用于纯化L‑α‑甘油磷酰胆碱的工艺的制作方法
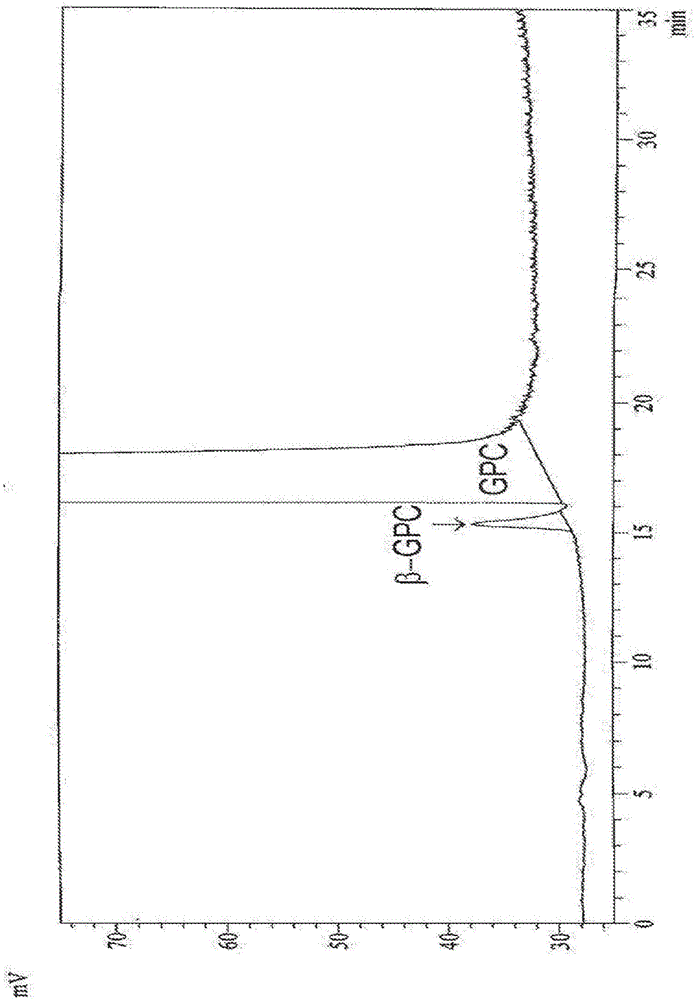
本发明的目的是用于纯化L-α-甘油磷酰胆碱的工艺,其中使L-α-甘油磷酰胆碱从DMSO中或从DMSO与至少另一种溶剂的混合物中结晶,该至少另一种溶剂优选地选自水、醇、卤化的溶剂、醚、酯和/或酰胺。此类工艺允许获得也构成本发明的目的的具有大于99.5%、优选地大于99.7%、甚至更优选地大于或等于99.9%的纯度的L-α-甘油磷酰胆碱;特别地,此类工艺允许获得被小于0.1%的其β-GPC异构体和/或被小于0.1%的环状物质cGP污染的GPC。本发明的另外的目的由用于确定L-α-甘油磷酰胆碱的纯度的方法来代表,所述方法包括使L-α-甘油磷酰胆碱通过具有氨基固定相的HPLC柱洗脱,以及随后借助于蒸发光散射检测器类型(EvaporativeLightScatteringDetectortype)检测L-α-甘油磷酰胆碱自身和其任何杂质。当前的经济和人口趋势已经导致人群的一般老龄化,并且此现象在全球的最富裕地区中特别地明显。个体能力随着老龄化的下降因此已经变成非常重要的问题,并且存在确定帮助减缓此下降的方法的渐增的需求。特别地,精神能力的弱化是老龄化的最负面结果中的一个,并且许多研究努力致力于寻找斗争精神能力的弱化的有效的疗法。在该领域中,最感兴趣的产品中的一种是L-α-甘油磷酰胆碱(GPC),其结构在下文被示出。GPC在认知紊乱(cognitivedisorder)领域中作为治疗剂具有明确的用途,并且基于医学实践中和大量临床试验中的效力的广泛确证,GPC的全世界的消耗是明确的。制备GPC的方法可以分成两种主要类型:基于天然来源的磷脂酰胆碱的脱乙酰化反应的类型,以及其中GPC从化学中间体的市场中可商购的原材料合成的类型。第一类型的方法具有使用例如已经在食品工业中广泛使用的大豆卵磷脂作为原材料产品的优点,并且其特征在于,第一类型方法的操作不产生任何特别的安全风险。基于GPC合成方法的程序可以反而引起对管理所使用的物质并且特别地管理手性合成前体的关注。这些手性合成前体主要是R-缩水甘油以及R-氯丙二醇(分别地下文概述的结构I和结构II)并且在两种情况下,这些手性合成前体是具有相当大的毒性特性的烷基化分子。因此,清楚的是,手性合成前体的使用可以如何在该使用和操作期间产生危险的问题,以及在残余痕量的情况下或如果其他物质存在(例如中间体或副产物)从而保持其毒性特性时,甚至如何产生消耗从这些手性合成前体获得的GPC时的危险。无论GPC制备方法是什么,与其纯化和分离有关的部分将对产物的质量具有很大影响,特别是其化学纯度可能影响其毒性和治疗概况。这样的纯度传统上通过“薄层色谱法”(TLC)来证实。此类型的分析具有良好的通用特性并且由于从最大极性至最小极性的所有组分是同时可见的而允许总体评估样品。然而,关于任何杂质的定量确定以及具有类似结构特征的物质例如比如具有相当的极性的异构体的分离,此分析技术不是完全令人满意的。HPLC是为了克服这些TLC缺点而最频繁地采用的分析技术;然而在GPC的情况下,由于分析物的高极性特性(这限制与固定性的相互作用)以及不存在发色团,HPLC的使用造成没有往常容易,这使得使用UV检测器即在质量控制实验室中更普遍的检测器成为可能。公布的方法对于灵敏度以及对于具有放大的或不对称的峰的色谱信号的形状是不令人满意的;在这些条件中,存在分离功率的降低,并且存在具有对应于隐藏在其他色谱信号下的某些物质的信号的风险,从而导致突出任何存在的杂质的能力的损失。见例如:J.Am.OilChem.Soc(2012)89:1155-1163;Talanta(2012)94:22-29;JournalofChromatographyA(2012)1220:108-114,通过引用并入本文。这种情况与药物领域中当前的指南差地相关,该指南提供在产生意图用于人类用途的活性成分时的杂质概况的详述。因此,用于确定不同的GPC制备的质量的新的分析技术的开发是最重要的,并且如果在GPC制备中杂质的存在用此类分析技术来突出,那么关于GPC的纯度的新标准的定义,以及新的纯化程序如此以致能够根据当前的质量标准生产GPC。附图说明图1通过程序1获得的GPC的HPLC-ELSD色谱图图2从实施例2获得的GPC的HPLC-ELSD色谱图图3从实施例1(比较)获得的GPC的HPLC-ELSD色谱图图4通过程序2获得的GPC的31P-NMR光谱图5从实施例3获得的GPC的31P-NMR光谱图6通过程序1获得的GPC的HPLC-ELSD色谱图图7从实施例3获得的GPC的HPLC-ELSD色谱图图8通过程序3获得的GPC的HPLC-ELSD色谱图图9从实施例4获得的GPC的HPLC-ELSD色谱图图10从实施例5(比较)获得的GPC的HPLC-ELSD色谱图图11通过程序4获得的GPC的HPLC-ELSD色谱图图12从实施例6获得的GPC的HPLC-ELSD色谱图图13从实施例7(比较)获得的GPC的HPLC-ELSD色谱图发明描述我们已经出乎意料地发现,在GPC的HPLC分析中,通过合适地使用氨基HPLC色谱柱类型,即其中氨基官能性结合至固定相的表面的色谱柱类型克服上文描述的问题是可能的。此类型的若干柱是可商购的:例如SupelcosilTMLC-NH2、HypersilGoldTM氨基、NH2、YMCTM多胺II、NH2、NH2。固定相可以由具有涂层的支撑材料的颗粒例如比如二氧化硅的颗粒组成,该涂层由包含氨基、优选地仲氨基和叔氨基的聚合物涂层组成。此类型的固定相的描述在YMCEuropeGmbH的出版物“HPLCColumnsYMCClassics”的第22页和第23页上被报告,其通过引用并入本文。同时,将必须使用蒸发光散射检测器(ELSD)类型。此类检测器例如在通过引用并入本文的Anal.Chem(1997)69:561A-563A和US6229605中被描述。这些检测器是其中将洗脱剂送到蒸发室中的工具,在蒸发室中,溶剂被蒸发以留下使光束散射的微小样品颗粒的薄雾。检测器响应与取决于存在的样品的量的扩散成比例;此系统使得检测没有发色团的物质例如GPC、以及同时不使用等度洗脱法成为可能。在固定相和检测器的此组合下,可以使用各种洗脱剂,并且特别地合适地由含水缓冲剂体系例如比如乙酸铵缓冲剂组成的体系可能与有机溶剂例如比如甲醇和/或乙腈组合。此类洗脱剂体系可以以等度模式使用,即通过保持相同的洗脱剂持续色谱运行的全部持续时间;或以梯度模式使用,其中洗脱剂的组成在色谱法期间根据预先确定的程序变化。分析方法的优化将遵循本领域的教导进行。因此,本发明的目的是用于确定L-α-甘油磷酰胆碱的纯度的方法,所述方法包括使L-α-甘油磷酰胆碱通过具有氨基固定相的HPLC柱洗脱,以及随后借助于蒸发光散射检测器类型检测L-α-甘油磷酰胆碱自身和其任何杂质。一种或多种洗脱剂相可以由水溶液、极性有机溶剂、或其混合物组成;水溶液优选地具有范围在3和6之间、甚至更优选地4和5之间的pH。极性有机溶剂可以是C1-C4醇、乙腈、或其混合物;C1-C4醇优选地是甲醇。此组合允许克服对应于GPC的色谱峰的扩大的缺点,并且同时允许在高灵敏度下进行确定GPC浓度。同时,发现在GPC制备中检测其他物质的存在是可能的,该其他物质来源于制备起始于大豆卵磷脂的GPC的工艺。在这些物质中,包括L-α-甘油磷酸乙醇胺(GPE)、糖及其类似物,这些物质来源于大豆卵磷脂中的其存在物或其前体中的一种的存在物,大豆卵磷脂在GPC生产工艺中用作原料。已经获得此重要的结果,以比在过去更灵敏且选择性的方式证实GPC样品中的杂质概况(如果存在)是立即感兴趣的,GPC样品通过最适合于用于人类用途的药物产品的生产方法获得。为此目的,基于大豆卵磷脂脱乙酰化的半合成工艺被主要考量,鉴于其中在这些程序中使用的高度毒性原材料例如缩水甘油或氯丙二醇的可能的污染,判定全合成程序对于用于人类用途的GPC的产生是不合适的。同时考量与通过全合成路线的GPC的产生有关的所述禁忌症,将在此方法下产生的中国来源的GPC样品提交至新的分析程序,用于完整评估。来源于半合成程序的样品根据在WO9013552(具体地实施例2)和EP575717(具体地实施例1)中报告的程序来产生。在这些文献中,报告了基于用离子交换树脂纯化GPC的程序,并且该程序不包括将GPC作为与氯化镉的加合物(GPC·CdCl2的沉淀和二氧化硅色谱法;因此这些程序是避免使用毒性镉盐以及特征是低生产率的程序的方法,真正可适用于GPC大规模生产。分析结果示出,在所用样品中,洗脱前峰的存在以及其中保留时间非常接近于GPC的保留时间,具有等于约0.94的RRT(相对保留时间)。具有明显强度的此信号不对应于鉴于制备工艺将已经被可合理预计的物质,例如缩水甘油、GPE、甘油磷酸、或蔗糖。考虑到新检测的物质的色谱性能与GPC的色谱性能如此类似,至于直到现在未曾被突出,并且甚至在本发明的新的HPLC方法目的下,新检测的物质的色谱性能与GPC的色谱性能区别很小,那么我们假定新检测的物质对应于其位置异构体。GPC的β异构体(β-GPC)的化学结构在下文被报告。β-GPCβ-GPC是不可商购的,并且为了检查β-GPC是否可能是用新的方法可检测的物质,β-GPC的合成根据下文图示的方案进行。由此获得的β-GPC通过NMR分析以确证结构,并且通过HPLC示出不仅其纯度是高的,而且还示出其保留时间与通过分析不同的GPC样品揭示的未知物质的保留时间是相容的。因此,将未知的信号归属于对应于β-GPC结构是可能的,并且此归属(attribution)通过HPLC“掺加(spiking)”实验被确证,其中向在HPLC中示出具有在约0.94处的RRT的物质的存在的GPC样品添加校正量的β-GPC,观察到具有在约0.94处的RRT的信号的对应的增强,而没有其分裂。作为结构归属的另外的确证,进行31P-NMR实验,该实验允许样品中的在约-0.6ppm处的信号的确定,其中具有在约0.94处的RRT的物质的存在更高。根据上文报告的方案合成的β-GPC的信号在相同的场值(fieldvalue)处被揭示。因此,31P-NMR方法是HPLC程序的可能的可选物,然而由于31P-NMR方法较低的灵敏度以及HPLC仪器在质量控制实验室中的更广泛的可用性,本发明的HPLC方法目的对于确定具有约0.94的RRT的物质(β-GPC)仍然是优选的。借助于31P-NMR分析,还可能的是,在根据EP575717获得的产物的光谱中确定在约18.4ppm处的另外的信号。这些值与下文以钠盐的形式图示的)环状磷酸盐cGP的值是相容的。cGP(钠盐)此环状磷酸盐的形成在根据EP575717的程序的GPC的分离条件中是可想象的。为了支持此假定,根据下文图示的方案制备cGP的真正的样品。此产物的31P-NMR分析确证归属(assignment),示出在约18.4ppm处的相同的信号以及在合适的掺加实验中的信号分裂的不存在。鉴于代表本发明的一个实施方案的创新性分析方法所发现的,我们已经实验地证实,已知的GPC纯化方法是否可以充分地除去突出的杂质。由于已经表明的原因,排除通过与氯化镉络合的纯化和二氧化硅色谱法,我们已经确定,在现有技术中报告的那些(见例如WO9013552),在工业适用性方面,从乙醇中结晶作为最好的程序。使根据WO9013552的程序获得的样品从乙醇中结晶,但HPLC分析示出,纯化仅仅是部分的,大量的β异构体(0.48%)仍然存在于产物中。以下表1总结了根据不同的制备程序获得的GPC样品的分析结果。表1用于纯化GPC的新的体系已经被确定,并且此发现还代表本发明的实施方案。新的纯化是基于二甲基亚砜(DMSO)的使用。为了实现本发明,此溶剂可以相对于被纯化的GPC以不同的条件和以不同定量比被使用。在本发明的实施方案中,除了DMSO之外,其他溶剂可以存在,例如极性溶剂、中等极性溶剂或非极性溶剂。这些另外的溶剂可以属于不同的溶剂类别,例如但不限于卤化的溶剂、醇、醚、酯以及酰胺。可以被用于实现本发明的溶剂的实例是但不限于水、甲醇、乙醇、异丙醇、正丁醇、二氯甲烷、四氢呋喃、乙酸乙酯、以及DMF。根据本发明的方面,以范围在每重量份的L-α-甘油磷酰胆碱2体积份和100体积份之间、优选地3体积份和70体积份之间的量、更优选地范围在4体积份和30体积份之间的比率、更优选地范围在5体积份和15体积份之间的比率使用DMSO。根据本发明的另外的方面,以范围在每体积DMSO0.01体积和10体积之间、优选地从0.05体积至5体积的量、更优选地范围在0.1体积和1体积之间的比率使用任何溶剂或任何另外的溶剂。取决于工艺参数,可以获得GPC的合适的结晶,或包含杂质的GPC晶体的纯化还可以通过使用时间和温度的合适的组合使包含杂质的GPC晶体悬浮在DMSO中并且然后从液相中分离晶体来进行;还在此情况下,其他溶剂可以与DMSO组合地存在。纯化可以在不同的温度下进行;通常,在纯化工艺的不同阶段中采用不同的温度可以是便利的,在GPC的最终分离的相中具有较低的温度。用于进行纯化的合适的温度可以是在100℃和0℃之间、优选地在70℃和5℃之间、更优选地在50℃和15℃之间。在选择温度中,考虑DMSO的冻结温度将是优选的,该冻结温度可以主要取决于另外的溶剂的存在、以及GPC和存在的其他物质的浓度而变化。对于GPC的分离,可以使用对本领域技术人员已知的设备,例如关闭或打开的离心机或过滤系统。可以使用用于纯化的相同溶剂或相同的溶剂混合物使晶体直接经历洗涤程序;可选择地,对于洗涤产物,可以使用不同于在结晶或悬浮步骤中使用的溶剂或溶剂混合物的溶剂或溶剂混合物。被使用的DMSO相比于在本发明的纯化工艺目的中使用的GPC的量的比率可以相当大地变化,并且可以根据本领域的教导方便地选择。在实践中,为了不降低多于所需要的工艺生产率,不使用太高数量的DMSO将是便利的,并且另一方面,这些数量不应被过度地减少以避免在结晶产物的分离操作中产生困难。对于该实施方案,范围在每重量份(kg)GPC2体积份(升)和100体积份之间的、更优选地范围在每重量份(kg)GPC3体积份和70体积份之间的比率、更优选地范围在4体积份和30体积份之间的比率、更优选地范围在5体积份和15体积份之间的比率的DMSO与GPC比率可以被认为是便利的。进行本发明的特别有利的模式是使GPC从GPC在水、或其他溶剂、或溶剂混合物中的溶液中结晶。通过将DMSO添加至此类溶液,发现获得稳定持续足以进行蒸馏操作而不使GPC同时沉淀的时间的溶液。利用相对于常用的其他溶剂具有较高沸点的DMSO,因此可能的是,在没有由产物的同时沉淀引起的障碍的情况下,使用于进行结晶的溶剂的混合物的组合物改性。在已经实现溶剂混合物组合物之后,通过添加将充当引发剂的等份结晶的GPC诱导GPC的结晶是可能的。此选择显得不是必需的;在其应用是期望的情况下,GPC晶体的样品可以被用作引发剂,引发剂应当不一定具有特别高的纯度,因为在所使用的引发剂的品质和在纯化结束时分离的GPC晶体的品质之间没有观察到密切的相依性。令人惊讶的是,观察到使用DMSO的这些纯化程序如何允许获得高纯度GPC,即使是从包含大量的其他分子物质的制品中,包括先前描述的其他物质。特别地,本发明的纯化体系目的除去GPCβ异构体的能力显得是明显的且不可预测的,因为GPCβ异构体具有与产物的化学物理特性非常类似的化学物理特性,具有高纯度的产物的分离是期望的。不同于从乙醇中结晶,还发现,本发明的纯化方法目的允许从由卵磷脂中获得的GPC制品中最常发现的物质中的某些纯化GPC。特别地,发现在本发明的实施方案中,有效地除去GPC杂质例如蔗糖和GPE是可能的。下表2总结相比于与使用乙醇作为结晶溶剂进行的比较实验有关的结果在这些实验中实现的主要结果。表2*31P-NMR测量值表2中示出的结果突出本发明的实施方案在GPC的纯化中的有用性。关于这一点,值得注意的是以下事实:如在实施例6中,本发明的实现证明在糖(在此情况下蔗糖)的存在下也使GPC结晶的可能性以及甚至在这些情况下可实现的高的纯化效率。相反地,从乙醇中的GPC结晶不允许令人满意地除去蔗糖。此外,以及在甚至较大测量糖例如蔗糖中,当乙醇被用作结晶溶剂时,GPC表现为结晶的抑制剂;因此,在这些情况下,结晶收率是如此低以至于致使该程序在用于GPC的工业生产的工艺中是不能实行的。本发明的另外的实施方案包括其纯度大于99.5%、优选地大于99.7%、甚至更优选地具有大于或等于99.9%的纯度的GPC。特定的特征表示为被小于0.1%的其β-GPC异构体污染的GPC和被小于0.1%的环状物质cGP污染的GPC。为了本发明的目的,上文指示的纯度被测量为在HPLC方法和/或31P-NMR并且优选地分别在实施例8和实施例9中描述且示例的HPLC方法和/或31P-NMR下的百分比面积。以下实施例意图说明本发明的方法中的某些,而不以任何方式限制本发明。实施例程序1根据WO9013552的实施例2制备GPC向125g的大豆卵磷脂添加500mL的甲醇并且在搅拌下保持直到完全溶解。添加在甲醇中的20mL的30%甲醇钠。搅拌在室温下继续持续3小时。在过滤之后,将残余物用甲醇(3x10mL)洗涤。将滤液用冰醋酸中和(约6的pH),然后将滤液浓缩成约125mL的残余的体积,分离上层油相。将富集的下层相在包含设定在甲醇中的140mL的Amberlyst15树脂(呈酸形式)的柱上洗脱。将洗脱用375mL的甲醇、随后是300mL的水进行。将水性洗脱液以由此制备的三个色谱柱的顺序洗脱:首先用设定在水中的呈OH-形式的30mL的IR93树脂,其次用设定在水中的呈OH-形式的30mL的IR401树脂,再次用设定在水中的呈酸形式的12mL的IRC50树脂。将最终的洗脱液浓缩成具有等于15%含水量的残余的粘稠流体。HPLC-ELSD分析示出等于0.8%(面积百分比)的β-GPC含量。程序2根据EP575717的实施例1制备GPC将商业上通过大豆卵磷脂的萃取获得的200g富含磷脂酰胆碱的级分溶解在600mL的甲醇中。将获得的溶液在包含设定在甲醇中的、呈碱形式的DuoliteA147树脂的色谱柱上洗脱。在装载富集溶液之后,GPC洗脱用甲醇完成。将洗脱液用乙酸中和,并且浓缩成300mL的残余体积,获得两相的急剧的分层。分离富集的下层相,用甲醇洗脱,并且用200mL的正庚烷萃取两次。将800mL的正丁醇添加至富集的下层相,并且将溶液浓缩成约300mL的残余的体积,冷却至T~5℃,并且过滤。将GPC晶体溶解在30mL的除盐水中,并且将溶液浓缩成直至其中残余含水量等于15%的粘稠的液体。HPLC-ELSD分析示出等于0.1%(面积百分比)的β-GPC含量。31P-NMR分析示出等于0.2%(面积百分比)的cGP含量。程序3制备被GPE污染的GPC将呈晶体的GPE添加至GPC在水中的溶液,并且将溶液浓缩成直至其中残余含水量等于15%的粘稠的液体。HPLC-ELSD分析示出等于0.2%(作为面积百分比)的GPE含量。程序4制备被蔗糖污染的GPC将呈晶体的蔗糖添加至GPC在水中的溶液,将溶液浓缩成直至其中残余含水量等于15%的粘稠的液体。HPLC-ELSD分析示出等于2%(作为面积百分比)的蔗糖含量。实施例1(比较)使用从乙醇中结晶纯化通过程序1获得的GPC将呈粘稠流体的形式的2.5g的GPC(通过程序1获得)在搅拌下在T~50℃下溶解在5mL的乙醇中。将溶液冷却下来至T~10℃并且用一点GPC晶体引发,观察到在几分钟内,形成沉淀物。将其冷却下来至T~5℃并且在搅拌下保持在0℃和5℃之间持续1.5小时,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得1.4g的GPC。HPLC-ELSD分析示出等于0.48%(作为面积百分比)的β-GPC含量。实施例2使用从DMSO中结晶纯化通过程序1获得的GPC向呈粘稠流体形式的2.5g的GPC(通过程序1获得)添加14mL的DMSO,将混合物在搅拌下加温多达T~50℃,将其冷却下来至T~25℃并且用一点GPC晶体引发。将混合物在搅拌下保持在20℃和25℃之间持续16小时;在此时间期间,观察到沉淀物的形成,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得1.9g的GPC。HPLC-ELSD分析示出等于0.16%(面积百分比)的β-GPC含量。实施例3使用从DMSO中结晶纯化通过程序2获得的GPC向呈粘稠流体形式的11.2g的GPC(通过程序2获得)添加60mL的DMSO,将混合物在搅拌下加温多达T~50℃,将其冷却下来至T~25℃并且用一点GPC晶体引发。将混合物在搅拌下保持在20℃和25℃之间持续16小时;在此时间期间,观察到沉淀物的形成,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得8.2g的GPC。HPLC-ELSD分析示出不存在β-GPC。31P-NMR分析示出不存在cGP。实施例4使用从DMSO中结晶纯化(被GPE污染的)GPC向呈粘稠流体形式的10.7g的GPC(通过程序3获得)添加20mL的DMSO,将混合物在搅拌下加温多达T~70℃,添加80mL的DMSO,将其冷却下来至T~25℃并且用一点GPC晶体引发。将混合物在搅拌下保持在20℃和25℃之间持续16小时;在此时间期间,观察到沉淀物的形成,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得8.7g的GPC。HPLC-ELSD分析示出不存在GPE。实施例5(比较)使用从乙醇中结晶纯化(被GPE污染的)GPC将呈粘稠流体的形式的11.2g的GPC(通过程序3获得)在搅拌下在T~45℃下溶解在20mL的乙醇中。将溶液冷却下来至T~5℃并且用一点GPC晶体引发。将混合物在搅拌下保持在0℃和5℃之间持续3小时;在此时间期间,观察到沉淀物的形成,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得3.4g的GPC。HPLC-ELSD分析示出等于0.02%(作为面积百分比)的GPE含量。实施例6使用从DMSO中结晶纯化(被蔗糖污染的)GPC向呈粘稠流体形式的11g的GPC(通过程序4获得)添加20mL的DMSO,将混合物在搅拌下加温多达T~70℃,添加80mL的DMSO,将其冷却下来至T~25℃;在此时间期间,观察到沉淀物的形成。将混合物在搅拌下保持在20℃和25℃之间持续16小时,将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得7.8g的GPC。HPLC-ELSD分析示出不存在蔗糖。实施例7(比较)使用从乙醇中结晶纯化(被蔗糖污染的)GPC将呈粘稠流体的形式的11.0g的GPC(通过程序4获得)在搅拌下在T~45℃下溶解在20mL的乙醇中。将溶液冷却下来至T~5℃并且用一点GPC晶体引发。将溶液冷却下来至T~5℃并且在搅拌下保持在0℃和5℃之间持续2.5小时;在此时间期间,观察到沉淀物的形成。将晶体通过布氏漏斗过滤,用乙醇洗涤。在真空下干燥之后,获得1.1g的GPC。HPLC-ELSD分析示出等于0.14%(作为面积百分比)的蔗糖含量。实施例8通过HPLC-ELSD的GPC分析方法操作参数:HPLC柱:YMCTM多胺II250x4.6mm(5μm)烘箱温度:35℃流速:0.7mL/min洗脱相:相A:85%(乙腈75%,甲醇25%)15%(50mM乙酸铵缓冲剂pH4.5)相B:65%(乙腈75%,甲醇25%)35%(50mM乙酸铵缓冲剂pH4.5)梯度程序:分钟相A%相B%0982598227298322983398245982检测器ELSD检测器参数:氮气流速=1.4mL/min;温度=90℃;增益=1样品分离:在甲醇中将GPC样品稀释至约20mg/mL。注射体积:20-40μL注解:观察到多达RT~5分钟的基线波动在空白中存在(不是由于样品中存在的物质)。在下表中,示出关于感兴趣的物质的洗脱的参考值。使用此方法分析的杂质的检测限(LOD)小于相对于GPC的0.01%。实施例9通过31P-NMR的GPC分析方法在下表中,示出关于感兴趣的物质的化学位移的参考值。使用此方法分析的杂质的检测限(LOD)小于相对于GPC的0.01%。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1