:三羟甲基丙烷三丙烯酸酯(新中村化学工业(株)制,制品名:A-TMPT)
AD-TMP:双三羟甲基丙烷四丙烯酸酯(新中村化学工业(株)制,制品名:AD-TMP)
Irg.907:2-甲基-1[4-(甲硫基)苯基]-2-吗啉代丙烷-1-酮(チバ·ジャパン(株)制,制品名:Ciba IRGACURE 907)
Irg.651:2,2’-二甲氧基-1,2-二苯基乙烷-1-酮(チバ·ジャパン(株)制,制品名:IRGACURE 651)
Irg.184:1-羟基环己基苯基酮(チバ·ジャパン(株)制,制品名:IRGACURE 184)
Dar.1173:2-羟基-2-甲基-1-苯基丙烷-1-酮(チバ·ジャパン(株)制,制品名:DAROCURE 1173)
SP-170:阳离子系聚合引发剂(ADEKA(株)制,制品名:アデカオプトマーSP-170)
CP-77:阳离子系聚合引发剂(ADEKA(株)制,制品名:アデカオプトマーCP-77)
ESACURE1720:阳离子系聚合引发剂(ラムバーティ社制,制品名:ESACURE1720)
MCHDC:4-甲基环己烷-1,2-二甲酸酐(东京化成工业(株)制)
PLA:聚乳酸(三井化学(株)制,制品名:LACEA,Mw(GPC):160,000)
PMMA:聚甲基丙烯酸甲酯(和光纯药工业(株)制,Mw(GPC):111,000)
CEL2021P:3,4-环氧环己烯基甲基-3’,4’-环氧环己烯甲酸酯(ダイセル化学工业(株)制,制品名:セロキサイド2021P)
P22S:热塑性聚氨酯弹性体(日本ミラクトラン(株)制,制品名:P22SRNAT)
N5257:涂布用聚氨酯溶液(日本ポリウレタン工业(株)制,制品名:ニッポランN5257)
KR-400:室温固化型有机硅树脂(信越シリコーン(株)制,制品名:KR-400)
EHPE3150:脂环式固体环氧树脂(ダイセル化学工业(株)制,制品名:EHPE3150)
157S70:酚醛清漆型环氧树脂(三菱化学(株)制,制品名:157S70)
PAA清漆:由1,2,3,4-环丁烷四甲酸二酐/4,4’-二氨基二苯基醚=1/1(摩尔比)获得的聚酰胺酸的NMP溶液(浓度12质量%,粘度358mPa·s(20℃))
SPI:由3,4-二羧基-1,2,3,4-四氢-1-萘琥珀酸二酐/对苯二胺/1,3-二氨基-4-十八烷氧基苯=10/9/1(摩尔比)获得的可溶性聚酰亚胺(酰亚胺化率86%)
F-552:市售氟系表面改性剂(DIC(株)制,制品名:メガファックF-552)
F-554:市售氟系表面改性剂(DIC(株)制,制品名:メガファックF-554)
此外,本实施例中使用的有机溶剂如下所示。
甲苯:关东化学(株)制,一级
己烷:关东化学(株)制,一级
THF(四氢呋喃):关东化学(株)制,一级
PGMEA(丙二醇单甲基醚乙酸酯):东京化成工业(株)制
EGME(2-甲氧基乙醇):纯正化学(株)制,特级
HFE(氢氟醚):住友3M(株)制,ノベックHFE-7600(制品名)
IPE(二异丙基醚):纯正化学(株)制,特级
丙酮:关东化学(株)制
MEK:关东化学(株)制,特级
MIBK(4-甲基-2-戊酮):纯正化学(株)制,特级
DMF(N,N-二甲基甲酰胺):关东化学(株)制,一级
DMAc(N,N-二甲基乙酰胺):纯正化学(株)制,特级
甲醇:关东化学(株)制,特级
乙醇:关东化学(株)制,关东化学保证的一级
IPA(2-丙醇):关东化学(株)制,关东化学保证的一级
氯仿:关东化学(株)制,关东化学保证的一级
环戊酮:关东化学(株)制
NMP(N-甲基-2-吡咯烷酮):关东化学(株)制
BA(乙二醇单丁基醚):东京化成工业(株)制
[实施例1]
<使用了EGDMA、C4FHM和MAIB的高支化聚合物1的合成>
在300mL的反应烧瓶中加入甲苯87g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一200mL的反应烧瓶中加入EGDMA 7.9g(40毫摩尔)、含氟单体C4FHM 6.0g(20毫摩尔)、MAIB 4.6g(20毫摩尔)和甲苯87g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C4FHM和MAIB的上述200mL的反应烧瓶向上述300mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
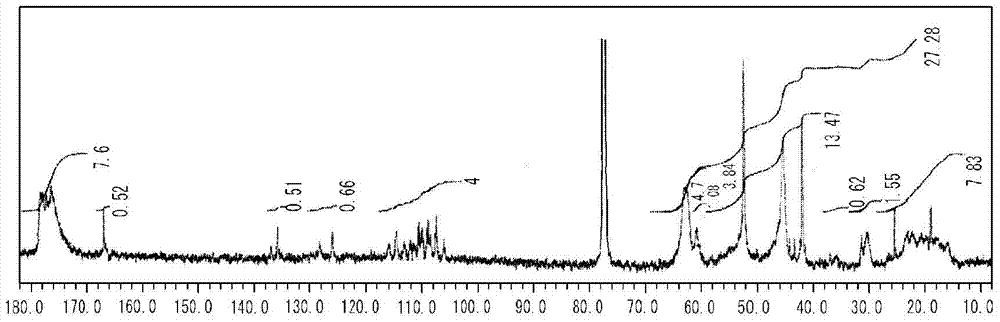
接下来,将该反应液添加至己烷/甲苯(质量比4:1)555g中使聚合物以浆料状态沉淀。将该浆料减压过滤,使用THF 42g再溶解,将该聚合物的THF溶液添加至己烷555g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物1)9.5g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图1和图2。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为21,000,分散度:Mw(重均分子量)/Mn(数均分子量)为3.5。
[实施例2]
<使用了EGDMA、C4FHM和MAIB的高支化聚合物2的合成>
在实施例1中,使C4FHM的加入量为12g(40毫摩尔),除此以外,与实施例1同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物2)8.3g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图3和图4。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw/Mn为2.9。
[实施例3]
<使用了EGDMA、C4FHA和MAIB的高支化聚合物3的合成>
在200mL的反应烧瓶中加入甲苯32g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入EGDMA 4.0g(20毫摩尔)、含氟单体C4FHA 5.7g(20毫摩尔)、MAIB 2.3g(10毫摩尔)和甲苯32g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C4FHA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,将该反应液添加至己烷/甲苯(质量比4:1)277g中使聚合物以浆料状态沉淀。将该浆料减压过滤,使用THF 36g再溶解,将该聚合物的THF溶液添加至己烷277g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物3)8.0g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图5和图6。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为22,000,分散度:Mw/Mn为2.9。
[实施例4]
<使用了EGDMA、C6FHA和MAIB的高支化聚合物4的合成>
在实施例3中,使用C6FHA 3.9g(10毫摩尔)作为含氟单体,除此以外,与实施例3同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物4)4.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图7和图8。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为15,000,分散度:Mw/Mn为2.2。
[实施例5]
<使用了EGDMA、C4FA和MAIB的高支化聚合物5的合成>
在实施例3中,使用C4FA 6.4g(20毫摩尔)作为含氟单体,除此以外,与实施例3同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物5)7.4g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图9和图10。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为16,000,分散度:Mw/Mn为2.2。
[实施例6]
<使用了EGDMA、C6FM和MAIB的高支化聚合物6的合成>
在实施例3中,使用C6FM 4.3g(10毫摩尔)作为含氟单体,除此以外,与实施例3同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物6)5.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图11和图12。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw/Mn为2.1。
[实施例7]
<使用了EGDMA、C6FM和MAIB的高支化聚合物7的合成>
在实施例6中,使C6FM的加入量为8.6g(20毫摩尔),除此以外,与实施例6同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物7)10.1g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图13和图14。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为16,000,分散度:Mw/Mn为1.8。
[实施例8]
<使用了EGDMA、C6FA和MAIB的高支化聚合物8的合成>
实施例3中,使用C6FA 4.2g(10毫摩尔)作为含氟单体,除此以外,与实施例3同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物8)4.9g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图15和图16。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw/Mn为2.2。
[实施例9]
<使用了EGDMA、C6FA和MAIB的高支化聚合物9的合成>
在实施例8中,使C6FA的加入量为8.4g(20毫摩尔),除此以外,与实施例8同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物9)9.7g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图17和图18。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为20,000,分散度:Mw/Mn为2.0。
[实施例10]
<使用了EGDMA、C6FA和MAIB的高支化聚合物10的合成>
在实施例8中,使C6FA的加入量为0.84g(2毫摩尔),除此以外,与实施例8同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物10)4.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图19和图20。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为16,000,分散度:Mw/Mn为3.8。
[实施例11]
<使用了EGDMA、C6FA和MAIB的高支化聚合物11的合成>
在实施例8中,使C6FA的加入量为2.5g(6毫摩尔),除此以外,与实施例8同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物11)5.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图21和图22。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为14,000,分散度:Mw/Mn为3.0。
[比较例1]
<使用了EGDMA和MAIB的高支化聚合物12的合成>
在300mL的反应烧瓶中加入甲苯79g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一200mL的反应烧瓶中加入EGDMA 9.9g(50毫摩尔)、MAIB5.8g(84毫摩尔)和甲苯79.2g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经90分钟从加入有EGDMA和MAIB的上述200mL的反应烧瓶向上述300mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,将该反应液添加至己烷748g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物12)10.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图23和图24。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw/Mn为4.8。
[实施例18]
<使用了EGDMA、C6FA和MAIB的高支化聚合物13的合成>
在200mL的反应烧瓶中加入甲苯43.6g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入EGDMA 4.0g(20毫摩尔)、作为含氟单体的C6FA 4.2g(10毫摩尔)、MAIB 2.8g(12毫摩尔)和甲苯43.6g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C6FA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯75.2g,然后添加至己烷278g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物13)4.4g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图26和图27。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为6,800,分散度:Mw/Mn为1.9。
[实施例19]
<使用了DVB、C6FA和MAIB的高支化聚合物14的合成>
在200mL的反应烧瓶中加入甲苯29.6g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DVB 1.6g(12毫摩尔)、作为含氟单体C6FA 7.5g(18毫摩尔)、MAIB 2.5g(11毫摩尔)和甲苯29.6g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB、C6FA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,在该反应液中添加己烷59.2g,使聚合物沉淀,倾析出上清液。在残留的沉淀物中添加THF 29.6g,在60℃下进行加热搅拌,使聚合物溶解。将该聚合物溶液的THF用旋转蒸发器减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物14)10.1g。将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图28和图29。
再沉淀纯化后的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为23,000,分散度:Mw/Mn为2.0。
[实施例20]
<使用了DCP、C6FA和MAIB的高支化聚合物15的合成>
在200mL的反应烧瓶中加入甲苯58g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DCP 7.3g(20毫摩尔)、作为含氟单体的C6FA 4.2g(10毫摩尔)、MAIB 2.8g(12毫摩尔)和甲苯58g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DCP、C6FA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯51.1g后,添加至己烷510g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物15)5.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图30和图31。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为12,000,分散度:Mw/Mn为1.9。
[实施例21]
<使用了ADM、C6FA和MAIB的高支化聚合物16的合成>
在200mL的反应烧瓶中加入甲苯31.7g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入ADM 6.7g(20毫摩尔)、作为含氟单体的C6FA 4.2g(10毫摩尔)、MAIB 2.3g(10毫摩尔)和甲苯31.7g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有ADM、C6FA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯31.7g后,添加至己烷465g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物16)5.9g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图32和图33。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为24,000,分散度:Mw/Mn为2.6。
[实施例22]
<使用了DCP、C6FA和DCHC的高支化聚合物17的合成>
在200mL的反应烧瓶中加入甲苯58.3g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DCP 7.3g(20毫摩尔)、作为含氟单体的C6FA 4.2g(10毫摩尔)、DCHC 3.7g(12毫摩尔)和甲苯58.3g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DCP、C6FA和DCHC的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯55.4g后,添加至己烷510g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物17)5.2g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图34和图35。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为20,000,分散度:Mw/Mn为2.4。
[实施例23]
<使用了DCP、C6FA和DCHC的高支化聚合物18的合成>
在实施例22中,使甲苯的加入量为66.3g,除此以外,与实施例22同样地进行聚合、纯化,获得了白色粉末的目标物质(高支化聚合物18)3.8g。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为7,200,分散度:Mw/Mn为2.9。
[实施例24]
<使用了EGDMA和AF1的高支化聚合物19的合成>
在200mL的反应烧瓶中加入MIBK15.9g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入EGDMA 2.0g(10毫摩尔)、AF12.2g(5毫摩尔)和MIBK 15.9g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有EGDMA和AF1的上述50mL的反应烧瓶向上述200mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 19.8g后,添加至甲醇139g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物19)2.2g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图36和图37。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为24,000,分散度:Mw/Mn为2.4。
[实施例25]
<使用了EGDMA、C6FA和AF1的高支化聚合物20的合成>
在100mL的反应烧瓶中加入MIBK 12.7g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入EGDMA 1.6g(8毫摩尔)、作为含氟单体C6FA 1.7g(4毫摩尔)、AF11.8g(4毫摩尔)和MIBK 12.7g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有EGDMA、C6FA和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 15.9g后,添加至己烷/丙酮(质量比10:1)174g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF 12g中。将该聚合物的THF溶液添加至己烷/丙酮(重量比10:1)174g中,再次使聚合物沉淀。采用倾析除去上清液后,使该残渣再溶解于THF 12g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物20)2.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图38和图39。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw/Mn为2.3。
[实施例26]
<使用了EGDMA、C6FA和AF1的高支化聚合物21的合成>
在100mL的反应烧瓶中加入MIBK 15.9g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入EGDMA 1.9g(10毫摩尔)、作为含氟单体的C6FA 6.3g(15毫摩尔)、AF12.2g(5毫摩尔)和MIBK 15.9g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有EGDMA、C6FA和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 19.9g后,添加至己烷/丙酮(质量比10:1)200g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF19g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)200g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF12g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物21)2.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图40和图41。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为19,000,分散度:Mw/Mn为3.4。
[实施例27]
<使用了DVB和AF1的高支化聚合物22的合成>
在200mL的反应烧瓶中加入MIBK 26.1g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、AF13.6g(8毫摩尔)和MIBK 26.1g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB和AF1的上述50mL的反应烧瓶向上述200mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 39.1g后,添加至己烷/丙酮(质量比10:1)200g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物22)2.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图42和图43。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为11,000,分散度:Mw/Mn为2.1。
[实施例28]
<使用了DVB、C6FA和AF1的高支化聚合物23的合成>
在100mL的反应烧瓶中加入MIBK13.1g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 0.7g(5毫摩尔)、作为含氟单体的C6FA 1.1g(2.5毫摩尔)、AF11.8g(4毫摩尔)和MIBK 26.1g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB、C6FA和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 19.5g后,添加至己烷/丙酮(质量比10:1)72g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF6.5g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)72g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF6.5g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物23)1.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图44和图45。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为12,000,分散度:Mw/Mn为1.9。
[实施例29]
<使用了DVB、C6FA和AF1的高支化聚合物24的合成>
在200mL的反应烧瓶中加入MIBK 26.1g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、作为含氟单体的C6FA 6.3g(15毫摩尔)、AF13.6g(8毫摩尔)和MIBK 26.1g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB、C6FA和AF1的上述50mL的反应烧瓶向上述200mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 39.1g后,添加至己烷/丙酮(质量比10:1)200g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF13g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)200g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF13g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物24)6.0g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图46和图47。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为11,000,分散度:Mw/Mn为2.3。
[实施例30]
<使用了EGDMA、C6FA和AF6的高支化聚合物25的合成>
在100mL的反应烧瓶中加入MIBK17.8g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入EGDMA 1.0g(5毫摩尔)、作为含氟单体的C6FA 1.1g(2.5毫摩尔)、AF61.4g(1.5毫摩尔)和MIBK 17.8g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有EGDMA、C6FA和AF6的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 29.7g后,添加至己烷/丙酮(质量比10:1)109g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF10g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)109g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF10g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物25)1.2g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图48和图49。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为12,000,分散度:Mw/Mn为1.6。
[实施例31]
<使用了DVB和AF6的高支化聚合物26的合成>
在100mL的反应烧瓶中加入MIBK 23.4g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 0.5g(4毫摩尔)、AF61.9g(2毫摩尔)和MIBK 23.4g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB和AF6的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 41.7g后,添加至己烷/丙酮(质量比10:1)67g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF5g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)67g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF5g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物26)0.5g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图50和图51。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为16,000,分散度:Mw/Mn为1.4。
[实施例32]
<使用了DVB、C6FA和AF6的高支化聚合物27的合成>
在200mL的反应烧瓶中加入MIBK23.4g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 0.5g(4毫摩尔)、作为含氟单体的C6FA 0.8g(2毫摩尔)、AF61.9g(2毫摩尔)和MIBK 23.4g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB、C6FA和AF6的上述50mL的反应烧瓶向上述200mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中几乎全量蒸馏除去MIBK后,使该残渣再溶解于THF5g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)67g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF5g中。将该聚合物的THF溶液添加至己烷/丙酮(质量比10:1)67g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF5g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物27)0.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图52和图53。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为13,000,分散度:Mw/Mn为1.2。
[实施例33]
<使用了EGDMA、C6FA和AVCA的高支化聚合物28的合成>
在300mL的反应烧瓶中加入EGME 47.6g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度125℃)。
在另一100mL的反应烧瓶中加入EGDMA 2.4g(12毫摩尔)、作为含氟单体的C6FA 2.5g(6毫摩尔)、AVCA 1.7g(9.6毫摩尔)和EGME 47.6g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经60分钟从加入有EGDMA、C6FA和AVCA的上述100mL的反应烧瓶向上述300mL反应烧瓶中的回流着的EGME中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去EGME 71.4g后,添加至己烷/乙醇(质量比10:1)262g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,使所得的固体再溶解于THF24g中。将该聚合物的THF溶液添加至己烷/乙醇(质量比10:1)262g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物28)2.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图54和图55。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为7,000,分散度:Mw/Mn为2.3。
[实施例34]
<使用了DVB、C6FA和AVCA的高支化聚合物29的合成>
在300mL的反应烧瓶中加入EGME 31.3g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度125℃)。
在另一100mL的反应烧瓶中加入DVB 1.6g(12毫摩尔)、作为含氟单体C6FA 2.5g(6毫摩尔)、AVCA 2.7g(9.6毫摩尔)和EGME 31.3g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经30分钟从加入有DVB、C6FA和AVCA的上述100mL的反应烧瓶向上述300mL反应烧瓶中的回流着的EGME中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去EGME 46.9g后,添加至己烷/乙醇(质量比10:1)172g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,使所得的固体再溶解于THF16g中。将该聚合物的THF溶液添加至己烷/乙醇(质量比10:1)172g中使聚合物以浆料状态再沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物29)1.9g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图56和图57。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为47,000,分散度:Mw/Mn为3.9。
[实施例35]
<使用了高支化聚合物32和C4FOH的高支化聚合物30的合成>
在100mL的反应烧瓶中,使后述的比较例2中获得的高支化聚合物322.6g溶解于DMF 26g中,加入作为氟代醇的C4FOH 2.9g(11毫摩尔)、DMAP 1.3g(10毫摩尔),一边搅拌一边流入氮气5分钟。在该溶液中滴加DIC 1.4g(11毫摩尔),在室温(大约25℃)下搅拌20小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去几乎全量的DMF后,使其再溶解于THF13g中。将该聚合物的THF溶液添加至己烷/IPA(质量比10:1)282g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF13g中。将该聚合物的THF溶液添加至己烷/IPA(质量比10:1)282g中,再次使聚合物沉淀。倾析出上清液后,使该残渣再溶解于THF13g中,减压蒸馏除去,真空干燥,获得了白色粉末的目标物质(高支化聚合物30)2.9g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图58和图59。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为34,000,分散度:Mw/Mn为1.8。
[实施例36]
<使用了高支化聚合物32和C6FOH的高支化聚合物31的合成>
在实施例35中,使用C6FOH 5.5g(15毫摩尔)作为氟代醇,除此以外,与实施例35同样地进行反应、纯化,获得了白色粉末的目标物质(高支化聚合物31)3.0g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图60和图61。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为46,000,分散度:Mw/Mn为1.6。
[比较例2]
<使用了DVB和AVCA的高支化聚合物32的合成>
在500mL的反应烧瓶中加入DMAc 200g,一边搅拌一边流入氮气5分钟,加热使内液的温度变为110℃。
在另一300mL的反应烧瓶中加入DVB 13g(100毫摩尔)、AVCA28g(100毫摩尔)和DMAc 200g,一边搅拌一边流入氮气5分钟进行氮气置换。
使用滴液泵经60分钟从加入有DVB和AVCA的上述300mL的反应烧瓶向上述的500mL反应烧瓶中的加热着的DMAc中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去DMAc 270g后,添加至氯仿1,300g中使聚合物沉淀。倾析出上清液后,使该残渣再溶解于甲醇130g中。将该聚合物的甲醇溶液添加至IPE 1,300g中,再次使聚合物沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物32)18g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图62和图63。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为22,000,分散度:Mw/Mn为2.1。
[比较例3]
<使用了MMA和C6FA的直链状聚合物1的合成>
在100mL的反应烧瓶中加入MMA 8.0g(80毫摩尔)、C6FA 8.4g(20毫摩尔)、MAIB 0.92g(4毫摩尔)和MEK 26.9g,一边搅拌一边进行5分钟的氮气鼓泡后,在内液的温度80℃下聚合7小时。
将该反应液添加至己烷662g中使聚合物沉淀,倾析出上清液后,使该残渣再溶解于THF 54g中。将该聚合物的THF溶液添加至己烷662g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(直链状聚合物1)6.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图64和图65。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为21,000,分散度:Mw/Mn为2.0。
[比较例4]
<使用了MMA和C6FA的直链状聚合物2的合成>
在比较例3中,使MAIB的加入量为1.8g(8毫摩尔),除此以外,与比较例3同样地进行聚合、纯化,获得了白色粉末的目标物质(直链状聚合物2)6.9g。
将所得的目标物质的13C NMR光谱的测定结果示于图66。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为11,000,分散度:Mw/Mn为1.8。
[比较例5]
<使用了MMA和C6FA的直链状聚合物3的合成>
在比较例3中,使MAIB的加入量为2.8g(12毫摩尔),除此以外,与比较例3同样地进行聚合、纯化,获得了白色粉末的目标物质(直链状聚合物3)5.7g。
将所得的目标物质的13C NMR光谱的测定结果示于图67。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为8,000,分散度:Mw/Mn为1.9。
[实施例45]
<使用了EGDMA、C6FA、VEEA和MAIB的高支化聚合物33的合成>
在200mL的反应烧瓶中加入甲苯59g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入EGDMA 4.0g(20毫摩尔)、C6FA5.2g(12.5毫摩尔)、VEEA 1.9g(10毫摩尔)、MAIB 2.8g(12毫摩尔)和甲苯59g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C6FA、VEEA和MAIB的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,将该反应液添加至己烷277g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物33)6.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图75和图76。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为8,400,分散度:Mw(重均分子量)/Mn(数均分子量)为2.5。
[实施例46]
<使用了DCP、C6FA、GMA和DCHC的高支化聚合物34的合成>
在200mL的反应烧瓶中加入甲苯66g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DCP 7.3g(20毫摩尔)、C6FA5.4g(12.5毫摩尔)、GMA 1.4g(10毫摩尔)、DCHC 3.7g(12毫摩尔)和甲苯66g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DCP、C6FA、GMA和DCHC的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,将该反应液添加至己烷510g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物34)6.4g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图77和图78。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为8,000,分散度:Mw(重均分子量)/Mn(数均分子量)为1.9。
[实施例47]
<使用了PGHM、C6FA和DCHC的高支化聚合物35的合成>
在200mL的反应烧瓶中加入甲苯50g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入PGHM 4.6g(20毫摩尔)、C6FA4.2g(10毫摩尔)、DCHC 3.7g(12毫摩尔)和甲苯50g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有PGHM、C6FA和DCHC的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,将该反应液添加至己烷320g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物35)6.2g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图79和图80。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为7,000,分散度:Mw(重均分子量)/Mn(数均分子量)为3.1。
[实施例48]
<使用了EGDMA、C6FA、TESMA和MAIB的高支化聚合物36的合成>
在100mL的反应烧瓶中加入甲苯16g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一50mL的反应烧瓶中加入EGDMA 2.0g(10毫摩尔)、C6FA2.1g(5毫摩尔)、TESMA 1.5g(5毫摩尔)、MAIB 1.2g(5毫摩尔)和甲苯16g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C6FA、TESMA和MAIB的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯28g后,将该反应液添加至己烷198g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物36)3.4g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图81和图82。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为18,000,分散度:Mw(重均分子量)/Mn(数均分子量)为2.2。
[实施例49]
<使用了DVB、C6FA、TESMA和MAIB的高支化聚合物37的合成>
在300mL的反应烧瓶中加入甲苯37g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DVB 2.0g(15毫摩尔)、C6FA 3.1g(7.5毫摩尔)、TESMA 2.4g(8.2毫摩尔)、MAIB 3.1g(14毫摩尔)和甲苯37g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA、TESMA和MAIB的上述100mL的反应烧瓶向上述300mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯59g后,将该反应液添加至甲醇195g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物37)2.0g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图83和图84。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为23,000,分散度:Mw(重均分子量)/Mn(数均分子量)为1.4。
[实施例50]
<使用了EGDMA、C6FA、TESMA和AF1的高支化聚合物38的合成>
在100mL的反应烧瓶中加入MIBK16g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入EGDMA 2.0g(10毫摩尔)、C6FA5.0g(12毫摩尔)、TESMA 1.5g(5毫摩尔)、AF11.1g(2.5毫摩尔)和MIBK16g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有EGDMA、C6FA、TESMA和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 28g后,将该反应液添加至己烷/乙醇(质量比10:1)150g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物38)5.4g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图85和图86。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为36,000,分散度:Mw(重均分子量)/Mn(数均分子量)为2.7。
[实施例54]
<使用了DVB、C6FA、CHMI和AF1的高支化聚合物39的合成>
在100mL的反应烧瓶中加入MIBK 25g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、C6FA 3.1g(7.5毫摩尔)、CHMI 0.9g(5毫摩尔)、AF13.6g(8毫摩尔)和MIBK25g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA、CHMI和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 42g后,添加至甲醇130g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物39)4.3g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图87和图88。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为21,000,分散度:Mw(重均分子量)/Mn(数均分子量)为1.8。
[实施例55]
<使用了DVB、C6FA、CHMI和AF1的高支化聚合物40的合成>
在100mL的反应烧瓶中加入MIBK 25g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、C6FA 4.2g(10毫摩尔)、CHMI 1.8g(10毫摩尔)、AF13.6g(8毫摩尔)和MIBK 25g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA、CHMI和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK42g后,添加至甲醇130g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物40)7.2g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图89和图90。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为32,000,分散度:Mw(重均分子量)/Mn(数均分子量)为2.5。
[实施例56]
<使用了DVB、C6FA、BMI和AF1的高支化聚合物41的合成>
在100mL的反应烧瓶中加入MIBK25g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一50mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、C6FA 3.1g(7.5毫摩尔)、BMI 0.9g(5毫摩尔)、AF13.6g(8毫摩尔)和MIBK 25g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA、BMI和AF1的上述50mL的反应烧瓶向上述100mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 42g后,添加至甲醇130g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物41)4.8g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图91和图92。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为22,000,分散度:Mw(重均分子量)/Mn(数均分子量)为1.9。
[实施例57]
<使用了DVB、C6FA、CHMI和AMBN的高支化聚合物42的合成>
在200mL的反应烧瓶中加入MIBK 39g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度116℃)。
在另一100mL的反应烧瓶中加入DVB 1.3g(10毫摩尔)、C6FA 4.2g(10毫摩尔)、CHMI 1.8g(10毫摩尔)、AMBN 1.9g(10毫摩尔)和MIBK 39g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA、CHMI和AMBN的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的MIBK中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去MIBK 70g后,添加至甲醇130g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物42)5.6g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图93和图94。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为13,000,分散度:Mw(重均分子量)/Mn(数均分子量)为2.2。
[实施例58]
<使用了DVB、C6FA和AMBN的高支化聚合物43的合成>
在200mL的反应烧瓶中加入甲苯62g,一边搅拌一边流入氮气5分钟,加热直到内液回流(大约温度110℃)。
在另一100mL的反应烧瓶中加入DVB 3.3g(25毫摩尔)、C6FA5.2g(12.5毫摩尔)、AMBN 3.8g(20毫摩尔)和甲苯62g,一边搅拌一边流入氮气5分钟进行氮气置换,采用冰浴冷却至0℃。
使用滴液泵经30分钟从加入有DVB、C6FA和AMBN的上述100mL的反应烧瓶向上述200mL反应烧瓶中的回流着的甲苯中滴加内容物。滴加结束后,熟化1小时。
接下来,使用旋转蒸发器从该反应液中蒸馏除去甲苯104g后,添加至己烷130g中使聚合物以浆料状态沉淀。将该浆料减压过滤,真空干燥,获得了白色粉末的目标物质(高支化聚合物43)6.9g。
将所得的目标物质的1H NMR和13C NMR光谱的测定结果示于图95和图96。
此外,目标物质的由GPC测得的以聚苯乙烯换算测定的重均分子量Mw为17,000,分散度:Mw(重均分子量)/Mn(数均分子量)为2.6。
将实施例1~11、18~36、45~50和54~58、以及比较例1~5中调制的高支化聚合物1~43和直链状聚合物1~3的重均分子量和分散度、以及各高支化聚合物的由13C NMR光谱求出的含氟单体导入量、和由元素分析求出的氟原子含量等示于表1。
表1
[实施例12]
<高支化聚合物1~43和直链状聚合物1~3在有机溶剂中的溶解性>
评价实施例1~11、18~36、45~50和54~58、以及比较例1~5中调制的高支化聚合物1~43和直链状聚合物1~3在有机溶剂中的溶解性。使高支化聚合物1~43或直链状聚合物1~3各10mg溶解在表2所示的各有机溶剂90mg中而评价。将所得的结果示于表2。
[评价基准]
○···完全溶解的状态
×···有未溶解的状态
表2
[实施例13]
<高支化聚合物1~43和直链状聚合物1~3的薄膜制作和物性评价>
将由实施例1~11、18~36、45~50和54~58、以及比较例1~5中获得的各高支化聚合物1~43或直链状聚合物1~3的0.25g溶解在表3所记载的溶剂4.75g中,进行过滤器过滤,调制出各高支化聚合物溶液或直链状聚合物溶液。将该高支化聚合物溶液或直链状聚合物溶液旋涂(slope 5秒,1,500rpm 30秒,slope 5秒)在硅晶片上,通过在100℃下进行30分钟的热处理而使溶剂蒸发,成膜。
对所得的薄膜的波长633nm下的折射率、以及水和二碘甲烷的接触角进行评价。此外,由接触角的结果计算出表面能。此外,测定各高支化聚合物粉末或直链状聚合物粉末的玻璃化转变温度(Tg)和5%重量减少温度(Td5%)。将所得的结果示于表3。
表3
[实施例14]
<使用了高支化聚合物2、4、8的光固化树脂的表面改性>
按照表4中记载的配合量配合作为光聚合性化合物(单体)的A-DCP、作为高支化聚合物的上述高支化聚合物2、4或8、作为光聚合引发剂的Irg.907、和作为有机溶剂的PGMEA,将其进行过滤器过滤,调制出光聚合性组合物。将该组合物旋涂(slope 5秒,1,500rpm 30秒,slope 5秒)在硅晶片上,通过在60℃下进行1分钟的热处理而进行预干燥,制作出应用例1~12的薄膜。作为比较例,制作出不添加高支化聚合物的薄膜(应用例13)、代替高支化聚合物而添加比较例3中调制的直链状聚合物1的薄膜(应用例14)、和代替高支化聚合物而添加市售的氟系表面改性剂F-552的薄膜(应用例15)。
对于所得的各薄膜(应用例1~15),采用UV照射装置以曝光量16mW/cm2曝光10分钟。将曝光后的各薄膜在150℃下进行20分钟的热处理,制作出光固化薄膜。
对于所得的各光固化薄膜,通过高精度微细形状测定机测定膜厚、通过椭圆光度法测定波长633nm下的折射率、通过雾度计测定全光透射率和浊度、以及测定水和二碘甲烷的接触角。将所得的结果示于表5。
表4
※表示树脂全体(A-DCP和高支化聚合物)相对于树脂组合物全部质量的添加量(质量%)。
表5
如表5所示,添加了高支化聚合物2、4或8的应用例1~12的光固化薄膜,与未配合高支化聚合物的应用例13的光固化薄膜同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的A-DCP单体的光固化薄膜(应用例13)中,水的接触角为64.8度,二碘甲烷的接触角为28.4度,与此相对,添加了高支化聚合物的光固化薄膜(应用例1~12)中,水的接触角为73.7~108.1度,二碘甲烷的接触角为46.7~78.8度,均显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水防油性。
此外,与添加了直链状聚合物1(应用例14)和F-552(应用例15)的情况相比,添加了高支化聚合物8的光固化薄膜(应用例12)显示出高的接触角。由该结果明确了,主动导入了分支结构的本发明的高支化聚合物显示出微粒的行为,容易在光固化薄膜的表面集中,并显示出高的接触角。
[实施例15]
<使用了高支化聚合物2、4、8的热塑性树脂的表面改性>
按照表6中记载的配合量配合作为热塑性树脂的PMMA或PLA、上述高支化聚合物2、4或8、和作为有机溶剂的THF(使用PMMA树脂时)或氯仿(使用PLA树脂时),将其进行过滤器过滤,调制出热塑性树脂组合物。将该组合物在玻璃基板上流延,在20℃下进行16小时干燥,制作出应用例16~31的流延膜。作为比较例,制作出未添加高支化聚合物的流延膜(应用例32、33)。
对于所得的各流延膜,在真空下在50℃下进行8小时的热处理。
对于进行了热处理的各膜,通过高精度微细形状测定机测定膜厚、通过雾度计测定全光透射率和浊度、以及测定水和二碘甲烷的接触角。将结果示于表7。
表6
※表示树脂全体(热塑性树脂和高支化聚合物)相对于树脂组合物全部质量的添加量(质量%)。
表7
如表7所示,添加了高支化聚合物2、4或8的应用例16~31的流延膜,与未配合高支化聚合物的应用例32和33的流延膜同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的PMMA单体的流延膜(应用例32)中,水的接触角为75.5度,二碘甲烷的接触角为39.6度,与此相对,添加了高支化聚合物的PMMA流延膜(应用例16~23)中,水的接触角为83.8~103.9度,二碘甲烷的接触角为53.8~79.2度,均显示出高的接触角。此外,未添加高支化聚合物的PLA单体的流延膜(应用例33)中,水的接触角为75.7度,二碘甲烷的接触角为43.7度,与此相对,添加了高支化聚合物的PLA流延膜(应用例24~31),水的接触角为83.7~104.5度,二碘甲烷的接触角为59.0~81.9度,均显示出高的接触角。由这些结果明确了,PMMA、PLA的任一树脂均通过添加高支化聚合物而赋予了防水防油性。
[实施例16]
<高支化聚合物3/PMMA掺混薄膜表面的分析>
将上述高支化聚合物3和PMMA(Polymer Source社制,制品编号P88-MMA,Mw:19,300,Mw/Mn:1.06)以质量比计为5/95进行混合,再将该混合物4.5质量份溶解在甲苯95.5质量份中(基质浓度4.5质量%)。将所得的甲苯溶液旋涂(1,300rpm 60秒)在硅晶片上后,在25℃下干燥12小时,制作出膜厚约200nm的高支化聚合物3/PMMA掺混薄膜(应用例34)。此外,作为比较例,制作出未添加PMMA的高支化聚合物3单体的薄膜(应用例35)。
对于所得的各薄膜,在真空中在150℃下进行24小时热处理。
对于热处理前和热处理后的各薄膜,对薄膜的最表面(距离表面大约深度10nm左右的范围)的C原子、O原子、F原子进行XPS测定。由所得的结果计算出各薄膜中的F原子/C原子的强度比,求出应用例34的掺混薄膜最表面中的高支化聚合物3的比率。将结果示于表8。
表8
※表示相对于应用例35的F/C比的比例。
如表8所示,高支化聚合物3/PMMA掺混薄膜的最表面中的高支化聚合物3的比率在热处理的前后大幅度变化,确认了经过了热处理的上述高支化聚合物3/PMMA掺混薄膜的最表面更大量存在高支化聚合物3。
[实施例17]
<高支化聚合物3/PMMA块体膜的剥离强度试验>
将上述高支化聚合物3和聚甲基丙烯酸甲酯(Aldrich社制,Cas No.9011-147,Mw:350,000)以质量比计为5/95进行混合,再将该混合物2质量份溶解在甲苯98质量份中(基质浓度2质量%)。将该溶液使用以体积计为80倍量的己烷进行再沉淀,将析出的固体过滤,干燥。将所得的固体在40MPa、210℃下进行10分钟加压成型后,再在140℃下保持10分钟,制作出厚度100μm的高支化聚合物3/PMMA块体膜。此外,作为比较例,同样地制作出未添加高支化聚合物3的厚度100μm的PMMA单独膜。
另外,将市售的聚碳酸酯在40MPa、255℃下进行10分钟加压成型,制作出厚度40μm的聚碳酸酯膜。
将制作出的高支化聚合物3/PMMA块体膜与制作的聚碳酸酯膜贴合,使用加压机一边在140℃下施加30MPa的压力一边热压接。同样地将PMMA单独膜与上述聚碳酸酯膜贴合,进行热压接。
对于所得的各个叠层膜,基于使用了拉伸试验机的180度剥离试验(JIS K6854),将切出的8mm宽度的叠层膜以300mm/分钟的速度拉开,评价剥离强度。将结果示于图25。
如图25所示,与PMMA单独膜的情况相比,高支化聚合物3/PMMA块体膜中,剥离强度降低。即,得到了通过添加高支化聚合物3而提高了PMMA膜的剥离性的结果。
[实施例37]
<对多官能丙烯酸系单体的溶解性>
评价由实施例8、18和45、以及比较例3~5获得的各聚合物对多官能丙烯酸系单体TPTA和AD-TMP的溶解性。此外,也同样地评价市售的氟系表面改性剂F-552和F-554对多官能丙烯酸系单体的溶解性。分别将各聚合物、F-552或F-5540.1g添加至各多官能丙烯酸系单体9.9g中,在螺纹管内在80℃下进行3小时加热搅拌,评价溶解性。将所得的结果示于表9。
[评价基准]
○···完全溶解的状态
△···虽然溶解但为白浊的状态
×···有未溶解的状态
表9
如表9所示,确认了高支化聚合物13和33对TPTA、AD-TMP显示良好的溶解性,即使不使用溶剂也可以直接将高支化聚合物添加在树脂中。另一方面,直链状聚合物1~3均不溶解。此外,市售的氟系表面改性剂也同样地不溶解。
[实施例38]
<使用了高支化聚合物13、33的光固化树脂的表面改性>
在作为多官能丙烯酸系单体的TPTA或AD-TMP中分别按照丙烯酸系单体和高支化聚合物的合计质量为10.0g的方式添加表10所示的添加量的高支化聚合物13或33,在80℃下进行3小时加热搅拌,使高支化聚合物溶解。然后,冷却至室温(大约25℃)后,添加表10中记载的光聚合引发剂0.1g,通过在室温(大约25℃)下进行1小时搅拌使光聚合引发剂溶解。将其注入到以1mm厚度的有机硅树脂为衬垫的玻璃基板上,采用UV照射装置以曝光量16mW/cm2曝光10分钟,制作出厚度1mm的光固化树脂。作为比较例,同样地制作出未添加高支化聚合物的光固化树脂。
对于所得的各光固化树脂,通过雾度计测定全光透射率和浊度、以及分别测定水的接触角。将所得的结果示于表10。
表10
如表10所示,在TPTA中添加了高支化聚合物13或33的光固化树脂(应用例44~47),与未添加高支化聚合物的光固化树脂(应用例53)同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的TPTA单体的光固化树脂(应用例53)中,水的接触角为74.0度,与此相对,添加了高支化聚合物的光固化树脂(应用例44~47)中,水的接触角为94.7~105.3度,均显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
另一方面,使用Irg.907作为光聚合引发剂,在AD-TMP中添加了高支化聚合物13或33的光固化树脂(应用例50和52)中,也与使用相同光聚合引发剂Irg.907且未添加高支化聚合物的光固化树脂(应用例54)同样地,显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的AD-TMP单体的光固化树脂(应用例54)中,水的接触角为73.9度,与此相对,添加了高支化聚合物的光固化树脂(应用例50和52)中,水的接触角为103.3~105.3度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
此外,在使用了其它光聚合引发剂的情况下(应用例48、49和51),显示出高的水的接触角,为104.2~105.5度。
[实施例39]
<对环氧树脂的溶解性>
评价由实施例8、18、20~23、46和47、以及比较例3~5获得的各聚合物对环氧树脂CEL2021P的溶解性。此外,也同样地评价市售的氟系表面改性剂F-552和F-554对CEL2021P的溶解性。向CEL2021P 9.9g中分别添加各聚合物、F-552或F-5540.1g,在螺纹管内在70℃下进行3小时加热搅拌,评价溶解性。将所得的结果示于表11。
[评价基准]
○···完全地溶解的状态
△···虽然溶解但白浊的状态
×···有未溶解的状态
表11
如表11所示,高支化聚合物17、18、34和35(应用例59~62)对CEL2021P显示出良好的溶解性。另一方面,直链状聚合物1~3(应用例63~65)均不溶解。此外,市售的氟系表面改性剂(应用例66和67)也同样地不溶解。
[实施例40]
<使用了高支化聚合物15~18、34、35的光固化环氧树脂的表面改性>
在作为环氧树脂的CEL2021P中分别以10.0g的方式添加表12所示的添加量的高支化聚合物15~18、34或35,在70℃下进行3小时加热搅拌,使各高支化聚合物溶解。然后,冷却至室温(大约25℃)后,添加作为阳离子系聚合引发剂的CP-770.6g,在室温(大约25℃)下进行1小时搅拌,从而使CP-77溶解。将其注入以1mm厚度的有机硅树脂为衬垫的玻璃基板上,采用UV照射装置以曝光量16mW/cm2曝光10分钟,制作出厚度1mm的光固化环氧树脂。作为比较例,同样地制作出未添加高支化聚合物的光固化环氧树脂。
对于所得的各光固化环氧树脂,通过雾度计测定全光透射率和浊度、以及测定水的接触角。将所得的结果示于表12。
表12
如表12所示,在CEL2021P中添加了高支化聚合物17、18、34或35的光固化环氧树脂(应用例70~75),与未添加高支化聚合物的光固化环氧树脂(应用例76)同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的CEL2021P单体的光固化树脂(应用例76)中,水的接触角为63.0度,与此相对,添加了高支化聚合物的光固化环氧树脂(应用例68~75)中,水的接触角为87.7~104.4度,均显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
[实施例41]
<使用了高支化聚合物8、23的热塑性聚氨酯弹性体的表面改性>
按照表13中记载的配合量配合作为热塑性聚氨酯弹性体的P22S、作为高支化聚合物的上述高支化聚合物8或23、和作为有机溶剂的THF,将其进行过滤器过滤,调制出热塑性树脂组合物。将该组合物在玻璃基板上流延,在20℃下进行24小时干燥,制作出应用例77~82的流延膜。作为比较例,制作出添加了市售的氟系表面改性剂F-552代替高支化聚合物的流延膜(应用例83~85)、和未添加高支化聚合物的流延膜(应用例86)。
通过棱镜耦合器测定膜厚、通过雾度计测定全光透射率和浊度、以及测定水的接触角。将结果示于表14。
表13
※表示树脂全体(P22S和高支化聚合物或表面改性剂)
相对于树脂组合物全部质量的添加量(质量%)。
表14
如表14所示,添加了高支化聚合物23的流延膜(应用例80~82),与未添加高支化聚合物的流延膜(应用例86)同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的P22S单体的流延膜(应用例86)中,水的接触角为88.9度,与此相对,添加了高支化聚合物的流延膜(应用例77~82)中,水的接触角为105.0~106.9度,均显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
此外,与添加了F-552代替高支化聚合物的流延膜(应用例83~85)相比,添加了各高支化聚合物的流延膜(应用例77~82)也显示出高的接触角。
[实施例42]
<使用了高支化聚合物8、23的利用涂布用聚氨酯溶液进行的聚氨酯树脂的表面改性>
按照表15中记载的配合量配合作为涂布用聚氨酯溶液的ニッポランN5257、作为高支化聚合物的上述高支化聚合物8或23、和作为稀释溶剂的MEK,调制出聚氨酯树脂组合物。利用刮刀将该组合物在玻璃基板上展开,在80℃下进行1小时干燥,制作出应用例87~92的聚氨酯涂膜。作为比较例,制作出添加了市售的氟系表面改性剂F-552代替高支化聚合物的聚氨酯涂膜(应用例93~95),和未添加高支化聚合物的聚氨酯涂膜(应用例96)。
通过椭圆光度法测定膜厚,通过雾度计测定全光透射率和浊度,以及测定水的接触角。将结果示于表16。
表15
※表示树脂全体(N5257和高支化聚合物或表面改性剂)
相对于树脂组合物全部质量的添加量(质量%)。
表16
如表16所示,添加了高支化聚合物8或23的涂膜(应用例87~92),与未添加高支化聚合物的涂膜(应用例96)同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的涂膜(应用例96)中,水的接触角为69.8度,与此相对,添加了高支化聚合物的涂膜(应用例87~92)中,水的接触角为78.4~104.8度,均显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
此外,与代替高支化聚合物而添加了F-552的涂膜(应用例93~95)相比,添加了各高支化聚合物的涂膜(应用例87~92)也显示出高的接触角。
[实施例43]
<添加了高支化聚合物8的光固化纳米压印试验>
将作为多官能丙烯酸系单体的A-DCP 9.9g、作为内部脱模剂的高支化聚合物8的0.1g、作为光聚合引发剂的Irg.907的0.1g、和作为溶剂的PGMEA 20.0g混合,制作出光聚合性树脂组合物的清漆。将该清漆旋涂(500rpm,30秒)在石英基板上,用60℃的电热板进行1分钟干燥,从而成膜(厚度约1μm)。在该石英基板上的膜上载置约2cm×2cm的硅制模件,设置于纳米压印装置的平台。然后,在25℃、1,000N下进行90秒加压后,接着在25℃、1,000N的加压下进行45mW、30秒的曝光,利用光纳米压印形成微细图案。
用光学显微镜观察所得的微细图案,结果如图68所示,图74所示的模件的图案完全被转印。
[比较例6]
<未添加高支化聚合物8的光固化纳米压印试验>
将作为多官能丙烯酸系单体的A-DCP 10.0g、作为光聚合引发剂的Irg.9070.1g、和作为溶剂的PGMEA 20.0g混合,制作出不含高支化聚合物8的光聚合性树脂组合物的清漆。使用该清漆,在与实施例43同样的条件下,在石英基板上利用光纳米压印形成微细图案。
用光学显微镜观察所得的微细图案,结果如图69所示,图74所示的模件的图案完全没有被转印。
[实施例44]
<添加了高支化聚合物8的热纳米压印试验>
将作为热塑性树脂的PMMA 0.99g、作为内部脱模剂的高支化聚合物80.01g、和作为溶剂的邻二氯苯9.0g混合,制作出热塑性树脂组合物的清漆。将该清漆旋涂(2,000rpm,60秒)在硅晶片上,用150℃的电热板进行1分钟干燥,从而成膜(厚度约1μm)。在该硅晶片上的膜上载置约2cm×2cm的硅制模件,设置于纳米压印装置的平台。然后,在25℃、1,500N下进行300秒加压后,接着在1,500N的加压下进行150℃、300秒的加热,利用热纳米压印形成微细图案。
用光学显微镜观察所得的微细图案,结果如图70和71所示,图74所示的模件的图案被完全转印。
[比较例7]
<未添加高支化聚合物8的热纳米压印试验>
将作为热塑性树脂的PMMA1.0g、和作为溶剂的邻二氯苯9.0g混合,制作出不含高支化聚合物8的热塑性树脂组合物的清漆。使用该清漆,在与实施例44同样的条件下,在硅晶片上利用热纳米压印形成微细图案。
用光学显微镜观察所得的微细图案,结果如图72和73所示,如各附图左侧所示,图案被切割,图74所示的模件的图案未被完全转印。
[实施例51]
<使用了高支化聚合物35的利用酸酐进行的热固化环氧树脂的表面改性>
添加作为环氧树脂的CEL2021P 4.95g、作为酸酐的MCHDC 4.95g和高支化聚合物350.1g,在70℃下进行3小时加热搅拌,使高支化聚合物溶解。然后,冷却至室温(大约25℃)后,在玻璃基板上滴加该溶液。将该玻璃基板用100℃的电热板加热1小时后,再升温至150℃加热3小时,从而制作出热固化环氧树脂。作为比较例,同样地制作出未添加高支化聚合物的热固化环氧树脂。
对于所得的热固化环氧树脂,通过雾度计测定全光透射率和浊度,以及测定水的接触角。将所得的结果示于表17。
表17
如表17所示,在CEL2021P和MCHDC中添加了高支化聚合物35的热固化环氧树脂(应用例97),与未添加高支化聚合物的热固化环氧树脂(应用例98)同样地,均显示出高的全光透射率和低的浊度。
此外,未添加高支化聚合物的仅含CEL2021P和MCHDC的热固化树脂(应用例98)中,水的接触角为76.5度,与此相对,添加了高支化聚合物的热固化环氧树脂(应用例97)中,水的接触角为104.0度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
[实施例52]
<对有机硅树脂的溶解性>
评价由实施例8、48和49获得的高支化聚合物8、36和37对有机硅树脂KR-400的溶解性。此外,也同样地评价市售的氟系表面改性剂F-552对KR-400的溶解性。向KR-4009.9g中分别添加各聚合物或F-5520.1g,在螺纹管内在室温(大约25℃)下进行3小时搅拌,评价溶解性。将所得的结果示于表18。
[评价基准]
○···完全溶解的状态
△···虽然溶解但白浊的状态
×···有未溶解的状态
表18
如表18所示,高支化聚合物36和37(应用例100和101)对KR-400显示出良好的溶解性。
[实施例53]
<使用了高支化聚合物36、37的有机硅树脂的表面改性>
在有机硅树脂KR-4009.9g中添加高支化聚合物36或370.1g,在室温(大约25℃)下进行3小时搅拌,使各高支化聚合物溶解。将该溶液旋涂(slope5秒,接着500rpm,30秒,接着slope5秒)在玻璃基板上。将该涂膜在室温(大约25℃)下静置1小时静置进行固化,制作出有机硅树脂膜。作为比较例,同样地制作出添加F-5520.1g代替高支化聚合物的有机硅树脂膜、和未添加高支化聚合物的有机硅树脂膜。
对于所得的有机硅树脂膜,分别测定水和十六烷的接触角。将所得的结果示于表19。
表19
如表19所示,未添加高支化聚合物的有机硅树脂(应用例106)中,水的接触角为78.5度,与此相对,添加了高支化聚合物的有机硅树脂(应用例103和104)中,水的接触角为94.5~96.5度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
此外,未添加高支化聚合物的有机硅树脂(应用例106)中,十六烷的接触角为40.3度,与此相对,添加了高支化聚合物的有机硅树脂(应用例103和104)中,十六烷的接触角为55.0~56.9度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物也赋予了防油性。此外,添加了F-552代替高支化聚合物的有机硅树脂(应用例105)中,十六烷的接触角比未添加高支化聚合物的有机硅树脂(应用例106)的十六烷的接触角显著降低,如果是F-552,则未赋予防油性。
[实施例59]
<环氧树脂EHPE3150的表面改性>
将环氧树脂EHPE31502.5g、高支化聚合物18、34或3525mg、SP-1700.1g和MIBK 2.5g混合,调制出环氧树脂组合物的清漆。将该清漆旋涂(slope5秒,接着500rpm,30秒,接着slope5秒)在硅晶片上。将该涂膜用90℃的电热板加热1分钟。利用UV照射装置将所得的薄膜以曝光量16mW/cm2曝光10分钟后,再用120℃的电热板加热5分钟,制作出环氧树脂光固化膜。作为比较例,同样地制作出未添加高支化聚合物的环氧树脂光固化膜。
对于所得的光固化膜,分别测定水的接触角。将所得的结果示于表20。
表20
如表20所示,未添加高支化聚合物的环氧树脂光固化膜(应用例110)中,水的接触角为62.1度,与此相对,添加了高支化聚合物的环氧树脂光固化膜(应用例107~109)中,水的接触角为97.0~100.6度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
[实施例60]
<环氧树脂157S70的表面改性>
按照表21中记载的配合量混合环氧树脂157S70、高支化聚合物18、34或35、ESACURE1720和环戊酮,将其进行过滤器过滤,调制出环氧树脂组合物的清漆。将该清漆旋涂(slope5秒,接着500rpm,30秒,接着slope5秒)在硅晶片上。将该涂膜用90℃的电热板加热1分钟。利用UV照射装置将所得的薄膜以曝光量16mW/cm2曝光10分钟后,再用120℃的电热板加热5分钟,制作出环氧树脂光固化膜。作为比较例,同样地制作出未添加高支化聚合物的环氧树脂光固化膜。
对于所得的光固化膜,分别测定水的接触角。将所得的结果示于表21。
表21
如表21所示,未添加高支化聚合物的环氧树脂光固化膜(应用例120~122)中,水的接触角为56.4~73.8度,与此相对,添加了高支化聚合物的环氧树脂光固化膜(应用例111~119)中,水的接触角为96.2~107.0度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
[实施例61]
<聚酰亚胺树脂的表面改性>
在高支化聚合物39~435mg中分别混合PAA清漆4.1g、NMP 6.5g和BA 1.6g,调制出聚酰胺酸组合物的清漆。将该清漆旋涂(slope5秒,接着500rpm,30秒,接着slope5秒)在玻璃基板上。将该涂膜用210℃的电热板加热30分钟,制作出聚酰亚胺树脂膜。作为比较例,同样地制作未添加高支化聚合物的聚酰亚胺树脂膜。
对于所得的聚酰亚胺树脂膜,分别测定水的接触角。将所得的结果示于表22。
表22
如表22所示,未添加高支化聚合物的聚酰亚胺树脂膜(应用例128)中,水的接触角为53.2度,与此相对,添加了高支化聚合物的聚酰亚胺树脂膜(应用例123~127)中,水的接触角为92.5~102.2度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
[实施例62]
<聚酰亚胺树脂的表面改性>
在高支化聚合物39~435mg中分别混合SPI 0.50g、NMP 7.6g和BA1.9,调制出可溶性聚酰亚胺树脂组合物的清漆。将该清漆旋涂(slope5秒,接着500rpm,30秒,接着slope5秒)在玻璃基板上。将该涂膜用210℃的电热板加热30分钟,制作出聚酰亚胺树脂膜。作为比较例,同样地制作未添加高支化聚合物的聚酰亚胺树脂膜。
对于所得的聚酰亚胺树脂膜,分别测定水的接触角。将所得的结果示于表23。
表23
如表23所示,未添加高支化聚合物的聚酰亚胺树脂膜(应用例134)中,水的接触角为78.2度,与此相对,添加了高支化聚合物的聚酰亚胺树脂膜(应用例129~133)中,水的接触角为97.4~100.7度,显示出高的接触角。由该结果明确了,通过添加高支化聚合物而赋予了防水性。
- 还没有人留言评论。精彩留言会获得点赞!