作为用于丙交酯聚合物的聚合催化剂的包含至少一个Cp环的第IVB族过渡金属催化剂的制作方法
聚(乳酸)(PLA)由于它们的生物可降解性和生物相容性已在过去几十年期间被强烈地研究。PLA具有多样性的物理性质并且已经在医学应用和组织工程例如用于受控制的药物释放的介质中被使用。丙交酯(LA)的通过单位点引发剂的开环聚合(ROP)对于具有受控制的分子量和窄的分子量分布的PLA是最有效的路线。在一个丙交酯分子中的两个立体中心产生三种有区别的构型异构体(S,S)-LA、(L-LA);(R,R)-LA、(D-LA)以及(R,S)-LA、(内消旋)-LA。(S,S)-LA和(R,R)-LA的1:1混合物被称为外消旋-LA。
对于引发丙交酯的开环聚合有用的金属络合物是已知的。
Wenshan Ren等人,Inorganic Chemistry Communications,30,(2013),26-28报道了苄基钍金属茂[η5-1,3-(Me3C)2C5H3]2Th(CH2Ph)2(1)和[η5-1,2,4-(Me3C)3C5H2]2Th(CH2Ph)2(2)在温和的条件下可以引发外消旋-丙交酯(外消旋-LA)的开环聚合(ROP)。500当量的丙交酯的完全转化在5小时内在40℃下在二氯甲烷中以[外消旋-LA]=1.0mol L-1发生,并且分子量分布在全部的单体比引发剂的范围内是非常窄的(约1.15),这指示单位点的催化剂体系。
Yalan Ning等人,Organometallics 2008,27,5632-5640报道了用于L-丙交酯(L-LA)和ε-己内酯(ε-CL)的开环聚合和链转移聚合的四种中性二茂锆双(酯烯醇化物)和非二茂锆双(烷氧基)络合物。
AJ Chmura等人,Chem.Commun.2008,1293发现,锆胺三(酚盐)醇盐和铪胺三(酚盐)醇盐是极其活性的,产生高度杂同立构的聚丙交酯(heterotactic polylactide)。
本发明基于不同类别的化合物的发现,所述化合物具有作为用于丙交酯单体的聚合的引发剂的用途。
本发明提供具有下式的化合物,
La M(OR1)b R2c Xd
其中
M是选自Ti、Zr以及Hf的金属;
L是选自以下的配体:全甲基并环戊二烯(Pn*=C8Me6)、(氢)全甲基并环戊二烯(Pn*(H)=C8Me6H)、(氢)并环戊二烯(Pn(H)=C8H8)、环戊二烯(Cp=C5H5)、茚(C7H7)以及亚乙基桥接的茚或硅烷桥接的茚;
R1是1-6C烷基、被取代的或未被取代的苯基、或被取代的或未被取代的苯基亚烷基;
R2是Me或Et
X是卤素
a=1至3,b=1至3,c=0或1并且d=0、1、2或3;
及其二聚体。
上文提到的并环戊二烯具有以下示出的结构:
硅烷桥可以任选地被一系列烷基取代。SBI配体指的是二甲基硅烷桥接的茚基配体。
本发明还提供了本发明的化合物作为在丙交酯单体的聚合中的引发剂的用途。
本发明还另外提供了用于产生聚丙交酯的工艺,所述工艺包括使丙交酯单体与本发明的化合物接触。
如上文所陈述的,本发明的化合物具有以下的通式,
La M(OR1)b R2c Xd
其中L、M、R1、R2、X、a、b、c以及d为如上文所定义的,及其二聚体。
M是选自钛、锆以及铪的第IV族过渡金属。根据一个优选的实施方案,M是钛。根据另一个优选的实施方案,M是锆。根据不同的实施方案,M是铪。
L是选自以下的配体:全甲基并环戊二烯(Pn*)、(氢)全甲基并环戊二烯(Pn*(H))、(氢)并环戊二烯(Pn(H))、环戊二烯(Cp)、茚以及亚乙基桥接的茚(EBI)和二甲基硅烷桥接的茚(SBI)。根据一个优选的实施方案,配体基团L是全甲基并环戊二烯。根据不同的实施方案,当M是Zr时,配体基团L是茚或EBI。
本发明的化合物是基于配体基团L的金属醇盐络合物、金属酚盐络合物或苯基亚烷基氧化金属络合物(phenylalkyleneoxide metal complex)。被附接至金属M的OR1基团的R1选自1-6C烷基、被取代的或未被取代的苯基以及被取代的或未被取代的苯基亚烷基。优选地,对于R1,1-6C烷基是叔丁基。优选地,当R1是苯氧基(phenyloxide group)时,其是式–C6H3(R3)2的二烷基苯氧基,其中R3是1-4C烷基,尤其是Me、iPr或tBu。根据优选的实施方案,R1选自2,6-二甲基苯基、2,6-二异丙基苯基以及2,6-二叔丁基苯基。
根据不同的实施方案,R1可以是如上文提及的被取代的或未被取代的苯基亚烷基。实例包括–CH2C6H5和–CH(Me)C6H5。
本发明的金属络合物可以包含被附接至金属M的一个R2基团。如果存在,那么R2选自Me和Et。如果R2在络合物中存在,那么其优选地是甲基。
本发明的金属络合物可以包含被附接至M的卤素基团X。优选地,如果存在,那么X是Cl。典型地,如果X在络合物中存在,那么d的值是1或2。
根据优选的实施方案,本发明的化合物是为以下的(半)金属茂络合物:
η5-Pn*(H)Ti(OtBu)3;或
η5-Pn*(H)Zr(OtBu)3;或
η5-Pn*(H)Zr(O-CH2C6H5)3;或
η5-Pn*(H)Zr(O-S-CH(CH3)C6H5)3;或
外消旋-η5-Pn*(H)Zr(O-外消旋-CH(CH3)C6H5)3;或
η5-Pn*(H)Zr(O-2,6-Me-C6H3)3;或
η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3;或
η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3);或
η5-Pn*(H)Hf(O-2,6-Me-C6H3)3;或
η5-Pn*(H)HfCl(O-2,6-iPr-C6H3)2;或
η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)。
上文化合物可以例如通过使η5-Pn*(H)SnMe3与对应的金属氯化物例如TiCl4、ZrCl4或HfCl4在苯中在80℃下反应持续2-72小时并且然后与合适的醇钾盐、酚钾盐或苯基亚烷基氧化钾在室温下在苯或甲苯中反应来制备。然而,用于制备本领域已知的化合物的任何合适的工艺都可以被用于其制备。
根据不同的优选的实施方案,本发明的(半)金属茂络合物是
[(Pn*)Ti(O-2,6-Me-C6H3)Cl];或
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl];或
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2];或
[η8-(Pn*)Ti(OtBu)Cl];或
[η8-(Pn*)Ti(OtBu)2;或
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2];或
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]。
上文化合物可以例如通过使一当量的[η8-(Pn*)TiCl(μ-Cl)]2与二当量的KOR(其中R=2,6-MeC6H3或2,4-tBuC6H3)在室温下在甲苯中反应持续24-48小时来产生。然而,用于制备本领域已知的化合物的任何合适的工艺都可以被用于其制备。
根据又另外的优选的实施方案,本发明的(半)金属茂络合物是
[(EBI)Zr(O-2,6-Me-C6H3)Cl];或
[Ind2Zr(O-2,6-Me-C6H3)Me];或
[Ind2Zr(O-2,6-Me-C6H3)Cl];或
[Ind2Zr(O-2,6-Me-C6H3)2];或
[Cp2Zr(O-2,6-Me-C6H3)2];或
[Cp2Zr(O-2,6-Me-C6H3)Cl];或
[Cp2Zr(O-2,6-Me-C6H3)Me]。
上文EBI Zr化合物可以通过使化学计量的量的[K(O-2,6-Me-C6H3)]和外消旋-[(EBI)ZrCl2]在室温下在甲苯中在搅拌下反应持续18小时来产生。用于制备[(Ind)2Zr(OtBu)Me]的修改的程序包括使化学计量的量的[(Ind)2ZrMe2]和叔丁醇在室温下在甲苯中在搅拌下反应持续18小时,随后是在真空中浓缩。[(Ind)2Zr(O-2,6-Me-C6H3)Me]可以通过使化学计量的量的[(Ind)2ZrMe]和2,6-二甲基苯酚在室温下在甲苯中在搅拌下反应持续18小时、随后在真空中浓缩来制备。然而,用于制备本领域已知的化合物的任何合适的工艺都可以被用于其制备。
本发明的化合物作为在丙交酯单体的聚合中的引发剂是有用的。因此,本发明还涉及如前文描述的化合物作为在丙交酯单体的聚合中的引发剂的用途。
根据另外的方面,本发明提供了用于产生聚丙交酯的工艺,所述工艺包括使丙交酯单体与如前文描述的化合物接触。
在该工艺的优选的实施方案中,丙交酯单体是L-丙交酯并且产生的聚丙交酯是等规立构聚丙交酯。在该工艺的另一个优选的实施方案中,丙交酯单体是外消旋-丙交酯并且产生的聚丙交酯是无规立构聚丙交酯。
实验细节I-涉及全甲基并环戊二烯
一般程序
所有有机金属合成都在氮气的惰性气氛下利用标准Schlenk技术在双重真空-进口气体歧管(dual vacuum-inlet gas manifold)或Braun手套箱上进行。在必要时,溶剂被SPS干燥体系(己烷、戊烷、甲苯)干燥。氘代的NMR溶剂在使用之前,经NaK(苯-d6、甲苯-d8)或CaH2(氯仿-d1)干燥、真空转移并且冷冻-抽吸-解冻-脱气三次。元素分析由Stephen Boyer先生在伦敦都市大学(London Metropolitan University)的元素分析服务部进行。NMR光谱使用杨氏分接头NMR管(Young’s tap NMR tubes)被记录在Varian Mercury VX-Works 300MHz光谱仪上。1H光谱和13C{1H}NMR光谱通过残余的质子溶剂峰(protio-solvent peak)作参考。叔丁醇钾购自Sigma-Aldrich并且按原样使用。L-丙交酯和外消旋-丙交酯购自Alfa Aesar并且在使用之前被重结晶并且升华(10-2毫巴,50℃)。[K(O-2,6-Me-C6H3)]和[K(O-2,4-tBu-C6H3)]通过在室温下搅拌在THF中的双(三甲基甲硅烷基)酰胺钾与合适的醇来制备。
X射线结晶学
使用全氟聚醚油将晶体固定在玻璃纤维上,转移至在衍射计上的测角计头并且在冷的氮气的流中使用Oxford Cryosystems CRYOSTREAM单元迅速冷却至150K。数据收集使用Enraf-Nonium FR590KappaCCD衍射计、利用石墨-单色化的Mo KαX射线辐射来进行。强度数据使用DENZO-SMN包来处理。结构使用直接法程序SIR92来求解,并且使用CRYSTALS程序组对所有F2数据应用全矩阵最小二乘法求精来求精。
聚合程序
所有聚合在包含0.4mL的具有[LA]0=0.104M的初始丙交酯浓度的丙交酯的苯-d6溶液和0.1mL的催化剂的苯-d6溶液(具有用于确保[LA]0/[初始]0=50的浓度)的杨氏分接头NMR管中进行。丙交酯转化率随后通过比较在1H NMR光谱中的PLA和丙交酯单体的甲烷信号的积分值来计算。进行给定的聚合的温度在80℃至100℃的温度范围内变化并且在合适的部分中被注明。
本发明的主题的另外的优点和特征可以结合附图取自以下详述,在附图中:
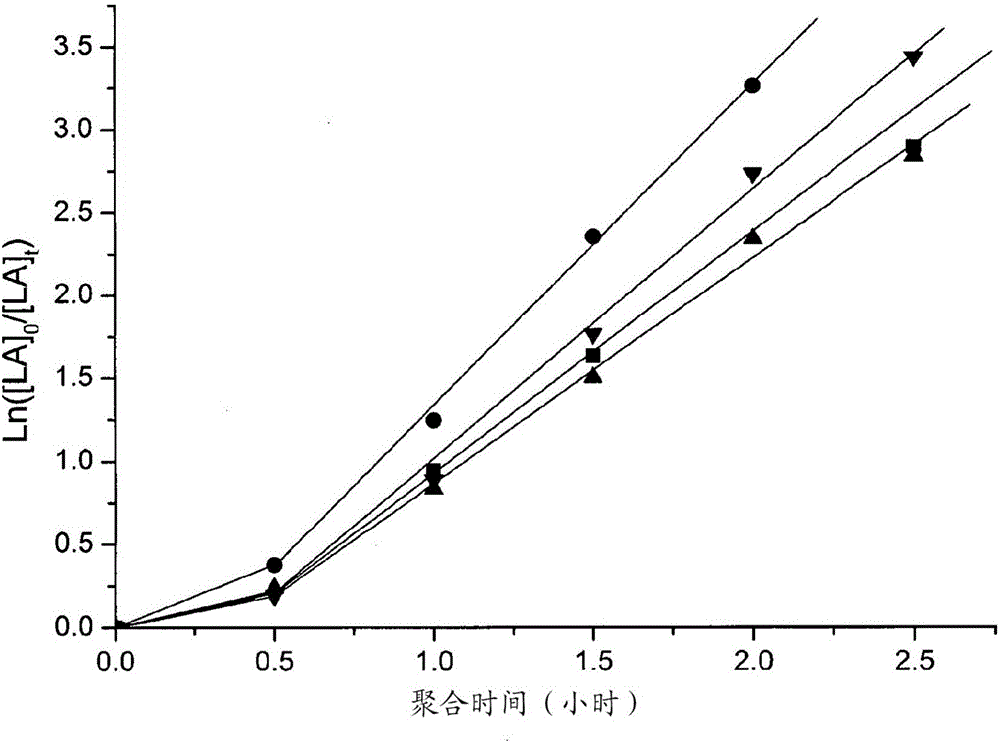
图1:使用以下络合物的L-丙交酯聚合:η5-Pn*(H)Ti(O-2,6-Me2C6H3)32(黑色正方形,k观察=0.113±0.014h-1)、η5-Pn*(H)Zr(O-2,6-Me2C6H3)3 7(红色圆形,k观察=0.479±0.032h-1)、η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8(粉色三角形,k观察=0.391±0.022h-1)、η5-Pn*(H)ZrCl2(O-2,6-iBu2C6H3)9(深蓝色三角形,k观察=0.043±0.003h-1)、η5-Pn*(H)Hf(O-2,6-Me2C6H3)3 10(蓝色三角形,k观察=0.364±0.027h-1)、η5-Pn*(H)HfCl(O-2,6-iPr2C6H3)2 11(绿色菱形,k观察=0.463±0.029h-1)以及η5-Pn*(H)HfCl2(O-2,6-iBuC6H3)12(紫色三角形,k观察=0.086±0.020h-1)。聚合条件:氯仿-d1,在100℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
图2:丙交酯聚合:L-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(黑色正方形,k观察=1.166±0.068h-1);L-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(红色圆形,k观察=1.954±0.063h-1);外消旋-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(粉色三角形,k观察=1.667±0.053h-1);外消旋-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(蓝色三角形,k观察=1.342±0.055h-1)。聚合条件:氯仿-d1,在80℃下,其中[LA]0/[M]0=50、[LA]0=0.5M。
图3:丙交酯聚合:L-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(黑色正方形,k观察=0.484±0.037h-1);L-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(红色圆形,k观察=0.850±0.063h-1);外消旋-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(粉色三角形,k观察=0.767±0.037h-1);外消旋-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(蓝色三角形,k观察=0.491±0.031h-1)。聚合条件:氯仿-d1,在60℃下,其中[LA]0/[M]0=50、[LA]0=0.5M。
图4:使用S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3 5的L-丙交酯聚合的艾林标绘图(Eyring plot)。斜率=-5610±488,且R2=0.993。聚合条件:氯仿-d1,其中[LA]0/[Zr]0=50并且[LA]0=0.5M。
图5:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合:[LA]0/[Zr]0=25(黑色正方形,k观察=0.521±0.021h-1);[LA]0/[Zr]0=50(红色圆形,k观察=0.391±0.022h-1);[LA]0/[Zr]0=100(蓝色三角形,k观察=0.377±0.015h-1);[LA]0/[Zr]0=200(粉色三角形,k观察=0.235±0.004h-1)。聚合条件:氯仿-d1,在100℃下,其中[LA]0=0.5M。
图6:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的Ln(k观察)相对于Ln([Zr]0)的标绘图。斜率=0.350±82,R2=0.901。聚合条件:氯仿-d1,在100℃下,并且[LA]0=0.5M。
图7:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合:T=100℃(黑色正方形,k观察=0.391±0.022h-1);T=90℃(红色圆形,k观察=0.151±0.008h-1);T=80℃(蓝色三角形,k观察=0.092±0.006h-1)。聚合条件:氯仿-d1,其中[LA]0/[M]0=50、[LA]0=0.5M。
图8:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合的艾林标绘图。斜率=-9133±1881,且R2=0.959。聚合条件:氯仿-d1,其中[LA]0/[Zr]0=50并且[LA]0=0.5M。
图9:L-丙交酯转化率相对于时间的半对数标绘图,[LA]0/[初始]0=50,[LA]0=0.104M,T=80℃,苯-d6(0.5mL),使用[η8-(Pn*)Ti(O-2,6-Me2C6H3)Cl](绿色点划线)、[η8-(Pn*)Ti(O-2,4-tBu2C6H3)Cl](黑色短划线)、[η8-(Pn*)Ti(O-2,6-Me2C6H3)2](红色线)以及[η5-(Pn*H)Ti(O-2,6-Me2C6H3)Cl2](蓝色点虚线)。省略诱导期。
图10:丙交酯单体转化率相对于时间的半对数标绘图。省略诱导期。[LA]0=0.104M,[LA]0/[初始]0=50,T=90℃,苯-d6。使用[η8-(Pn*)Ti(O-2,6-Me2C6H3)2]、L-丙交酯(红色线)和外消旋-丙交酯(黑色短划线)的聚合。
图11:L-丙交酯转化率相对于时间的半对数标绘图,[LA]0/[初始]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL),使用[(EBI)Zr(OC6H3Me2-2,6)Cl]1(点划线)、[(Ind)2Zr(OtBu)Me]2(实线)、[(Ind)2Zr(OC6H3Me2-2,6)Me]3(短划线)的聚合。
图12:增长速率相对于温度的倒数的半对数标绘图,[LA]0/[2]0=50,[LA]0=0.50M,氯仿-d1,并且
图13:丙交酯转化率相对于时间的半对数标绘图,[LA]0/[2]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL);L-丙交酯(实线),外消旋-丙交酯(短划线)。
实施例1
[(Pn*)Ti(O-2,6-Me-C6Me2)Cl]的合成将[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.26mmol)和[K(O-2,6-MeC6H3)](78mg,0.50mmol)在甲苯(30mL)中合并并且留下在室温下搅拌持续25小时。将产生的溶液过滤,然后在真空中浓缩。在-35℃下储存甲苯浓缩的溶液持续24小时之后获得了X射线质量单晶(X-ray quality single crystal)。收率=52%。1H NMR(苯-d6,25℃,300MHz):δ7.04(d,2H,3JHH=7.3Hz,Ar-H),6.82(t,1H,3JHH=7.4Hz,Ar-H),2.11,2.06,1.68(s,每6H,Pn-CH3),1.61(s,6H,Ar-CH3)。13C{1H}NMR(甲苯-d8,25℃,75.1MHz):δ130.69,126.16,14.36,123.41,120.41(季碳),18.09(Ar-CH3)13.20,12.47,11.11(Pn-CH3)。四级桥头碳原子没有观察到并且某些季碳信号被溶剂共振遮蔽。对于C22H27ClTiO(%)的分析计算值:C,67.62,H,6.98;实测值:C,67.70;H,7.03。
实施例2
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl]的合成将[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(O-2,4-tBu-C6H3)](120mg,0.50mmol)在甲苯(30mL)中在室温下搅拌持续48小时。将产生的溶液过滤并且在真空中除去溶剂。随后溶解在最少的热的苯中并且在室温下储存24小时导致X射线质量单晶的形成。收率=55%。1H NMR(苯-d6,25℃,300MHz):δ7.54(d,1H,4JHH=2.4Hz,间位-Ar-H),7.18(dd,1H,3JHH=8.1Hz,4JHH=2.4Hz,间位-Ar-H),6.41(d,1H,3JHH=8.3Hz,邻位-Ar-H),2.07,1.80,1.64(s,每6H,Pn-CH3),1.59(s,9H,C(CH3)),1.35(s,9H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ161.21(本位-Ar),142.66,140.76,139.1,0 135.5,7 130.28,124.62(季碳),123.82,123.77(间位-Ar)122.82(季碳)121.22(邻位-Ar),35.55,34.55(Ar-CM3),31.95,30.88(Ar-CH3),13.04,12.48,10.84(Pn-CH3)。对于C28H39TiClO(%)的分析计算值:C,70.8;H,8.29;实测值:C,70.65;H,8.23。
实施例3
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]的合成将[η8-Pn*TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(O-2,6-Me-C6H3)](160mg,0.50mmol)在甲苯(30mL)中在25℃下搅拌持续24小时。将产生的溶液过滤并且随后在真空中浓缩。在-35℃下储存甲苯浓缩的溶液持续24小时之后获得了X射线质量单晶。收率=62%。1H NMR(苯-d6,25℃,300MHz):δ6.96(d,4H,3JHH=7.5Hz,间位-Ar-H),6.69(t,2H,3JHH=7.3Hz,对位-Ar-H),2.13(s,12H,Pn-CH3),1.81(s,12H,Ar-CH3),1.77(s,6H,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ162.52(本位-Ar),139.73 130.97 125.22 120.47 118.89(季碳),17.73(Ar-CH3),11.90 10.89(Pn-CH3)。四级桥头碳原子没有观察到。对于C30H36TiO2(%)的分析计算值:C,75.62;H,7.63;实测值:C,75.48;H,7.77。
实施例4
[η8-(Pn*)Ti(OtBu)Cl]的合成将[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(OtBu)](53mg,0.50mmol)在甲苯(30mL)中合并并且留下在室温下搅拌持续2小时。将产生的溶液过滤并且在减压下除去溶剂。添加最少的热的己烷并且在冷却至室温并且在-35℃下储存24小时之后获得了X射线质量单晶。收率=54%。1H NMR(苯-d6,25℃,300MHz):δ2.09,1.90,1.62(s,每6H,Pn-CH3),1.31(s,9H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ138.41,136.38(Pn-桥头),128.86,122.16,121.91(Pn),81.16(C(CH3)),32.48(C(CH3)),13.09,12.85,10.86(Pn-CH3)。
实施例5
[η8-(Pn*)Ti(OtBu)2]的合成将[η8-(Pn*)TiCl(μ-Cl)]2(20mg,0.035mmol)和[K(OtBu)](16mg,0.14mmol)在苯-d6(1mL)中合并。将获得的深红色溶液过滤并且允许经历缓慢蒸发,这导致X射线质量单晶的形成。1H NMR(苯-d6,25℃,300MHz):δ2.08(s,12H,Pn-CH3),1.76(s,6H,Pn-CH3),1.31(s,18H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):134.38,128.71,116.58(Pn)76.44(C(CH3))33.60(C(CH3))13.07,10.81(Pn-CH3)。四级桥头碳原子没有观察到。
实施例6
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl)2]的合成将[η8-Pn*TiCl(μ-Cl)]2(350mg,0.57mmol)和[H(O-2,6-Me-C6H3)](139mg,1.14mmol)在甲苯(50mL)中在85℃下搅拌持续24小时,然后过滤并且在真空下浓缩。在-35℃下储存浓缩的溶液持续24小时之后形成X射线质量单晶。收率=68%。1HNMR(苯-d6,25℃,300MHz):δ6.82-6.72(m,3H,Ar-H),3.19(q,1H,3JHH=7.3Hz,Pn-H),2.24(s,6H,Ar-CH3),2.23,2.12,2.08,1.79,1.44(s,每3H,Pn-CH3),0.84(d,3H,3JHH=7.5Hz,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz)δ162.73(本位-Ar),150.62,147.79,141.18,136.86,129.80,129.07(季碳),128.13(间位-Ar)123.70(季碳)123.06(对位-Ar),44.19(sp3Pn),17.22(Ar-CH3),15.53,14.08,13.53,13.23,11.75,11.59(Pn-CH3)。四级桥头碳原子没有观察到。对于C22H28Cl2TiO(%)的分析计算值:C,61.84;H,6.62;实测值:C,61.71;H,6.70。
实施例7
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]的合成将[η5-(Pn*H)Ti(OC6H3Me2-2,6)Cl2](50mg,0.12mmol)和[K(O-2,6-Me2C6H3)](37.5mg,0.24mmol)在甲苯(10mL)中的溶液在100℃下搅拌持续30分钟。将产生的亮橙色溶液过滤并且在减压下除去溶剂。随后溶解在最少的热的己烷中、随后在-35℃下储存持续24小时导致X射线质量单晶的形成。收率=65%。1H NMR(苯-d6,25℃,300MHz):δ6.90(d,6H,3JHH=7.2Hz,间位-Ar-H),6.73(t,3H,3JHH=7.3Hz,对位-Ar-H)3.59(q,1H,3JHH=7.7Hz,Pn-H)2.28(s,18H,Ar-CH3)2.21,2.14,1.96,1.58,1.53(s,每3H,Pn-CH3)1.08(d,3H,3JHH=7.32Hz,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz)δ164.05(本位-Ar),147.30,144.11,138.88,130.71(Pn),129.06(Ar),128.93(Pn),127.41(Ar),122.49(Pn),120.79(Ar),116.96(Pn),44.46(sp3Pn),18.15(sp3Pn-CH3),15.33,13.37,12.49,11.99,11.75,11.58(Pn-CH3)。
结晶学细节
[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]单晶在-35℃下从甲苯溶液生长,C22H27ClOTi,Mr=390.81,三斜晶系,P-1,α=77.0104(7)°,β=89.3195(7)°,γ=85.2957(8)°,Z=4,T=150K,棱晶,红棕色,9129独立反射,R(初始)=0.038,R1=0.046wR2=0.133[I>2σ(I)]。
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl]单晶在-35℃下从苯溶液生长,C28H39ClOTi,Mr=474.97,单斜晶系,P21/n,α=102.4652(5)°,β=90°,γ=90°,Z=4,T=150K,块状,紫色,5884独立反射,R(初始)=0.029,R1=0.038wR2=0.091[I>2σ(I)]。
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]单晶在-35℃下从甲苯溶液生长,C30H36O2Ti,Mr=476.51,正交晶系,Pbca,α=90°,β=90°,γ=90(8)°,Z=8,T=150K,块状,深红色,5807独立反射,R(初始)=0.036,R1=0.047wR2=0.092[I>2σ(I)]。
[η8-(Pn*)Ti(OtBu)Cl]单晶在-35℃下从己烷溶液生长,C18H27ClOTi,Mr=342.76,三斜晶系,P-1,α=79.8812(7)°,β=78.4070(7)°,γ=73.9368(7)°,Z=2,T-150K,块状,深红色,4073独立反射,R(初始)=0.016,R1=0.034wR2=0.085[I>2σ(I)]。
[η8-(Pn*)Ti(OtBu)2]单晶在-35℃下从苯溶液生长,C22H36O2Ti,Mr=380.43,单斜晶系,P21/n,α=90°,β=93.2853(6)°,γ=90°,Z=4,T=150K,棱晶,红棕色,5062独立反射,R(初始)=0.022,R1=0.075wR2=0.188[I>2σ(I)]。
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2]单晶在-35℃下从己烷溶液生长,C22H28Cl2TiO,M=427.27,单斜晶系,P21/c,α=γ=90.00°,β=106.6445(11),T=150(2)K,Z=4,4844独立反射,R(初始)=0.019,R1=0.054wR2=0.119[I>2σ(I)]。
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]单晶在-35℃下从己烷溶液生长,C38H46TiO3,M=598.68,单斜晶系,P21/n,α=γ=90.00°,β=91.352(2),T=150(2)K,Z=4,6634独立反射,R(初始)=0.060,R1=0.056wR2=0.141[I>2σ(I)]。
实验细节II-涉及(氢)全甲基并环戊二烯
一般程序
所有有机金属合成都在氮气的惰性气氛下利用标准Schlenk技术在双重真空-进口气体歧管或Braun手套箱上进行。在必要时,溶剂通过经以下合适的干燥剂回流来干燥:NaK(Et2O)、钠(THF)以及SPS干燥体系(己烷、戊烷、甲苯)。将溶剂在氮气的流动的流下从干燥剂蒸馏并且使用虹吸技术通过钢插管来转移并且在氮气的气氛下储存在火焰干燥的玻璃安瓿中。氘代的NMR溶剂在使用之前,经NaK(苯-d6、甲苯-d8)或CaH2(吡啶-d5)干燥、真空转移并且冷冻-抽吸-解冻-脱气三次。元素分析由Stephen Boyer先生在伦敦都市大学的元素分析服务部进行。NMR光谱使用杨氏分接头NMR管被记录在Varian Mercury VX-Works 300MHz光谱仪上。1H光谱和13C{1H}NMR光谱以残余的质子溶剂峰作参考。
X射线结晶学
使用全氟聚醚油将晶体固定在玻璃纤维上,转移至在衍射计上的测角计头并且在冷的氮气的流中使用Oxford Cryosystems CRYOSTREAM单元迅速冷却至150K。数据收集使用Enraf-Nonius FR590KappaCCD衍射计利用石墨-单色化的Mo KαX射线辐射来进行。强度数据使用DENZO-SMN包来处理。结构使用直接法程序SIR92来求解,并且使用CRYSTALS程序组对所有F2数据使用全矩阵最小二乘法求精来求精。
聚合程序
将丙交酯单体(40mg)和络合物遵循期望的单体:引发剂的比率引入NMR管中。然后,将0.57mL的氯仿-d1添加至化合物,产生[LA]0=0.5M的初始单体浓度。溶液通过1H NMR光谱学来监测。转化率通过将聚合物相对于单体的甲烷面积积分来确定。
实施例8
Pn*(H)SnMe3的合成。在-78℃下向Pn*(H)Li(2.09g,10.7mmol)在戊烷(20mL)中的浆料添加SnMe3Cl(2.14g,10.7mmol)在戊烷(10mL)中的溶液。将反应混合物加温至室温并且搅拌持续3小时以提供橙色溶液和LiCl的无色沉淀。将此过滤并且将挥发物在真空中除去以提供作为橙色油的Pn*(H)SnMe3(通过1H NMR光谱学判断的非对映异构体的50:50混合物)。收率:3.56g(97%)。1H NMR(苯-d6,23℃):δ2.98(q,1H,3JHH=7.2Hz,Pn*(H)),2.09 2.05 2.00(s,每3H,CH3-Pn*(H)),1.95(重叠的单峰,每3H,CH3-Pn*(H))1.93 1.83(s,每3H,CH3-Pn*(H)),1.70(重叠的单峰,每3H,CH3-Pn*(H)),1.59(s,3H,CH3-Pn*(H)),1.18(d,3H 3JHH=7.2Hz,1-CH3-Pn*(H)),0.94(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H)),-0.01(s,9H,2J1H-119Sn=25.2Hz,2J1H-117Sn=24.2Hz,5-SnMe3-Pn*(H))-0.03(s,9H,2J1H-119Sn=25.3Hz,2J1H-117Sn=24.3Hz,5-SnMe3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):150.2 147.2 144.6 144.4 129.8 129.4 121.3 119.6(6x重叠的共振,(q-Pn*(H)),44.3 41.8(1-Pn*(H)),13.5 13.4 12.8 12.4 121.3 12.2 12.1 12.0(2x重叠的共振CH3-Pn*(H)),18.2 17.6(1-CH3-Pn*(H)),-8.8(5-SnMe3-Pn*(H),2J1H-119Sn=153Hz,2J1H-117Sn=148Hz),-9.2(5-SnMe3-Pn*(H),2J1H-119Sn=157Hz,2J1H-117Sn=150Hz)。
实施例9
Pn*(H)TiCl3的合成。向TiCl4(thf)2(0.408g,1.44mmol)在苯(2mL)中的浆料添加Pn*(H)SnMe3(0.505g,1.44mmol)在苯(2mL)中的溶液以提供深紫色溶液。将反应混合物加热至80℃持续4小时。将挥发物在真空中除去以提供作为紫色粉末的Pn*(H)TiCl3。收率:0.363g(74%)。适于X射线衍射研究的单晶在-35℃下从饱和的Et2O溶液生长。1H NMR(苯-d6,23℃):δ0.85(d,3H,3JHH=7.5Hz,1-CH3-Pn*(H)),1.57 1.89 2.02 2.03 2.14(s,每3H,CH3-Pn*(H)),3.80(q,1H,3JHH=8.5Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ153.6 152.7 147.4 140.6 133.3 131.3 127.4(q-Pn*(H)),46.8(1-Pn*(H)),15.4(1-CH3-Pn*(H)),14.5,14.4,14.2,12.2,11.6(CH3-Pn*(H))。
实施例10
[Pn*(H)ZrCl3]2的合成。向ZrCl4(0.995g,4.27mmol)在苯(5mL)中的浆料添加Pn*(H)SnMe3(1.50g,4.27mmol)在苯(5mL)中的溶液。将反应混合物加热至80℃持续72小时以提供深绿色溶液。将挥发物在真空中除去以产生绿色固体。向此添加戊烷(15mL)并且将反应混合物声处理持续15分钟以提供细的橄榄绿色的粉末和浅黄色溶液。将反应混合物过滤并且将滤液在减压下干燥以提供作为橄榄绿色粉末的[Pn*(H)ZrCl3]2。收率:1.42g(87%)。单晶在23℃下从饱和的苯溶液生长。1H NMR(苯-d6,23℃):δ0.92(d,3H,3JHH=7.5Hz,1-CH3-Pn*(H)),1.81 2.01 2.06 2.17 2.19(s,每3H,CH3-Pn*(H)),3.50(q,3JHH=7.5Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ151.0 147.0 142.1 133.5 129.5 127.4 120.7(q-Pn*(H),46.0(1-Pn*(H)),15.6(1-CH3-Pn*(H)),14.2 13.6 13.5 12.3 12.2(CH3-Pn*(H))。
实施例11
[Pn*(H)HfCl3]2的合成。向HfCl4(0.164g,0.467mmol)在苯(2mL)中的浆料添加Pn*(H)SnMe3(0.149g,0.467mmol)在苯(2mL)中的溶液。将反应混合物加热至80℃持续2小时以提供橙色溶液。将挥发物在真空中除去以产生作为浅黄色固体的[Pn*(H)HfCl3]2。单晶在室温下从饱和的苯溶液生长。1H NMR(苯-d6,23℃):δ0.93(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H)),1.621.84 2.01 2.01 2.10(s,每3H,CH3-Pn*(H)),3.42(q,3JHH=7.4Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ150.0 142.3 137.2 123.9 117.4(q-Pn*(H)),45.5(1-Pn*(H)),15.9(1-CH3-Pn*(H)),12.2,12.1,12.0,11.9,11.6(CH3-Pn*(H))。
实施例12
η5-Pn*(H)Ti(OtBu)3的合成。将Pn*(H)TiCl3(0.020g,0.059mmol)和KOtBu(0.020g,0.18mmol)在苯-d6(0.5mL)中合并并且声处理持续5分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为浅黄色粉末的Pn*(H)Ti(OtBu)3,1。1H NMR(苯-d6,23℃):δ3.35(q,3JHH=7.2Hz,Pn*(H)),2.27 2.24 2.12 2.08 1.84(s,每3H,CH3-Pn*(H)),1.30(s,27H,OC(CH3)3),1.25(d,3H 3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ141.5 135.1 132.9 128.6 121.0 115.6 110.5(q-Pn*(H)),75.4(OC(CH3)3),43.7(1-Pn*(H)),33.4(OC(CH3)3),16.2(1-CH3-Pn*(H)),12.712.6 12.1 12.1 11.4(CH3-Pn*(H))。
实施例13
η5-Pn*(H)Zr(OtBu)3的合成。将[Pn*(H)ZrCl3]2(0.028g,0.036mmol)和KOtBu(0.024g,0.22mmol)在苯-d6(0.5mL)中合并并且声处理持续5分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为浅黄色粉末的Pn*(H)Zr(OtBu)3。1H NMR(苯-d6,23℃):δ3.33(q,3JHH=6.9Hz,Pn*(H)),2.25 2.22 2.09 2.05 1.83(s,每3H,CH3-Pn*(H)),1.35(s,27H,OC(CH3)3),1.23(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ142.8 137.5 134.4 129.3 123.4 117.8 112.9(q-Pn*(H)),80.2(OC(CH3)3),43.8(1-Pn*(H)),33.1(OC(CH3)3),15.9(1-CH3-Pn*(H)),13.7,12.8,12.7,12.3 12.2(CH3-Pn*(H))。
实施例14
η5-Pn*(H)Zr(O-CH2C6H5)3的合成。将[Pn*(H)ZrCl3]2(0.100g,0.131mmol)和KO-CH2C6H5(0.115g,0.786mmol)在C6H6(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,在室温下提供作为浅黄色油状固体的Pn*(H)Zr(O-CH2C6H5)3。1H NMR(苯-d6,23℃);δ7.37–7.03(重叠的多重峰,15H,CH2C6H5),5.10(s,6H,CH2C6H5),3.12(q,1H,3JHH=7.3Hz,Pn*(H)),2.13 2.09 1.99 1.90 1.68(s,每3H CH3-Pn*(H)),1.13(d,3H 3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ143.9(CH2-1-C6H5),143.0 135.4 133.2(q-Pn*(H)),128.5(CH2-2,3,4-C6H5),127.1(q-Pn*(H)),126.9 126.4(CH2-2,3,4-C6H5),122.0 116.5 111.3(q-Pn*(H)),71.7(CH2C6H5),43.2(1-Pn*(H)),16.2(1-CH3-Pn*(H)),12.3 11.8 11.5 11.0 10.4(CH3-Pn*(H))。
实施例15
η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3的合成。将Pn*(H)ZrCl3和S-KOCH{CH3}C6H5在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,在室温下提供作为浅黄色油状固体的η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3。1H NMR(苯-d6,23℃):两种非对映异构体:δ7.40(d,6H,3JHH=7.3Hz,2,6-C6H5),7.11(m,3H,4-C6H5),5.30(q,3H,3JHH=6.1Hz,CHMe),3.14 3.09(q,3JHH=6.9Hz,Pn*(H)),2.11 2.11 2.08 2.05 1.97 1.97 1.89 1.88 1.70 1.65(重叠的单峰,每3H,CH3-Pn*(H)),1.44(d,9H,3JHH=6.1Hz,CHMe),1.12(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):非对映异构体1:δ148.7142.7 135.5 133.3(q-Pn*(H)),128.6(3,5-C6H5),126.9(2,6-C6H5),125.7(q-Pn*(H)),125.7(4-C6H5),122.0 116.4 111.4(q-Pn*(H)),77.0(CHMe),43.4(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.8 11.2 10.6(CH3-Pn*(H))。非对映异构体2:δ148.7 142.7 135.3 133.2(q-Pn*(H)),128.5(3,5-C6H5),126.9(2,6-C6H5),125.9(4-C6H5),125.5(q-Pn*(H)),121.8 116.4 111.1(q-Pn*(H)),77.0(CHMe),43.3(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.7 11.2 10.5(CH3-Pn*(H))。
实施例16
η5-Pn*(H)Zr(O-外消旋-{CH3}C6H5)3的合成。将Pn*(H)ZrCl3和外消旋-KOCH{CH3}C6H5在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,在室温下提供作为浅黄色油状固体的η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3。1H NMR(苯-d6,23℃):非对映异构体的混合物:δ7.48-7.10(重叠的多重峰,15H,C6H5),5.24(重叠的四重峰,3H,CHMe),3.06(q,1H,3JHH=7.3Hz,Pn&(H)),2.12 2.12 2.08 2.07 1.98 1.98 1.90 1.69 1.67(重叠的单峰,每3H,CH3-Pn*(H)),1.50 1.45 1.44 1.41(重叠的双峰,9H 3JHH=6.4Hz,CHMe),1.12(d,3H,3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):非对映异构体的混合物:δ148.7 142.7 135.5 133.3(q-Pn*(H)),128.4(3,5-C6H5),127.3(q-Pn*(H)),126.9(2,6-C6H5),125.6(4-C6H5),122.0 116.4 111.4(q-Pn*(H)),77.0(CHMe),43.3(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.7 11.2 10.6(CH3-Pn*(H))。在13C{1H}NMR光谱中报道的值是观察到的多重重叠的共振的中心值。
实施例17
η5-Pn*(H)Zr(O-2,6-Me-C6H3)3的合成。将[Pn*(H)ZrCl3]2和KO-2,6-Me2C6H3在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为浅黄色粉末的Pn*(H)Zr(O-2,6-Me-C6H3)3。1H NMR(苯-d6,23℃):δ6.93(d,6H,3JHH=7.4Hz,3,5-C6H3),6.75(t,3H,3JHH=7.3Hz,4-C6H3,3.31(q,3JHH=7.4Hz,Pn*(H)),2.25(s,18H,O-2,6-CH3-C6H3),2.20 2.12 1.96 1.69 1.54(s,每3H,CH3-Pn*(H)),1.09(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ159.7(1-C6H3),145.3 139.7 135.6(q-Pn*(H)),128.9(3,5-C6H3),128.0(q-Pn*(H)),126.6(2,6-C6H3),125.7(q-Pn*(H)),120.4(4-C6H3),119.0 113.7(q-Pn*(H)),43.9(q-Pn*(H)),17.9(O-2,6-CH3-C6H3),15.7(1-CH3-Pn*(H)),12.5 11.8 11.7 11.0(CH3-Pn*(H))。
实施例18
η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3的合成。将[Pn*(H)ZrCl3]2(0.174g,0.225mmol)和KO-2,6-iPr2C6H3(0.2925g,1.35mmol)在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为黄绿色粉末的η5-Pn*(H)Zr(O-2,6iPr-C6H3)3。对于C50H70O3Zr的分析计算值:C,74.11;H,8.71。实测值:C,67.68;H,8.60。1H NMR(苯-d6,23℃):δ7.11-7.03(重叠的双峰,6H,3,5-C6H3),6.96(明显的三重峰,3H,3JHH=6.7Hz,4-C6H3),3.52(重叠的七重峰,6H 3JHH=6.7Hz,CH(CH3)2),3.11(q,1H,3JHH=7.4Hz,Pn*(H)),2.17 2.13 1.93 1.85 1.44(s,每3H,CH3-Pn*(H)),1.32(d,12H,3JHH=6.7Hz,CH(CH3)2,1.01(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ155.9 155.8(1-C6H3),146.8(q-Pn*(H)),140.6(q-Pn*(H)),137.9 137.7(2,6-C6H3),136.4 127.4(q-Pn*(H)),123.6 123.5(3,5-C6H3),122.1 122.1(4-C6H3)120.5 116.2(q-Pn*(H)),43.3(1-Pn*(H)),27.3 27.2(CH(CH3)2),24.9 24.8 24.0 23.9(CH(CH3)2),15.6(1-CH3-Pn*(H)),12.1 12.0 12.0 11.8 10.9(CH3-Pn*(H))。对Pn*(H)碳作出解释的一个四级共振与残余的质子溶剂共振重叠。
实施例19
η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)的合成。将[Pn*(H)ZrCl3]2(0.239g,0.310mmol)和KO-2,6-tBu2C6H3(0.152g,0.62mmol)在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为黄绿色粉末的η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)。收率:0.072g(42%)。1H NMR(苯-d6,23℃):δ7.13(d,2H,3JHH=7.7Hz,3,5-C6H3),6.78(t,1H,3JHH=7.7Hz,4-C6H3),2.96(q,1H,3JHH=7.4Hz,Pn*(H)),2.20 2.05 1.98 1.69(s,每3H,CH3-Pn*(H)),1.41(宽的单峰,18H,C(CH3)3),1.32(s,3H,CH3-Pn*(H)),0.86(d,6H,3JHH=7.4Hz,CH(CH3)2)。13C{1H}NMR(苯-d6,23℃):δ161.0(1-C6H3),148.2 145.9 134.4 132.3(q-Pn*(H)),125.4(3,5-C6H3),125.2(q-Pn*(H)),121.4(4-C6H3),119.6(q-Pn*(H)),44.7(1-Pn*(H)),35.6(C(CH3)3),32.1(C(CH3)3),15.5(1-CH3-Pn*(H)),14.0 12.9 12.4 12.4 116(CH3-Pn*(H))。对2,6-C6H3和Pn*(H)碳作出解释的两个四级共振与残余的质子溶剂共振重叠。
实施例20
η5-Pn*(H)Hf(O-2,6-Me-C6H3)3的合成。将[Pn*(H)HfCl3]2和KO-2,6-Me2C6H3在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为浅黄色粉末的Pn*(H)Hf(O-2,6-Me-C6H3)3。1H NMR(苯-d6,23℃):δ6.93(d,6H,3JHH=7.4Hz,3,5-C6H3),6.74(t,3H,3JHH=7.4Hz,4-C6H3),3.27(q,1H,3JHH=7.4Hz,4-C6H3),3.27(q,1H,3JHH=7.4Hz,Pn*(H)),2.24(s,18H,O-2,6-CH3-C6H3),2.24 2.16 1.99 1.68 1.56(s,每3H,CH3-Pn*(H)),1.10(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ159.4(1-C6H3),145.1 138.7 134.0(q-Pn*(H)),129.0(3,5-C6H3),127.8(q-Pn*(H)),126.9(2,6-C6H3),124.1(q-Pn*(H)),120.4(4-C6H3),117.6 112.1(q-Pn*(H)),43.9(1-Pn*(H)),17.8(O-2,6-CH3-C6H3),15.5(1-CH3-Pn*(H)),12.4 11.8 11.6 11.4 10.9(CH3-Pn*(H))。
实施例21
η5-Pn*(H)HfCl(O-2,6-iPr-C6H3)2的合成。将[Pn*(H)HfCl3]2和KO-2,6-iPr2C6H3在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。过滤,随后在真空中干燥滤液,提供作为浅黄色粉末的η5-Pn*(H)HfCl(O-2,6iPr-C6H3)2。1H NMR(苯-d6,23℃):δ7.10-6.86(重叠的多重峰,6H,3,4,5-C6H3),3.52(重叠的七重峰,4H,CH(CH3)2),3.11(q,1H,3JHH=7.4Hz,Pn*(H)),2.25 2.20 1.92 1.92 1.49(s,每3H,CH3-Pn*(H)),1.30(d,12H,3JHH=6.5Hz,CH(CH3)2),1.24 1.23(重叠的双峰,每6H,3JHH=6.5Hz,CH(CH3)2)1.05(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ155.7 155.6(1-C6H3),146.3 139.1(q-Pn*(H)),137.9 137.8(2,6-C6H3),134.7 127.6 125.6(q-Pn*(H)),123.9 123.5(3,5-C6H3),122.1 122.0(4-C6H3),118.5 114.5(q-Pn*(H)),43.5(1-Pn*(H)),27.1 27.0(CH(CH3)2),24.9 24.9 24.1 24.0(CH(CH3)2),15.5(1-CH3-Pn*(H)),11.9 11.8 10.8(CH3-Pn*(H))。
实施例22
η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)的合成。将[Pn*(H)HfCl3]2和KO-2,6-tBu-C6H3在苯(5mL)中合并并且搅拌持续10分钟以提供澄清的浅黄色溶液和无色沉淀。η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)通过NMR光谱学被鉴定为唯一的产物。NMR收率:(99.5%)。1H NMR(苯-d6,23℃):δ7.19(d,2H,3JHH=7.7Hz,3,5-C6H3),6.80(t,1H,3JHH=7.7Hz,4-C6H3),2.95(q,1H,3JHH=7.4Hz,Pn*(H)),2.29 2.16 2.06 1.71(s,每3H,CH3-Pn*(H)),1.42和1.44(宽的重叠的单峰,18H,C(CH3)3),1.36(s,3H,CH3-Pn*(H)),0.91(d,3H,3JHH=7.4Hz 1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ160.7(1-C6H3),147.5 143.3 133.0 129.6 127.6(q-Pn*(H)或2,6-C6H3),125.4(3,5-C6H3),123.0(q-Pn*(H)或2,6-C6H3),121.2(4-C6H3),117.7(q-Pn*(H)或2,6-C6H3),44.5(1-Pn*(H)),35.6(C(CH3)3),32.1(C(CH3)3,15.5(1-CH3-Pn*(H)),13.7,12.6 12.0 11.4(CH3-Pn*(H))。对2,6-C6H3或Pn*(H)碳作出解释的一个四级共振与残余的溶剂共振重叠。
实验细节III-涉及基于茚基的催化剂
一般细节
空气和水分敏感的化合物在氮气的惰性气氛下使用标准Schlenk管线技术在双重歧管真空/氮气管线或Braun Unilab手套箱上来操作。反应溶剂(戊烷、己烷、以及甲苯)使用MBraun SPS-800溶剂纯化系统来干燥。将己烷、甲苯、以及戊烷储存在预活化的分子筛上。将干燥的溶剂在氮气的气氛下储存在烘箱干燥的安瓿中,用Rotoflo或杨氏分接头密封。将在空气敏感的化合物的NMR分析中使用的氘代的溶剂在使用之前经合适的干燥剂干燥、冷冻-解冻脱气并且真空转移:将氯仿-d1(Sigma-Aldrich)储存在预活化的分子筛上。NMR光谱被记录在300MHz Varian Mercury VX-Works光谱仪上。除非另有陈述,否则1H(300.27MHz)光谱和13C{1H}(75.50MHz)光谱在25℃下被记录,并且在内部以在使用的氘代溶剂中的残余的质子溶剂峰作为参考。1H化学位移和13C{1H}化学位移δ以百万分率(ppm)被给出,相对于残余的溶剂峰被给出。空气敏感的样品在惰性气氛中在手套箱中使用在杨氏分接头NMR管中干燥的溶剂来制备。[(Ind)2ZrMe2]和络合物根据文献程序被合成。将[(EBI)ZrCl2](Strem Chemicals)在甲苯中热重结晶(hot re-crystallise)。
聚合程序
所有聚合都在包含40mg的在引发剂的氯仿-d1溶液(20mg的在4mL氯仿-d1中的引发剂)中的丙交酯的杨氏分接头NMR管中进行,确保丙交酯:引发剂的比率是50:1。然后,添加氯仿-d1以确保初始的丙交酯浓度是[LA]0=0.50M。包括添加叔丁醇的聚合照常被制备,并且将叔丁醇经由微注射器添加至氯仿-d1溶液,确保了丙交酯:引发剂:叔丁醇的比率是50:1:1。
X射线结晶学
使用全氟聚醚油将晶体固定在MiTeGen MicroMants上,并且在冷的氮气的流中使用Oxford Cryosystems CRYOSTREAM单元迅速冷却至150K。数据收集使用Enraf-Nonius FR590KappaCCD衍射计、利用石墨-单色化的Mo KαX射线辐射来进行。原始帧数据(Raw frame data)在150(2)K下使用Nonius KappaCCD衍射计收集,使用DENZO-SMN归纳并且使用SORTAV对吸收作出校正。使用SuperFlip解出结构并且使用CRYSTALS用全矩阵最小二乘法求精。
[EBI)Zr(O-2,6-Me-C6H3)Cl]的结晶学数据
单晶在室温下从甲苯溶液生长,C28H25BClOZr,Mr=504.17,正交晶系,Pcab,α=90°,β=90°,γ=90°,Z=6,T=150K,块状,黄色,10201独立反射,R(初始)=0.070,R1=0.057wR2=0.137[I>2σ(I)]。
实施例23
[EBI)Zr(O-2,6-Me-C6H3)Cl]的合成
在室温下向一当量的在甲苯(10mL)中的[EBI)ZrCl2](100mg,0.24mmol)添加一当量的在甲苯(10mL)中的2,6-二甲基苯酚(29mg,0.24mmol)。将黄色悬浮液留下搅拌持续18小时,产生澄清的黄色溶液。将溶剂在真空中除去,以提供作为黄色结晶固体的[(EBI)Zr(O-2,6-Me-C6H3)Cl],收率为80%(96mg,0.19mmol)。1H NMR(氯仿-d1,25℃,300MHz):δ7.84(1H,dd,ArH,3JHH=8.7Hz,4JHH=0.9Hz),7.61(1H,dd,ArH,3JHH=8.6Hz,4JHH=0.9Hz),7.30–7.27(2H,m,ArH),7.20–7.10(2H,m,ArH),7.03(1H,t,ArMe2H,3JHH=7.5Hz),6.78(2H,d,ArMe2H,3JHH=7.4Hz),6.63–6.55(2H,m,ArH),6.54(1H,d,CpH,3JHH=3.2Hz),6.41(1H,d,CpH,3JHH=3.2Hz),6.15(1H,d,CpH,3JHH=3.2Hz),6.02(1H,d,CpH,3JHH=3.2Hz),3.88-3.62(4H,m,桥),1.91(6H,s,ArMe2)。13C{1H}NMR(氯仿-d1,25℃,75.5Mhz):δ128.2,126.6,125.7 125.6,125.4,123.7,123.6,122.7 121.7,120.9,119.5,115.7,113.2,108.4,106.1(全部非四级环碳)29.6(C2H4),28.7(C2H4),18.0(2xArMe2)。季碳未赋值。
实施例24
[Ind2Zr(O-2,6-Me-C6H3)Me]的合成
在室温下向一当量的在甲苯(5mL)中的[Ind2ZrMe2](100mg,0.28mmol)添加一当量的在甲苯(5mL)中的2,6-二甲基苯酚(34mg,0.28mmol)。将澄清的稻草色溶液留下搅拌持续18小时。将溶剂在真空中除去,以提供作为无色油的[(Ind)2Zr(O-2,6-Me-C6H3)Me],收率为75%(0.21mmol,96mg)。1H NMR(氯仿-d1,25℃,300Mhz):δ7.48-7.43(2H,m,ArH),7.34-7.29(2H,m,ArH),7.07-7.00(2H,m,ArH),6.92-6.85(2H,m,ArH),6.82(2H,d,ArMe2H,3JHH=7.4Hz),6.59(1H,t,ArMe2H,3JHH=7.4Hz),6.20(2H,m,CpH),5.91(2H,m,CpH),5.76(2H,t,CpH,3JHH=3.4Hz),1.86(6H,s,ArMe2),0.22(3H,s,ZrMe)。13C{1H}NMR(氯仿-d1,25℃,75.5Mhz):δ159.4(CO,Ar),128.0(2x CH,Ar),125.7(2x C四级,Ar),124.6(2x CH,Ar),124.3(2x CH,Ar),124.0(2x C四级,Ar),123.9(2x CH,Ar),123.8(2x C四级,Ar),123.6(2x CH,Ar),118.9(CH,Ar),117.6(2x CH,Cp),100.6(2x CH,Cp),99.0(2x CH,Cp),27.8(ZrMe),17.6(2x ArMe2)。
实施例-丙交酯的聚合
(I)
L-丙交酯和外消旋-丙交酯的聚合
L-丙交酯单体和外消旋-丙交酯单体
为了研究芳氧基(aryloxide group)之间的差异,以50的单体:引发剂的比率在100℃下在氯仿-d1中进行丙交酯单体的聚合的伪一级动力学数据(pseudo-first order kinetic data)。结果在表1中被示出并且在图1中被图示。观察到的增长速率k观察通过分析1n([LA]0/[LA]t)相对于时间的半对数标绘图来确定,其中[LA]0=0.50mol/L。
表1. L-丙交酯聚合:芳氧基取代基的变化
聚合条件:100℃,[LA]0=50,[LA]0=0.5M,氯仿-d1。
图1图示使用以下络合物的L-丙交酯聚合:[η5-Pn*(H)Ti(O-2,6-Me-C6H3)3 2(黑色正方形,k观察=0.113±0.014h-1)、η5-Pn*(H)Zr(O-2,6-Me-C6H3)3 7(红色圆形,k观察=0.479±0.032h-1)、η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3 8(粉色三角形,k观察=0.391±0.022h-1)、η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)9(深蓝色三角形,k观察=0.043±0.003h-1)、η5-Pn*(H)Hf(O-2,6-Me-C6H3)3 10(蓝色三角形,k观察=0.364±0.027h-1)、η5-Pn*(H)HfCl(O-2,6iPr-C6H3)2 11(绿色菱形,k观察=0.463±0.029h-1)以及η5-Pn*(H)HfCl2()-2,6tBu-C6H3)12(紫色三角形,k观察=0.086±0.020h-1)。聚合条件:氯仿-d1,在100℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
如在图1中可以看到,基于二甲基和二异丙基芳族基团与锆和铪的四种络合物(7、8、10以及11)产生最高的增长常数(0.364<k观察<0.479h-1)。两种叔丁基络合物(9和12)均证明最低的聚合速率(对于η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)具有0.043h-1的k观察,而对于η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)具有0.086h-1的k观察))。当使用二甲基酚盐类型络合物改变金属时,出现的是,锆比铪比钛更迅速(分别地,0.479h-1、0.391h-1以及0.113h-1的k观察)。这些速率在文献的范围内。
当然由于聚合的高温度,多分散性是相对地高的(1.45<Mw/Mn<1.74)。分子量比理论的分子量略微较高(7,547g/mol<Mn<11,680g/mol)。
烷氧基取代基(alkoxide group substituent)的变化对L-丙交酯的聚合的影响已经在100℃下以50的单体:引发剂的比率在氯仿-d1中被研究。当使用络合物η5-Pn*(H)Ti(OtBu)3 1和η5-Pn*(H)Zr(OtBu)3 3时,在1小时之后未实现转化。然而,将叔丁基改变为苄基型取代基显著地增大了聚合的速率,对于η5-Pn*(H)Ti(O-CH2Ph)3 4、η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6,具有高于90%的转化率(分别地,93%、96%以及97%)。
为了研究手性对引发基团的影响,以50的单体:引发剂的比率在100℃下在氯仿-d1中进行丙交酯单体的聚合的伪一级动力学数据。结果在表2中被示出并且在图2-3和SI中被图示。
聚合条件:[LA]0/[M]0=50,[LA]0=0.5M,氯仿-d1。
图2示出丙交酯聚合:L-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(黑色正方形,k观察=1.166±0.068h-1);L-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(红色圆形,k观察=1.954±0.063h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(粉色三角形,k观察=1.667±0.053h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(蓝色三角形,k观察=1.342±0.055h-1)。聚合条件:氯仿-d1,在80℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
图3示出丙交酯聚合:L-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(黑色正方形,k观察=0.484±0.037h-1);L-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(红色圆形,k观察=0.850±0.063h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(粉色三角形,k观察=0.767±0.037h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(蓝色三角形,k观察=0.491±0.031h-1)。聚合条件:氯仿-d1,在60℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
两种络合物η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6对于L-丙交酯和外消旋-丙交酯在100℃下均证明非常高的聚合速率(1.885h-1<k观察<3.442h-1)。在三个温度(60℃、80℃以及100℃)下,对于两种丙交酯单体,外消旋络合物聚合得更迅速。在60℃下,当使用外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3 6时观察到的增长速率(对于L-丙交酯为0.850h-1的k观察并且对于外消旋-丙交酯为0.767h-1的k观察),比当使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5时(对于L-丙交酯为0.484h-1的k观察并且对于外消旋-丙交酯为0.491h-1的k观察)快约70%。L-丙交酯和外消旋-丙交酯似乎以类似的聚合速率被聚合。
多分散性随降低的温度而降低(对于100℃,1.27<Mw/Mn<1.44并且对于80℃,1.15<Mw/Mn<1.24)。分子量实验值是理论值的一半(2,693g/mol<Mn<4,549g/mol)。
使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6引发的用于L-丙交酯和外消旋-丙交酯的开环聚合的活化参数使用艾林标绘图来确定并且被发现是30.4kJ/mol<ΔH#<46.6kJ/mol且411.4J/(mol K)<ΔS#<640J/(mol K),图4和SI。
图4是使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5的L-丙交酯聚合的艾林标绘图。斜率=-5610±488,且R2=0.993。聚合条件:氯仿-d1,其中[LA]0/[Zr]0=50并且[LA]0=0.5M。
L-丙交酯的聚合的伪一级动力学数据用于使用η5-Pn*(H)Zr(O-2,6iPr-C6H3)3 8在100℃下在氯仿-d1中研究单体浓度:引发剂浓度的比率的变化的影响。结果在表3中被核对并且在图5中被图示。
表3. L-丙交酯聚合:浓度的变化
聚合条件:100℃,[LA]0=0.5M,氯仿-d1。
图5示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯聚合:[LA]0/[Zr]0–25(黑色正方形,k观察=0.521±0.021h-1);[LA]0/[Zr]0=50(红色圆形,k观察=0.391±0.022h-1);[LA]0/[Zr]0=100(蓝色三角形,k观察=0.377±0.015h-1);[LA]0/[Zr]0=200(粉色三角形,k观察=0.235±0.004h-1)。聚合条件:氯仿-d1,在100℃下,其中[LA]0=0.5M。
如所预计,L-丙交酯在100℃下在氯仿-d1中的聚合速率随着降低的单体:引发剂的比率而增大(分别地,对于200、100、50以及25的[LA]0/[Zr]0,0.235h-1、0.377h-1、0.391h-1以及0.521h-1的k观察)。此外,分别地,对于25至200的初始单体:引发剂的比率,分子量Mn随着增加的浓度从6,920g/mol增大至17,042g/mol。多分散性,Mw/Mn在1.40<Mw/Mn<1.74之间变化。
使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯的观察到的增长速率证明了作为引发剂的函数的伪一级动力学(图6)。
图6示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的Ln(k观察)相对于Ln([ZR]0)的标绘图。斜率=0.350±82,且R2=0.901。聚合条件:氯仿-d1,在100℃下并且[LA]0=0.5M。
L-丙交酯的聚合的伪一级动力学数据用于使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8在氯仿-d1中以50的单体:引发剂的比率研究温度的影响。结果在表4中被核对并且在图7中被图示。
表4. L-丙交酯聚合:温度的变化
聚合条件:[LA]0/[M]0=50,[LA]0=0.5M,氯仿-d1。
图7示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯聚合:T=100℃(黑色正方形,k观察=0.391±0.022h-1);T=90℃(红色圆形,k观察=0.151±0.008h-1);T=80℃(蓝色三角形,k观察-0.092±0.006h-1)。聚合条件:氯仿-d1,其中[LA]0/[M]0=50,[LA]0-0.5M。
如所预计,L-丙交酯在氯仿-d1中的聚合速率随着降低的温度而增大(分别地,对于100、90以及80的T,0.391h-1、0.151h-1以及0.092h-1的k观察)。分子量Mn在6,348g/mol至7,751g/mol之间保持不变,这与理论的分子量非常接近。然而,多分散性Mw/Mn分别随着从100℃至80℃的降低的温度而从1.72降低至1.58。
用于使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8引发的L-的开环聚合的活化参数使用艾林标绘图来确定并且被发现是ΔH#=75.9kJ/mol和ΔS#=1847J/(mol K),图8。
图8示出使用η5-Pn*(H)Zr(O-2,6-Pr2C6H3)3 8的L-丙交酯聚合的艾林标绘图。斜率=-9133±1881,且R2=0.959。聚合条件:氯仿-d1,其中[LA]0/[Zr]0=50并且[LA]0=0.5M。
合成的聚丙交酯通过1H、1H{1H}以及13C{1H}NMR光谱学来表征。NMR光谱证明了当L-丙交酯被聚合时没有差向异构并且当外消旋-丙交酯被聚合时等规立构偏向的PLA(an isotactic biased PLA)。
它们还已经通过MALDI-TOF和13C{1H}NMR光谱学来表征以确定链的终止。示出的是,丙交酯单体插入在金属-氧键中。
(II)
图9中示出了在80℃下在苯-d6中、以1:50的引发剂:单体的比率、使用被选择的全甲基并环戊二烯络合物的L-丙交酯的聚合的伪一级动力学数据。用于所有聚合的动力学数据展现出取决于引发的络合物而变化的诱导期。同样地,在图9中的数据从32小时被给出,直到那时所有诱导期已结束。
迄今为止展现出朝向丙交酯异构体的开环聚合的最高活性的络合物是[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]。其证明了与公布的钛络合物(k观察=69.9x10-3h-1)37类似并且在80℃下是络合物[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]和[η8-(Pn*)Ti(O-2,4-Bu-C6H3)Cl]的10倍的聚合速率,络合物[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]和[η8-(Pn*)Ti(O-2,4-Bu-C6H3)Cl]证明了类似的增长速率(分别地,k观察=7.2x 10-3h-1和7.0x 10-3h-1)。这些增长速率是当使用[η5-(Pn*)Ti(O-2,6-Me-C6H3)Cl2](k观察=1.9x 10-3h-1)时的3.5倍。
图9示出L-丙交酯转化率相对于时间的半对数标绘图,[LA]0/[初始]0=50,[LA]0=0.104M,T=80℃,苯-d6(0.5mL),使用[η8-(Pn*)Ti(O-2,6-MeC6H3)Cl](绿色点划线)、[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl](黑色短划线)、[η8-(Pn*)Ti(O-2,6-Me-C6H3)2](红色线)以及[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2](蓝色点虚线)。省略诱导期。
如在图10中所示出的,在90℃下,当通过[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]引发时,外消旋-丙交酯和L-丙交酯以相同的速率(k观察≈110x 10-3h-1)聚合。这可推测地是因为非手性引发剂不能在两种丙交酯对映异构体之间区分并且已经以相同的速率并入每个。此立体化学优先性的不存在表明,聚合通过链终止控制的机理进行。
图10示出丙交酯单体转化率相对于时间的半对数标绘图。省略诱导期。[LA]0=0.104M,[LA]0/[初始]0=50,T=90℃,苯-d6。使用[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]、L-丙交酯(红色线)和外消旋-丙交酯(黑色短划线)的聚合。
使用[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]的L-丙交酯的聚合在80℃至100℃的温度范围内进行并且结果与艾林标绘图核对。从艾林标绘图获得活化参数的估计:ΔH#=75.15kJ mol-1,ΔS#=-125.85J K-1mol-1,ΔG#(100℃)=87.74kJ mol-1。ΔH#的适度的值对于对与金属中心配位的羰基的攻击是典型的并且ΔS#的负的和相对高的值意味着在过渡态中的高的有序度。同样地,所有参数与展现出高度有序的过渡态的配位插入机理一致。L-丙交酯的聚合产生等规立构PLA并且外消旋-丙交酯产生无规立构PLA。
(III)
在图11中示出了使用[(EBI)Zr(O-2,6-Me-C6H3)Cl]、[(Ind)2Zr(OtBu)Me]和[(Ind)2Zr(O-2,6-Me-C6H3)Me]的L-丙交酯的聚合的伪一级动力学数据。所有最初的聚合研究在80℃下在氯仿-d1中、以50:1的L-LA:引发剂的比率进行,确保[LA]0–0.50M。
图12示出L-丙交酯转化率相对于时间的半对数标绘图,[LA]0/[初始]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL),使用[(EBI)Zr(O-2,6-Me-C6H3)Cl](点划线)、[(Ind)2Zr(OtBu)Me](实线)、[(Ind)2Zr(O-2,6-C6H3)Me](短划线)的聚合。
图12示出了络合物[(Ind)2Zr(OtBu)Me]在八个小时内展现出以84%转化率的最高活性(k观察=0.24h-1)并且没有引发周期。这与在文献中的锆引发剂的适中的活性是可比较的。明显地较低的活性通过[(Ind)2Zr(O-2,6-Me-C6H3)Me]展现,该[(Ind)2Zr(O-2,6-Me-C6H3)Me]在28小时之后、以4.5倍慢于[(Ind)2Zr(OtBu)Me]的速率实现74%转化率(k观察=0.05h-1)。感兴趣地,[(Ind)2Zr(O-2,6-Me-C6H3)Me]展现出约半小时的引发周期。相比于[(Ind)2Zr(OtBu)Me]的叔丁氧基取代基(tert-butoxide substituent),这些观察结果通过[(Ind)2Zr(O-2,6-Me-C6H3Me]的芳氧基取代基(aryl-oxidesubstituent)的增加的体积被合理说明。相比之下,[(EBI)Zr(O-2,6-Me-C6H3)Cl]展现出最低的催化活性,在28小时内达成4.2%转化率(k观察=0.002h-1),这与由Ning等人,Organometallics,2008,27,5632合成的外消旋-[(EBI)Zr(OC{OiPr}=CMe2)](在18小时内在80℃下在甲苯中的7%转化率)是可比较的。[(EBI)Zr(O-2,6-Me-C6H3)Cl]的速率比[(Ind)2Zr(OtBu)Me]和[(Ind)2Zr(O-2,6-Me-C6H3)Me]小两个数量级,可能是由于与叔丁氧基相比,芳氧基的增加的体积以及通过柄型-桥接的配体赋予的增强的刚度二者,防止了茚基部分的再定向。
因为[(Ind)2Zr(OtBu)Me]示出最高的聚合速率,所以进行另外的研究以调查其立体选择性并且评估其活化。使用[(Ind)2Zr(OtBu)Me]作为引发剂的聚合在60℃和100℃之间以相同的LA:引发剂的比率和如先前使用的[LA]0来进行。活化焓和活化熵从In(k观察/T)相对于(1/T)的标绘图(图12)来计算,给出和这些值与文献是一致的;对于双分子反应和配位-插入机理是典型的。
此外,经发现,L-LA的聚合(k观察=0.24h-1)是在80℃下以50:1的类似的[LA]0:[2]0的比率利用[(Ind)2Zr(OtBu)Me](图13)的外消旋-LA(k观察=0.11h-1)的两倍。这可能是由于在金属中心处翻转手性构型所需要的能量屏障,该能量屏障指示链终止的控制机制。
使用[(Ind)2Zr(OtBu)Me]作为引发剂的外消旋-LA的聚合的1H{1H}NMR光谱证明了朝向等规立构的PLA的偏向,72%的Pi。
在80℃下,在添加以化学计量的量的叔丁醇与[(Ind)2Zr(OtBu)Me]的情况下,重复L-LA和外消旋-LA的聚合。叔丁醇的添加对L-LA和外消旋-LA两者的k观察几乎不具有影响。对于没有醇的L-LA的聚合速率与具有醇的L-LA的聚合速率类似(分别地,0.24h-1的k观察和0.23h-1的k观察)。对于外消旋-LA的聚合的速率也是类似的(分别地,0.11h-1的k观察和0.10h-1的k观察)。具有和不具有叔丁醇的L-LA和外消旋-LA的聚合的分子量和多分散性在表5中被核对。作为引发剂,[(Ind)2Zr(OtBu)Me]证明了L-LA和外消旋-LA在80℃下以50:1的LA:引发剂的比率在氯仿-d1中的高度受控制的聚合,如通过低的多分散性(1.08<Mw/Mn<1.12)所示出的。叔丁醇的添加不影响多分散性;然而,在醇的存在下,实验分子量显得更受控制,如不死聚合(immortal polymerisation)所预期的。
表5.用于使用2a的L-丙交酯和外消旋-丙交酯的聚合数据
a聚合条件;[LA]0/[2]0=50,[LA]0=0.5M,80℃。
通过GPC利用聚苯乙烯标准物在THF中测量的。Mn,理论=[LA]0/[2]0x MLAx转化率。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂体系及其应用和烯烃聚合方法与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂体系及其应用和烯烃聚合方法与制造工艺
- 一种回收甲烷磺酸的装置的制造方法
- 一种用于生产高熔体流动速率聚烯烃的含杂环类化合物的烯烃聚合反应催化剂体系的制作方法