一种含有邻二酚羟基的类EDTA配体和非钆磁共振造影剂及其制备方法与流程
本发明属于分子影像学的生物医药材料技术领域,涉及造影剂及其制备方法,具体涉及一种含有邻二酚羟基的类EDTA配体和非钆磁共振造影剂及其制备方法。
背景技术:
磁共振成像(MRI,Magnetic Resonance Imaging)是一种非介入诊断技术,具有高空间分辨率和高软组织对比度的特点;同时,MRI能多参数(质子密度、弛豫、化学位移等)、多方位和高对比度成像,兼具结构成像和功能成像的特点,其中结构成像可以显示有形的实体病变,功能成像对脑、心、肝等功能性反应可以进行精确的判定。而相对于X-射线透视技术(X-Ray,CT(Computed Tomohtaphy))和放射造影技术(SPECT(Single Photo Emission Computed Tomography),PET(Positron Emission Tomography)),MRI对人体没有辐射影响,且具有更高的成像精度(特别是软组织成像);相对于超声探测技术(US,Ultrasound),核磁共振成像更加清晰,能够显示更多细节。因此,MRI是目前多种疾病诊断的重要手段。
MRI造影剂(Contrast agents)的使用可以为影像医生做出准确的诊断提供更多的物理、化学诊断数据,从而成为这一技术的重要组成部分。据统计,世界上大约30%MRI检查为增强检查,即需要使用造影剂。目前用于MRI的小分子造影剂主要为顺磁性金属离子配合物,如金属离子Fe3+、Mn2+、Gd3+和Dy3+等的配合物,且以钆(Gd)类造影剂为主,如Gd-DTPA,Gd-DOTA等。其中已经用于临床的Gd-DTPA(马根维显,Magnevist)、Gd-DTPA-BMA(钆二胺,Omniscan)、Gd-DO3A-HP(钆替醇,Prohance)、Gd-DOTA(Dotarem)和Gd-DO3A-butrol(Gadoburol)。
对人体而言,钆是外源性金属,游离的Gd3+是剧毒的,在体内分布于骨骼和肝脏,并可迅速导致肝脏坏死。2006年,美国食品和药品管理局(FDA,Food and Drug Administration)陆续发现肾功能不全患者(肾小球滤过率<30mL/(min·1.73m2)),在使用某些稳定性不高的钆类造影剂后,由于造影剂不能及时完全清除体外,从而出现全身皮肤硬化的肾源性系统纤维化(NSF,Nephrogenic Systemic Fibrosis)症状,严重者会出现组织器官受累。目前,对NSF尚无有效的治疗方法。
此外,单位浓度下,高弛豫效率(r1)的磁共振造影剂可以更有效地影响水分子的弛豫,在更小的剂量下达到同样的对比增强效果,从而降低患者接触有毒金属的剂量,降低风险。在低场下(≦1.5T),表征分子在溶液中的翻转速率的旋转相关时间(τR)是影响磁共振造影剂弛豫效率的最为重要的物理参数。临床使用的小分子钆类造影剂(MW=500左右)由于其在溶液中翻转速率过快(τR:~0.1ns,25℃),弛豫效率通常在2.5-5.0mmol-1s-1,远远低于理论预测值(~100.0mmol-1s-1,0.5T,25℃)。这也成为钆类造影剂发展的一个瓶颈。
鉴于钆类造影剂潜在的多方面副作用以及较低的弛豫效率,具有较高弛豫效率的非钆磁共振造影剂的研发具有重要的临床意义和社会意义。
技术实现要素:
本发明的目的旨在针对上述现有技术中钆类造影剂的所带来的巨大副作用,提供一种含有邻二酚羟基的类EDTA配体2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸,该配体为进一步制备非钆磁共振造影剂提供重要的结构基础。
本发明另一目的旨在提供上述2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的制备方法。
本发明再一目的旨在基于EDTA(乙二胺四乙酸)配体优良的螯合性能,提供一种非钆顺磁性金属配合物磁共振造影剂,满足用于临床的MRI造影剂水溶性好、弛豫效率高、毒副作用低等特点。
本发明再一目的旨在提供上述非钆顺磁性金属配合物磁共振造影剂的制备方法。
本发明再一目的旨在提供一种基于配位自组装的非钆金属簇集体磁共振造影剂,该造影剂以上述非钆顺磁性金属配合物磁共振造影剂为基础,再利用邻二酚羟基与金属离子配位形成中等分子量、致密的三聚体金属簇集体结构,以提高弛豫效率。
本发明再一目的旨在提供上述基于配位自组装的非钆金属簇集体磁共振造影剂的制备方法。
为达到上述目的,本发明采用以下技术方案来实现。
本发明提供了一种含有邻二酚羟基的类EDTA配体——2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸,结构式为:
基于上述2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸与金属M的+2价或+3价离子配位,制成相应的顺磁性金属螯合物,即得到如下结构式所示的非钆顺磁性金属配合物磁共振造影剂:
所述结构式中,M为顺磁性金属Mn、Fe、Eu或Dy的+2价离子,或Mn、Fe、Eu或Dy的+3价离子;M2为Na+或K+;当M为+2价离子时,a=2;当M为+3价离子时,a=3。
基于上述2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸先与金属M的+2价或+3价离子配位,再与金属M1的+2价、+3价或+4价离子配位组装制成相应的顺磁性金属螯合物,即得到如下结构式所示的非钆金属簇集体磁共振造影剂:
所述结构式中,M为顺磁性金属Mn、Fe、Eu或Dy的+2价离子,或Mn、Fe、Eu或Dy的+3价离子;M1为金属Sc、Ti、Mn或Fe的+2价离子,或Sc、Ti、Mn或Fe的+3价离子,或Sc、Ti、Mn或Fe的+4价离子;M2为Na+或K+;当M为+2价离子时,a=2;当M为+3价离子时,a=3;当M1为+2价离子时,b=2;当M1为+3价离子时,b=3;当M1为+4价离子时,b=4。
制备2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的化学反应式如下:
以上化学反应式中的结构式命名对应关系为:
结构式(2):化学物2,[(N-叔丁氧羰基)-氨基]-3-[(3,4-二羟基)-苯基]丙酸甲酯;
结构式(3):化合物3,2-[(N-叔丁氧羰基)-氨基]-3-[(3,4-二苄氧基)-苯基]丙酸;
结构式(4):化合物4,2-[(N-叔丁氧羰基)-氨基]-3-[(3,4-二苄氧基)-苯基]丙酰胺;
结构式(5):化合物5,2-氨基-3-[(3,4-苄氧基)-苯基]丙胺;
结构式(6),化合物6,2-[N,N-二(乙酸苄酯基)-氨基]-3-[(3,4-二苄氧基)-苯基]-N,N-二(乙酸苄酯基)丙胺;
结构式(7),化合物7,2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸。
本发明基于以上化学反应式来制备2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸,制备方法如下:
(一)制备结构式(2)的化合物2,[(N-叔丁氧羰基)-氨基]-3-[(3,4-二羟基)-苯基]丙酸甲酯:可以根据本领域中披露的常规方法制备(参考文献Miller,D.D.;Hamada,A.;Clark,M.T.;Adejare,A.;Patil,P.N.;Shams,G.;Romstedt,K.J.;Kim,S.U.;Intrasuksri,U.J.Med.Chem.1990,33(4),1138),也可以使用本发明中描述的制备方法来制备。本发明中使用的制备方法为:在冰浴条件下将二氯亚砜加入左旋多巴的甲醇溶液中,在搅拌下于室温反应24~72小时,反应完成后将反应液减压蒸馏去除溶剂甲醇得产物,将得到的产物用饱和碳酸氢钠溶液溶解形成产物溶液并用饱和碳酸氢钠溶液调整产物溶液的pH为7~8,再将二碳酸二叔丁酯的四氢呋喃溶液滴入所述产物溶液中,在搅拌下于室温反应2~12小时,反应完成后蒸除溶剂四氢呋喃,将所得水相的pH调节至2~4,再用二氯甲烷萃取至少三次,合并各次萃取所得有机相,然后用饱和食盐水将有机相清洗至中性得含水和有机相的多相液体,依次用无水硫酸镁去除所述多相液体中的水分、通过浓缩去除二氯甲烷即得到[(N-叔丁氧羰基)-氨基]]-3-[(3,4-二羟基)-苯基]丙酸甲酯;所述左旋多巴、二氯亚砜、二碳酸二叔丁酯的摩尔比为1:(1~3):(1~3);
(二)制备化合物3,2-[(N-叔丁氧羰基)-氨基]-3-[(3,4-二苄氧基)-苯基]丙酸:可以根据本领域中披露的常规方法制备(参考文献Miller,D.D.;Hamada,A.;Clark,M.T.;Adejare,A.;Patil,P.N.;Shams,G.;Romstedt,K.J.;Kim,S.U.;Intrasuksri,U.J.Med.Chem.1990,33(4),1138),也可以使用本发明中描述的制备方法来制备。本发明中使用的制备方法为:将步骤(一)得到的化合物2溶于无水DMF(二甲基甲酰胺)溶剂中,加入苄氯、K2CO3、KI,在搅拌下于40~60℃反应16~24小时,反应完成后将反应液过滤,并将滤液浓缩至干得残留物;之后将残留物进行后处理,所述残留物的后处理是用乙酸乙酯溶解残留物,再依次用稀盐酸洗涤残留物溶液去除K2CO3、用饱和食盐水将残留物溶液洗涤至中性、用无水硫酸镁去除中性残留物溶液中的水分、通过浓缩去除残留物溶液中的溶剂乙酸乙酯得后处理产物;将后处理产物用乙醇重结晶得到重结晶产物,再将重结晶产物混悬于NaOH的甲醇-水混合液中,回流1~2小时,回流结束后冷却至室温,再加入浓盐酸得到沉淀物,过滤出沉淀物并将沉淀物用乙醇重结晶即得到2-[(N-叔丁氧羰基)-氨基]]-3-[(3,4-二苄氧基)-苯基]丙酸;所述化合物2、苄氯、K2CO3、KI的摩尔比为1:(2~6):(2~6):0.1,所述重结晶产物与甲醇-水混合液中NaOH的摩尔比为1:(2.0~4.0);
(三)制备化合物4,2-[(N-叔丁氧羰基)-氨基]-3-[(3,4-二苄氧基)-苯基]丙酰胺:可以根据本领域中披露的常规方法制备(参考文献Miller,D.D.;Hamada,A.;Clark,M.T.;Adejare,A.;Patil,P.N.;Shams,G.;Romstedt,K.J.;Kim,S.U.;Intrasuksri,U.J.Med.Chem.1990,33(4),1138),也可以使用本发明中描述的制备方法来制备。本发明中使用的制备方法为:将步骤(二)得到的化合物3溶于无水DMF(二甲基甲酰胺)中,在冰盐浴条件下加入EDCI(碳化二亚胺)、HOBt(1-羟基苯并三唑)、DIPEA(N,N-二异丙基乙胺)并搅拌均匀,然后通入氨气在搅拌下反应12~24小时,反应完成后在室温下向反应液中加水沉淀得到初产物,过滤出初产物,再将初产物经二氯甲烷/正己烷重结晶得到2-[(N-叔丁氧羰基)-氨基]-3-[(3,4-二苄氧基)-苯基]丙酰胺;所述化合物3、EDCI、HOBt、DIPEA的摩尔比为1:(1~3):(1~3):(2~9),所述氨气的通入量应满足化合物3与HOBt作用生成的HOBt的活化酯完成酰化反应;
(四)制备化合物5,2-氨基-3-[(3,4-苄氧基)-苯基]丙胺:可以根据本领域中披露的常规方法制备(参考文献Miller,D.D.;Hamada,A.;Clark,M.T.;Adejare,A.;Patil,P.N.;Shams,G.;Romstedt,K.J.;Kim,S.U.;Intrasuksri,U.J.Med.Chem.1990,33(4),1138中披露的制备方法),也可以使用本发明中描述的制备方法来制备。本发明中使用的制备方法为:将步骤(三)得到的化合物4溶于二氯甲烷中,在搅拌下滴加三氟乙酸,三氟乙酸滴加完毕后在搅拌下于室温反应1~3小时,反应完成后将反应液旋干得第一残留物;将第一残留物溶于无水四氢呋喃,再在冰浴、氮气保护条件下加入BH3·THF的四氢呋喃溶液,回流16~24小时,回流结束后将所得液体旋干得第二残留物;将第二残留物用甲醇溶解,在搅拌下于室温向第二残留物的甲醇溶液中加入草酸的甲醇饱和溶液,草酸的甲醇饱和溶液加完后静置至少30分钟,再经过滤,得到的滤渣即为2-氨基-3-[(3,4-苄氧基)-苯基]丙胺;所述化合物4、三氟乙酸、BH3·THF、草酸的摩尔比为:1:(5~10):(3~6):(1~3);
(五)制备化合物6,2-[N,N-二(乙酸苄酯基)-氨基]-3-[(3,4-二苄氧基)-苯基]-N,N-二(乙酸苄酯基)丙胺:本发明中使用的制备方法为:将步骤(四)得到的化合物5混悬于无水乙腈溶剂中,加入溴乙酸苄酯、K2CO3和KI,在搅拌下于40~60℃反应16~24小时,反应完成后对得到的反应液过滤,并将所得滤液旋干得残留物;之后将残留物进行后处理,所述后处理是用二氯甲烷溶解残留物,再依次用稀盐酸洗涤残留物溶液去除K2CO3、用饱和食盐水将残留物溶液洗涤至中性、用无水硫酸镁去除中性残留物溶液中的水分、通过浓缩去除残留物溶液中的溶剂二氯甲烷得后处理产物;将后处理产物经硅胶柱色谱纯化,得到2-[N,N-二(乙酸苄酯基)-氨基]-3-[(3,4-二苄氧基)-苯基]-N,N-二(乙酸苄酯基)丙胺;所述化合物5、溴乙酸苄酯、K2CO3、KI的摩尔比为1:(4~8):(8~16):0.1;
(六)制备化合物7,2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸:将步骤(五)得到的化合物6溶于乙酸乙酯与甲醇按照体积比1:1混合得到的复合溶剂中,再加入化合物6质量10%~50%的钯碳并通入氢气在搅拌下于室温反应16~24小时,反应完成后对得到的反应液过滤,并将所得滤液旋干得残留物,再将残留物经水/乙腈重结晶得到2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸,所述钯碳为催化剂,所述氢气的通入量应满足化合物6完成氢化脱苄反应。
尽管上述制备2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的方法中,化合物2和化合物4可以采用本领域中披露的常规制备方法来制备,然而,常规制备方法中所使用的原料为易爆或者剧毒化合物,存在一些弊端,例如文献中化合物2的制备使用了易爆化学试剂Boc-N3(叠氮碳酸叔丁基酯)、化合物4中的制备使用了剧毒化学试剂氯甲酸乙酯作为偶联剂等。本发明对上述制备工艺进行改进,使用安全、无毒的Boc2O(二碳酸二叔丁酯)和常规多肽缩合剂EDCI作为原料,仍可制备出产率相当的化合物2和化合物4。
上述制备2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的方法中,涉及的重结晶方法为本领域的常规纯化操作,操作过程为:选择合适的良溶剂和不良溶剂,在加热回流条件下,将待处理的化合物用良溶剂溶解,然后滴加不良溶剂至浑浊,再冷却、析出晶体即可。只要选择合适的良溶剂和不良溶剂即可实现对某些化合物的重结晶纯化;上述二氯甲烷/正己烷重结晶方法中,二氯甲烷为良溶剂,正己烷为不良溶剂;上述水/乙腈重结晶方法中,水为良溶剂,乙腈为不良溶剂。另外还有一种用于重结晶的常规物质乙醇,在进行乙醇重结晶操作时,先用乙醇溶解待处理的化合物,加热得到其饱和溶液,然后降温冷却析出即得到重结晶纯化处理的化合物。
上述制备2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的方法中,各步骤溶剂的量以对应的溶质能完全溶解为限。
本发明所述非钆顺磁性金属配合物磁共振造影剂是利用2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸(化合物7)与金属M的+2价或+3价离子配位制成,化学反应式如下:
本发明基于以上化学反应式来制备非钆顺磁性金属配合物磁共振造影剂,步骤如下:
(1)将化合物7和金属M离子化合物分别溶于pH为6~8的Hepes缓冲液中,得到化合物7溶液和金属M离子化合物溶液;
(2)在氮气保护和搅拌下将步骤(1)配制的化合物7溶液和金属M离子化合物溶液加入反应容器,在氮气保护下于室温搅拌反应10~30分钟,反应完成后将所得反应液用凝胶柱过滤除去未参加反应的金属M离子,即得到非钆顺磁性金属离子配合物磁共振造影剂的Hepes溶液,将该溶液冷冻干燥,即得到非钆顺磁性金属离子配合物磁共振造影剂;所述化合物7与金属M离子化合物中的金属M离子的摩尔比为1:(0.8~1.5),可以通过NMR(Nuclear Magnetic Resonance)滴定实验确定;所述金属M离子化合物为Mn、Fe、Eu或Dy的+2价离子化合物,或Mn、Fe、Eu或Dy的+3价离子化合物。
本发明所述非钆金属簇集体磁共振造影剂是利用2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸(化合物7),先与金属M的+2价或+3价离子配位,再与金属M1的+2价、+3价或+4价离子配位组装制成,化学反应式如下:
本发明基于以上化学反应式来制备非钆金属簇集体磁共振造影剂,步骤如下:
(1)将化合物7、金属M离子化合物和金属M1离子化合物分别溶于pH为6~8的Hepes缓冲液中,得到化合物7溶液、金属M离子化合物溶液和金属M1离子化合物溶液;
(2)在氮气保护和搅拌下将步骤(1)配制的化合物7溶液和金属M离子化合物溶液加入反应容器,在氮气保护下于室温搅拌反应10~30分钟,再加入金属M1离子化合物溶液,在氮气保护下于室温搅拌反应10~30分钟,反应完成后,将所得反应液用凝胶柱过滤除去未参加反应的金属M离子和金属M1离子,即得到基于配位自组装的非钆金属簇集体磁共振造影剂的Hepes溶液,将该溶液冷冻干燥,即得到基于配位自组装的非钆金属簇集体磁共振造影剂;所述2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸、金属M离子化合物中的金属M离子、金属M1离子化合物中的金属M1离子的摩尔比为1:(0.8~1.5):0.33;所述金属M离子化合物为Mn、Fe、Eu或Dy的+2价离子化合物,或Mn、Fe、Eu或Dy的+3价离子化合物;所述M1离子化合物为金属Sc、Ti、Mn或Fe的+2价离子化合物,或Sc、Ti、Mn或Fe的+3价离子化合物,或Sc、Ti、Mn或Fe的+4价离子化合物。
上述制备非钆顺磁性金属配合物磁共振造影剂和非钆金属簇集体磁共振造影剂中采用的Hepes缓冲液用于提供合适的pH反应环境,可以从市场上外购(例如友康基业生物科技(北京)有限公司生产的型号为RS0004的Hepes缓冲液)或者由市场外购的Hepes(4-羟乙基哌嗪乙磺酸)按照缓冲液常规配置方法得到(可参考冷泉港实验室给出的制备方法);上述非钆顺磁性金属配合物磁共振造影剂和非钆金属簇集体磁共振造影剂中的阳离子M2来自于配制Hepes缓冲液时所使用的调节pH值的碱性溶液(NaoH或KOH)。
与现有技术相比,本发明具有以下有益效果:
1、本发明提供的非钆顺磁性金属配合物磁共振造影剂,具有较高的弛豫效率,一般Gd类造影剂的弛豫效率为2.5-5.0mmol-1s-1,本发明提供的配合物造影剂的弛豫效率可以达到5.93;上述配合物可进一步通过邻二酚羟基与金属M1配位,自组装形成中等分子量(1500左右)致密的三聚体金属簇集体结构([(MnLH2O)3M1],使得造影剂在溶液旋转变慢,有效延长了旋转恢复时间,从而进一步提高弛豫效率;金属簇集体磁共振造影剂的弛豫效率可以高达12.68mmol-1s-1,相比Gd类造影剂提高了一倍多;
2、由于本发明提供的顺磁性金属配合物磁共振造影剂及金属簇集体磁共振造影剂的配位核心结构为EDTA及邻二酚羟基(Catechol),从而表现出良好的水溶性和结构稳定性;
3、与临床使用的Gd类造影剂相比,使用配位自组装有序金属簇集体磁共振造影剂后,MRI成像效果更佳,能够显示更多细节,为多种疾病诊断提供更加准确的分析依据;
4、本发明所的提供的含有邻二酚羟基的类EDTA配体及非钆顺磁性金属配合物磁共振造影剂、非钆金属簇集体磁共振造影剂的制备方法安全、无毒,并便于工业化生产。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,以下将对实施例或现有技术描述中所需要使用的附图作简单的介绍,显而易见地,以下描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图所示实施例得到其它的实施例及其附图。
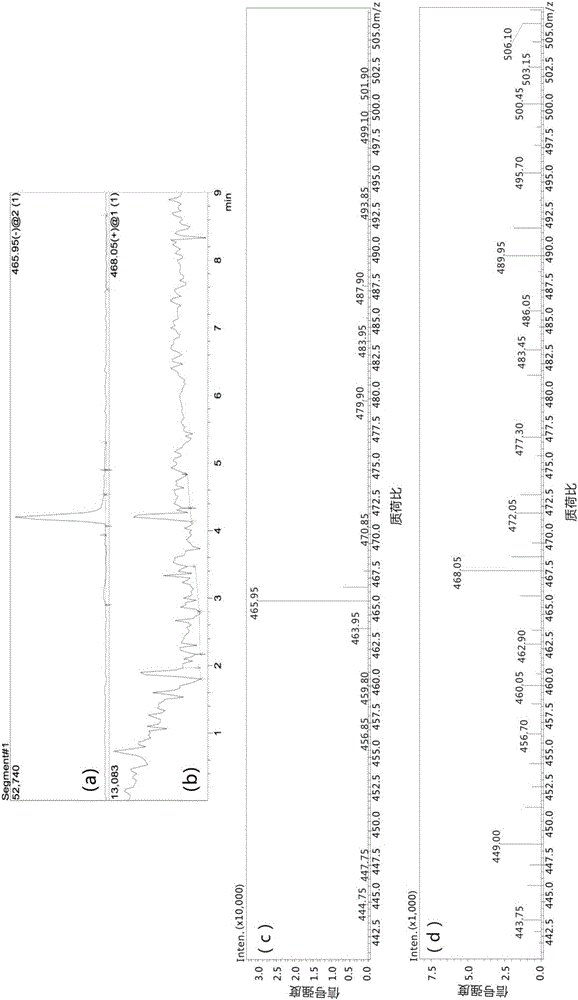
图1为本发明使用LCMS(Shimadzu LCMS2020,色谱质谱联用仪)对实施例2制备的锰配合物(MnL)进行结构表征的的色谱图;
图2为本发明使用LCMS(Shimadzu LCMS2020,色谱质谱联用仪)对实施例2制备的锰配合物(MnL)进行结构表征的的质谱信号总离子图和质谱图;其中(a)为阴离子模式[M+H]-下的质谱信号总离子图,(b)为阳离子模式[M+3H]+下的质谱信号总离子图,(c)为阴离子模式[M+H]-下的质谱图,(d)为阳离子模式[M+3H]+下的质谱图;配体造影剂MnL的阴离子以M记;
图3为本发明实施例2制备的锰配合物[MnL]在20℃、3.0T条件下的纵向弛豫率/锰浓度(mmol)曲线;其中弛豫率R1=1/T1,T1为弛豫时间(s),斜率为纵向弛豫效率r1(mmol-1s-1);
图4为本发明实施例3制备的Fe配位自组装金属簇集体磁共振造影剂[Fe(MnL)3]在20℃、3.0T条件下的纵向弛豫率/锰浓度(mmol)曲线;其中弛豫率R1=1/T1,T1为弛豫时(s))间,斜率为纵向弛豫效率r1(mmol-1s-1);
图5为本发明实施例4制备的Ti配位自组装金属簇集体磁共振造影剂[Ti(MnL)3]在20℃、3.0T条件下的纵向弛豫率/锰浓度(mmol)曲线;其中弛豫率R1=1/T1(s),T1为弛豫时间,斜率为纵向弛豫效率r1(mmol-1s-1);
图6为本发明应用例大鼠尾静脉注射造影剂[Ti(MnL)3](剂量:0.05mmol/Kg)和造影剂(剂量:0.1mmol/Kg)前后不同的时间点MRI图像对比;其中,上面一排为注射造影剂[Ti(MnL)3]注射液后的MRI成像图,下面一排为注射注射液后的MRI成像图;
图7为本发明应用例大鼠尾注射造影剂[Ti(MnL)3](0.05mmol/Kg)左心室、肝脏及左上肢肌肉时间-信号强度曲线(n=5);
图8为本发明应用例大鼠尾注射造影剂(0.1mmol/Kg)左心室、肝脏及左上肢肌肉时间-信号强度曲线(n=5)。
具体实施方式
以下将对本发明各实施例的技术方案结合附图进行清楚、完整的描述,显然,所描述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施例,都属于本发明所保护的范围。
实施例1含有邻二酚羟基的类EDTA配体(L):2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸的制备
化学反应式为:
依据上述化学反应式,本实施例采用的制备方法如下:
(1)制备化合物2,[(N-叔丁氧羰基)-氨基]]-3-[(3,4-二羟基)-苯基]丙酸甲酯:
在冰浴条件下将二氯亚砜(15mL,95.68mmol)加入L-Dopa(14.5g,73.6mmol)的100mL甲醇溶液中,在搅拌下于室温反应24小时,反应完成后将反应液减压蒸馏去除反应液中的溶剂甲醇得产物;将得到的产物用100mL饱和碳酸氢钠溶解且产物溶液pH为7~8,然后在1小时内将二碳酸二叔丁酯Boc2O(20g,91.6mmol)的120mL THF滴入产物溶液中,在搅拌下于室温反应2小时,反应完成后蒸除溶剂THF,将所得水相pH调节至2,再用二氯甲烷萃取三次(每次100mL),合并各次萃取所得有机相,然后用饱和食盐水清洗至中性(pH=7左右)得含水和有机相的多相液体,依次用无水硫酸镁去除多相液体中的水分、通过浓缩去除溶剂二氯甲烷即得到化合物2(18.9g,产率为85%)。
化合物2的结构表征为:1H-NMR(400MHz,CDCl3)δ:6.75(s,1H),6.67(s,1H),6.54(s,1H),5.12(s,1H),4.54(s,1H),3.73(s,3H),2.97(s,2H),1.43(s,9H);13C NMR(101MHz,CDCl3)δ172.94(s),171.18(s),155.07(s),144.49(s),142.85(s),127.85(s),121.18(s),115.74(s),114.98(s),77.01(s),60.48(s),54.57(s),51.81(s),37.84(s),28.29(s),27.64(s),20.98(s),14.17(s),1.00(s);MS:[M-H]-计算值:310.33,测定值:310.19。
(2)制备化合物3,2-[(N-叔丁氧羰基)-氨基]]-3-[(3,4-二苄氧基)-苯基]丙酸:
将(1)得到的化合物2(18.9g,60.7mmol)溶于无水DMF(150mL)中,加入苄氯(21mL,177mmol)、K2CO3(26.4g,191mmol)、KI(0.99g,6.0mmol),在搅拌下于60℃反应24小时;反应完成后将反应液过滤,并将滤液浓缩去除滤液中的溶剂DMF以及未参加反应的苄氯得残留物;之后将残留物进行后处理,残留物的后处理是用乙酸乙酯(150ml)溶解残留物,再依次用10%HCl(50mL)洗涤残留物溶液去除K2CO3,用饱和食盐水将残留物溶液洗涤至中性(pH=7左右),用无水硫酸镁去除中性残留物溶液中的水分,通过浓缩去除残留物溶液中的溶剂乙酸乙酯得后处理产物;将后处理产物用乙醇(120mL)重结晶得到重结晶产物;再将重结晶产物混悬于NaOH(6.4g,160.0mmol)的甲醇-水混合液(体积比1:1,200mL)中,回流1小时,回流结束后冷却至室温,再加入质量浓度为37%的浓盐酸(10mL)得到白色沉淀,过滤出沉淀物并将沉淀物用乙醇(80mL)重结晶得到化合物3(25.3g,产率85%)。
化合物3的结构表征为:1H-NMR(400MHz,CDCl3)δ:7.46(s,10H),6.88(s,1H),6.81(s,1H),6.72(d,J=7.7Hz,1H),5.15(s,4H),4.91(s,1H),4.54(s,1H),3.09(s,2H),1.45(s,9H);MS:[M-H]-计算值:476.55,测定值:476.88。
(3)制备化合物4,2-[(N-叔丁氧羰基)-氨基]]-3-[(3,4-二苄氧基)-苯基]丙酰胺:
将(2)得到的化合物3(25.00g,52.4mmol)溶于无水DMF(150mL)中,在冰盐浴条件下加入EDCI(10.2g,66mmol)、HOBt(8.91g,66mmol)、DIPEA(22mL,127mmol),搅拌1小时;然后通入氨气在搅拌下反应24小时,反应完成后再室温下向反应液中加水(100mL)沉淀得到初产物,过滤出初产物,并将初产物利用二氯甲烷(100mL)/正己烷(50mL)重结晶得到化合物4(12.5g,产率50%)。
化合物4的结构表征为:1H-NMR(400MHz,CDCl3)δ:7.51–7.30(m,10H),6.89(d,J=8.1Hz,1H),6.84(s,1H),6.75(d,J=8.1Hz,1H),5.56(s,1H),5.19(t,J=10.1Hz,5H),3.05(dd,J=13.6,5.7Hz,1H),1.66(s,3H),1.45(s,9H);13C NMR(101MHz,CDCl3)δ:173.41(s),155.36(s),148.78(s),148.04(s),137.25(s),129.71(s),128.55(s),128.52(s),127.85(s),127.83(s),127.42(s),127.32(s),122.27(s),115.99(s),115.12(s),80.31(s),77.36(s),77.25(s),77.05(s),76.73(s),71.28(s),71.01(s),55.45(s),38.00(s),28.32(s),1.05(s),0.03(s);MS:[M+H]+计算值:477.56,测定值:477.55;[M-H]-计算值:475.56,测定值:475.88。
(4)制备化合物5,2-氨基-3-[(3,4-苄氧基)-苯基]丙胺:
将(3)得到的化合物4(3.2g,6.7mmol)溶于二氯甲烷(75mL)中,在磁力搅拌下滴加三氟乙酸(5.0mL,65mmol),三氟乙酸滴加完毕后在搅拌下于室温反应3小时,反应完成后将反应液旋干除去反应液中的二氯甲烷和过量三氟乙酸得第一残留物;将第一残留物溶于无水THF(20mL);再在冰浴、氮气保护条件下加入BH3·THF的四氢呋喃溶液(1.0M,30mL),回流24小时;回流结束后将所得液体旋干除去所得液体中的THF得到第二残留物;将第二残留物用甲醇(5mL)溶解,在搅拌下于室温向第二残留物的甲醇溶液中加入草酸的甲醇饱和溶液(草酸1.0g溶于甲醇10mL制成),草酸的甲醇饱和溶液加完后静置至少30分钟,再经过滤,得到的滤渣即为化合物5(3.2g,产率45%)。
化合物5的结构表征为:1H-NMR(400MHz,CDCl3)δ:7.51–7.30(m,11H),6.90(d,J=8.1Hz,1H),6.80(s,1H),6.72(d,J=8.0Hz,1H),5.17(d,J=7.9Hz,4H),4.15(q,J=7.1Hz,1H),3.68(s,1H),2.90(s,1H),2.83–2.65(m,2H),2.57–2.37(m,2H),2.07(s,1H),1.27(s,1H);13C NMR(101MHz,DMSO-d6)δ:163.76(s),128.88(s),128.09(s),127.94(s),40.60(s),40.39(s),40.19(s),39.98(s),39.77(s),39.56(s),39.35(s);MS:[M+Na]+计算值:385.46,测定值:385.40。
(5)制备化合物6,2-[N,N-二(乙酸苄酯基)-氨基]-3-[(3,4-二苄氧基)-苯基]-N,N-二(乙酸苄酯基)丙胺:
将(4)得到的化合物5(2.4g,5.5mmol)混悬于无水乙腈(30mL)中,加入溴乙酸苄酯(5mL,32mmol)、K2CO3(7.2g,52.1mmol)和KI(0.083g,0.5mmol),在搅拌下于50℃反应24小时;反应完成后对得到的反应液液过滤,并将所得滤液旋干去除滤液中的溶剂乙腈以及未参加反应的溴乙酸苄酯得残留物;之后将残留物进行后处理,后处理是用二氯甲烷(100mL)溶解残留物;再依次用10%HCl(30mL)洗涤残留物溶液去除K2CO3,用饱和食盐水将残留物溶液洗涤至中性(pH=7左右),用无水硫酸镁去除中性残留物溶液中的水分、通过浓缩去除残留物溶液中的溶剂二氯甲烷得后处理产物;将后处理产物经硅胶柱色谱纯化,得到的淡黄色油状物即化合物6(1.2g,产率50%)。
化合物6的结构表征为:1H-NMR(400MHz,CDCl3)δ:7.44–7.27(m,40H),6.80(d,J=1.9Hz,1H),6.76(s,1H),6.74(s,1H),5.26–5.19(m,3H),5.13(s,1H),5.13(dd,J=10.4,6.2Hz,2H),5.11(d,J=4.4Hz,3H),5.07(d,J=4.1Hz,2H),5.05(d,J=3.9Hz,7H),4.73(d,J=16.7Hz,3H),3.58–3.46(m,8H),3.20–3.02(m,2H),2.90(dd,J=13.6,7.2Hz,2H),2.69(dd,J=13.7,6.0Hz,2H),2.64–2.43(m,4H);13C NMR(101MHz,CDCl3)δ:173.24(s),171.80(s),171.23(s)148.65(s),140.85(s),137.46(s),135.75(s),134.99(s),128.73(s),128.72(s),128.61(s),128.56(s),128.54(s),128.44(s),128.35(s),128.29(s),127.72(s),127.33(s),127.03(s),121.92(s),115.11(s),77.37(s),77.25(s),77.05(s),76.73(s),71.38(s),71.01(s),67.36(s),66.29(s),66.16(s),65.45(s),60.69(s),55.29(s),52.82(s),1.05(s),0.03(s);MS:[M+H]+计算值:956.10,测定值:956.26。
(6)制备化合物7,2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸:
将(5)得到的化合物6(200mg,0.2mmol)溶于乙酸乙酯-甲醇复合溶剂(体积比1:1,20mL)中,再加入钯碳(100mg)并通入氢气在搅拌下于室温反应24小时,反应完成后对得到的反应液过滤,并将所得滤液旋干去除滤液中的复合溶剂乙酸乙酯-甲醇得残留物,再将残留物经水(5mL)/乙腈(5mL)重结晶得到化合物7(100mg,产率50%)。
化合物7的结构表征为:1H-NMR(400MHz,D2O)δ:6.84(dd,J=8.3,4.6Hz,1H),6.76(s,1H),6.70–6.63(m,1H),3.79(dd,J=6.1,4.2Hz,2H),3.78–3.72(m,4H),3.62–3.58(m,2H),3.06(dd,J=13.8,5.1Hz,2H),2.90(dd,J=15.4,8.4Hz,1H),2.70–2.59(m,1H);13C NMR(101MHz,D2O)δ174.58(s),170.51(s),144.84(s),142.96(s),128.19(s),121.58(s),116.23(s),60.94(s),55.46(s),54.61(s),52.78(s),32.03(s);MS:[M+H]+计算值:414.36,测定值:414.20。
以下实施例2至实施例3中采用的Hepes缓冲液(50mM)的配制方法为:准确称取Hepes(4-羟乙基哌嗪乙磺酸,MW=238.31)5.967g,溶解于400ml蒸馏水中,再加入0.5M的NaOH水溶液调节pH至7.36,然后用蒸馏水定容至500ml,于4℃保存备用。
实施例2顺磁性金属配合物磁共振造影剂(MnL)的制备
化学反应式为:
依据上述化学反应式,本实施例采取的制备方法如下:
将实施例1制备的化合物7(0.1g,0.24mmol)溶于Hepes缓冲液(pH=7.36,5mL)中得到化合物7溶液,将MnCl2·4H2O(0.057g,0.28mmol)溶于Hepes缓冲液(pH=7.36,5mL)得到MnCl2溶液;在氮气保护和搅拌下,将化合物7溶液和MnCl2溶液加入反应容器,在氮气保护下于室温搅拌反应10min,反应完成后将所得反应液用Sephdex G-25凝胶柱过滤除去未参加反应的Mn2+,得到结构式(10)的Mn配合物造影剂(MnL)的Hepes溶液。
本实施例使用LCMS(Shimadzu LCMS2020,色谱质谱联用仪)对本实施例制备的配合物造影剂(MnL)进行色谱检测和质谱检测,进而对配合物造影剂(MnL)的结构进行表征。
(1)结构表征
色谱检测条件:尺寸为100mm×2mm的Luna C18色谱柱;洗脱液A:含有质量分数为0.1%甲酸的水溶液;洗脱液B:含有质量分数为0.1%甲酸的乙腈溶液;变化梯度为:在9min内,洗脱液B浓度从5%递变到95%;流速为0.8mL/min;检测波长为280nm。从图1中可以看出,配合物造影剂(MnL)的保留时间TR(retention time)=4.183min。
ESI质谱检测结果:如图2(a)和图2(b)所示,阴离子模式和阳离子模式下的质谱信号总离子图中保留时间与图1中给出的基本一致;由2(c)得出,阴离子模式下,质荷比(m/z)为465.95,对应[M+H]-(配合物带一个负电),这与[M+H]-的理论计算值466.04基本一致(其中M的理论计算值由配体与金属离子Mn2+理论推导得出);由图2(d)得出,阳离子模式下,质荷比(m/z)为468.05,对应[M+3H]+(配合物带一个正电),这与[M+3H]+理论计算值468.06基本一致。
由此,本实施例制备的配合物造影剂(MnL)阴离子具有如下结构式(11),其色谱检测保留时间在上述条件下TR为4.183min。
(2)弛豫性能表征
为了研究造影剂(MnL)的弛豫效率,对MnL造影剂进行弛豫性能测试(体外溶液测试),测试过程为:将造影剂MnL溶于水中,配制成5个不同Mn浓度梯度的水溶液,分别为0.1mmol/L、0.2mmol/L、0.3mmol/L、0.4mmol/L和0.5mmol/L(该浓度通过ICP-MS测定),分别置于5个2mL离心管中,在20℃条件下,用3.0T磁共振扫描仪进行扫描(采用快速自旋回波序列,回波时间TE=5ms,重复时间TR=300ms)并计算造影剂MnL的弛豫时间T1的弛豫效率,MnL的纵向弛豫率/锰浓度曲线如图3所示,从图中可以看出,R1与Mn浓度呈线性关系;其斜率即为纵向弛豫效率r1,为5.93mM-1s-1。
实施例3 Fe配位自组装金属簇集体[Fe(MnL)3]磁共振造影剂的制备
化学反应式为:
依据上述化学反应式,实施例采取的制备方法如下:
将实施例1制备的化合物7(0.1g,0.24mmol)溶于Hepes缓冲液(pH=7.36,5mL)中得到化合物7溶液,将MnCl2·4H2O(0.057g,0.28mmol)溶于Hepes缓冲液(pH=7.36,5mL)中得到MnCl2溶液,将FeCl3(0.013g,0.08mmol)溶于Hepes缓冲液(pH=6.0,2.0mL)得到FeCl3溶液;在氮气保护和搅拌下将化合物7溶液和MnCl2溶液加入反应容器,在氮气保护下于室温搅拌反应10min;再加入FeCl3溶液,在氮气保护下于室温搅拌反应10min,反应完成后,将所得反应液用Sephdex G-25凝胶柱过滤除去未参加反应的Mn2+和Fe3+,得到结构式(12)的造影剂[Fe(MnL)3]的Hepes溶液。
为了研究造影剂[Fe(MnL)3]的弛豫效率,对造影剂[Fe(MnL)3]进行弛豫性能测试(体外溶液测试),测试过程为:将造影剂[Fe(MnL)3]溶于水中,配制成5个不同Mn浓度梯度的水溶液,分别为0.1mmol/L、0.2mmol/L、0.3mmol/L、0.4mmol/L和0.5mmol/L(该浓度通过ICP-MS测定),分别置于5个2mL离心管中,在20℃条件下,用3.0T磁共振扫描仪进行扫描(采用快速自旋回波序列,回波时间TE=5ms,重复时间TR=300ms)并计算造影剂[Fe(MnL)3]的弛豫时间T1弛豫效率,[Fe(MnL)3]的纵向弛豫率/锰浓度曲线如图4所示,从图中可以看出,R1与Mn浓度呈线性关系;其斜率即为纵向弛豫效率r1,为12.68mM-1s-1。
实施例4 Ti配位自组装金属簇集体[Ti(MnL)3]磁共振造影剂的制备
化学反应式为:
依据上述化学反应式,实施例采取的制备方法如下:
将实施例1制备的化合物7(0.1g,0.24mmol)溶于Hepes缓冲液(pH=7.36,5mL)中得到化合物7溶液,将MnCl2·4H2O(0.057g,0.28mmol)溶于Hepes缓冲液(pH=7.36,5mL)中得到MnCl2溶液,将TiO(acac)2(乙酰丙酮氧钛0.021g,0.08mmol)溶于Hepes缓冲液(pH=7.36,2mL)中得到TiO(acac)2溶液;在氮气保护和搅拌下,将化合物7溶液和MnCl2溶液加入反应容器中,在氮气保护下于室温搅拌反应10min,再加入TiO(acac)2溶液,在氮气保护下于室温搅拌反应10min,反应完成后,将所得反应液用Sephdex G-25凝胶柱过滤除去未参加反应的Mn2+和Ti4+,得到结构式(13)的造影剂[Ti(MnL)3]的Hepes溶液。
为了研究造影剂[Ti(MnL)3]的弛豫效率,对造影剂[Ti(MnL)3]进行弛豫性能测试(体外溶液测试),测试过程为:将造影剂[Ti(MnL)3]溶于水中,配制成5个不同Mn浓度梯度的水溶液,分别为0.1mmol/L、0.2mmol/L、0.3mmol/L、0.4mmol/L和0.5mmol/L(该浓度通过ICP-MS测定),分别置于5个2mL离心管中,在20℃条件下,用3.0T磁共振扫描仪进行扫描(采用快速自旋回波序列,回波时间TE=5ms,重复时间TR=300ms)并计算造影剂[Ti(MnL)3]的弛豫时间T1弛豫效率值,[Ti(MnL)3]的纵向弛豫率/锰浓度曲线如图5所示,从图中可以看出,R1与Mn浓度呈线性关系;其斜率即为纵向弛豫效率r1,为9.52mmol-1s-1。
实施例2-实施例4得到的造影剂(MnL)、[Fe(MnL)3]、[Ti(MnL)3]均具有较高的弛豫效率,特别是配位自组装得到的金属簇集体磁共振造影剂[Fe(MnL)3]、[Ti(MnL)3的效率更高,[Fe(MnL)3]的弛豫效率高达12.68mmol-1s-1,是传统Gd类造影剂(相同条件下测定的为2.5~5mmol-1s-1)的两到五倍。
应用例大鼠MRI成像及定量分析
(1)实验准备
实验对象:SD大鼠;
麻醉剂:质量浓度为3%的戊巴比妥钠;
实验组造影剂[Ti(MnL)3]注射液:将实施例4制备的造影剂[Ti(MnL)3]的Hepes溶液冷冻干燥得到褐色固体90mg。将上述固体溶于注射用水(2.0mL),制得实验组造影剂注射液:0.03mmol/mL。
对照组造影剂注射液:注射液;的结构式如下:
准备过程:将16只SD大鼠随机分成2组,每组8只,雌雄各半。检查前禁食6~12小时,禁水4h。用3%戊巴比妥钠腹腔内注射进行麻醉,麻醉剂量为50mg/Kg。麻醉后,尾静脉植入头皮针,仰卧位固定。实验组注射通过实施例4制备的金属簇集体磁共振造影剂[Ti(MnL)3]注射液,对照组注射注射液。注射剂量为:实验组每公斤体重0.05mmol造影剂[Ti(MnL)3](0.05mmol/Kg BW);对照组每公斤体重0.1mmol造影剂(0.1mmol/Kg BW)。
(2)MRI成像参数及方法
采用GE 3.0T超导磁共振成像仪及动物线圈。平扫及增强T1WI:肝脏容积加速采集序列(liver acquisition with volume acceleration,LAVA),冠状位、轴位。扫描参数:TR=5ms,TE=2ms,层厚1.6mm,层间距-0.8mm,扫描视野(FOV)14×11.2mm,激励次数(NEX)0.69,矩阵(Matrix)192×192像素,激发角度(Flip)12°,带宽(Bandwidth)100Hz。
实验过程:注射造影剂前平扫2次,然后尾静脉注射造影剂后动态增强扫描,于1min、5min、10min、15min、20min、25min、30min、35min、40min时间点用相同参数序列进行MRI成像,动态观察造影剂在体内大鼠的显影情况;观察大鼠48h有无死亡。
(3)MRI定量分析
用GE ADW4.4后处理工作站分别测量两组大鼠左心室、脑实质、肝脏及左前肢肌肉第2次平扫和增强后1min、5min、10min、15min、20min、25min、30min、35min、40min时间点的T1WI信号强度(SI)。测量各组织器官时,选择同一层面不同时间点的扫描序列,尽量选取相同部位,感兴趣区(regions of interest,ROI)大小至少5mm2,测量时避开血管区。整理数据并绘制部分组织器官平扫及增强后的时间-信号强度曲线。分别比较实验组和对照组左心室、肝脏及左上肢肌肉平扫和增强后各个时间点的T1WI信号值。
图6给出了大鼠尾静脉注射造影剂[Ti(MnL)3]和造影剂前后不同的时间点MRI图像对比。如图所示,注射[Ti(MnL)3]造影剂后,大鼠心脏及血管信号在1.0min明显增强,清晰可见,随着造影剂循环分布心血管信号逐渐降低;肝脏在5.0min即出现强化,15.0min时强化更明显,且一直持续到40.0min;大鼠在注射造影剂后,48h未见大鼠死亡。由此,可以看出,注射本实施例4提供的造影剂[Ti(MnL)3]相比现有Gd造影剂,在剂量为其一半时(0.05Vs 0.1mmol/kg),其MRI可以达到或具有更佳的成像效果,尤其能够在肝脏显示更多细节,为多种疾病诊断提供更加准确的分析依据。
图7和图8分别给出大鼠尾注射造影剂[Ti(MnL)3]和造影剂左心室、肝脏及左上肢肌肉时间-信号强度曲线(n=5);从图中可以看出,注射造影剂[Ti(MnL)3]各检测部位的信号强度均与造影剂相当,特别是随着时间的延长,在15min以后,造影剂Ti(MnL)3表现出肝靶向性且持续增强,说明造影剂[Ti(MnL)3]在进行肝脏检查时具有明显的优势。
综上所述,本发明合成了具有邻苯二酚(catechol)结构的类EDTA配体2,2’,2”,2”’-((3-(3,4-二羟基苯基)丙烷-1,2二基)二胺四乙酸,基于该配体配位螯合得到的EDTA-Mn顺磁性配合物可与Fe3+、Ti4+等过渡金属通过强的邻苯二酚-金属配位作用自组装形成中等分子量(1500左右)致密的三聚体结构([(MnLH2O)3M],M=Fe,Ti)。该配位组装体在溶液中有着较慢的旋转恢复时间,同时保持了较快地配位水分子交换速率,从而表现出优异的弛豫性能。动物体内成像实验表明:以临床最常用的钆造影剂,DTPA-Gd为对照,三聚体结构([(MnLH2O)3Ti]注射剂量为DTPA-Gd的一半(0.05mmol/kg)),多组织器官对比增强效果明显如心脏、血管、肝脏和肾脏,且肝脏持续增强效果明显,可以作为一种潜在的肝靶向非钆磁共振造影剂。
本领域的普通技术人员将会意识到,这里的实施例是为了帮助读者理解本发明的原理,应被理解为本发明的保护范围并不局限于这样的特别陈述和实施例。本领域的普通技术人员可以根据本发明公开的这些技术启示做出各种不脱离本发明实质的其它各种具体变形和组合,这些变形和组合仍然在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!