双基因敲除的减毒单核细胞增生李斯特氏菌及其制备方法与流程
本发明属于微生物学领域,涉及一种李斯特氏菌,具体来说是一种双基因敲除的减毒单核细胞增生李斯特氏菌及其制备方法。
背景技术:
:
单核细胞增生李斯特氏菌(Listeria monocytogenes,Lm)是一种革兰氏阳性兼性厌氧胞内寄生条件致病菌。该菌可穿越肠道屏障、血脑屏障和胎盘屏障,引起人和动物的胃肠炎、脑膜炎、败血症。由于Lm对环境的耐受能力极强并可以形成菌膜,因此其广泛存在于自然界中,且容易通过生产和加工过程污染蔬菜、牛奶和肉制品等食品。其作为一种常见的食源性致病菌,所引起的食品安全问题已经引起全世界的广泛关注。然而,随着Lm全基因组测序工作的完成,Lm逐渐成为国外学者在基因工程疫苗方面的研究热点,而国内在Lm作为疫苗载体的研究方面鲜有报道。以“Listeria monocytogenes+vaccine”为关键词在PubMed进行检索发现,截止2015年09月01日为止,共有785篇关于Lm作为疫苗载体的研究报道,其中近五年相关研究近200篇。迄今为止减毒Lm活载体疫苗已应用于人类免疫缺陷病毒(HIV)、人类乳头瘤病毒(HPV)和转移性胰腺癌等人类复杂疾病的免疫治疗研究中。
Lm作为疫苗载体能够同时引起MHCⅠ和MHCⅡ类抗原递呈系统,具有显著激发强烈的CD8+和CD4+细胞免疫的能力。然而野生型Lm对宿主毒性太强,因此在Lm作为疫苗载体之前需要进行减毒。目前研究发现Lm的毒力主要由六个毒力因子编码基因(prfA-plcA-hly-mpl-actA-plcB)组成。plcA和plcB分别编码磷脂酰肌醇特异性磷脂酶(PI-PLC)和卵磷脂酶(PC-PLC),有助于Lm逃离吞噬小体;actA与基于Lm肌动蛋白的运动性有关;hly则与Lm的溶血素有关,参与单增李斯特氏菌一级和次级吞噬小体的逃离;prfA主要调控Lm的侵袭相关毒力因子,如inlA和inlB等;mpl是一个依赖锌的金属蛋白酶基因。
技术实现要素:
:
针对现有技术中的上述技术问题,本发明提供了一种双基因敲除的减毒单核细胞增生李斯特氏菌及其制备方法,所述的这种双基因敲除的减毒单核细胞增生李斯特氏菌及其制备方法要解决现有技术中的单核细胞增生李斯特氏菌由于对宿主毒性太强,影响作为疫苗载体的使用的技术问题。
本发明提供了一种双基因敲除减毒单核细胞增生李斯特氏菌,其中,在单核细胞增生李斯特氏菌中敲除了actA和inlB两个毒力因子。
进一步的,所述的双基因敲除减毒单核细胞增生李斯特氏菌的保藏号为:CCTCC NO:M 2016282。
本发明还提供了上述的一种双基因敲除减毒单核细胞增生李斯特氏菌的制备方法,包括如下步骤:
1)分别用SEQ ID NO:1所示的引物actA-P1、SEQ ID NO:2所示的引物actA-P2、SEQ ID NO:3所示的引物actA-P3、SEQ ID NO:4所示的引物actA-P4扩增Lm EGDe基因组actA基因的上游片段P1/P2、下游片段P3/P4;
2)采用限制性内切酶SmaI和XbaI、XbaI和SalI双酶切actA基因的上下游片段,然后使用T4DNA连接酶连接上下游片段,获得片段P1/P4;
3)使用SmaI和SalI同时对P1/P4片段和穿梭质粒pKSV7进行双酶切,T4DNA连接酶连接双酶切后的产物,得到重组质粒;
4)将获得的重组质粒导入感受态细胞DH5α中,同时导入空载体pKSV7作对照,PCR鉴定并使用SmaI和SalI对扩增得到的重组质粒进行双酶切鉴定,将测序正确的阳性克隆命名为重组质粒pKSV7-actA(P1/P4);
5)一个制备Lm-inlB感受态细胞的步骤;
6)采用终浓度为5μg/ml的青霉素G和3.5×SMHEM溶液处理Lm-inlB感受态细胞,将感受态细胞浓度控制在1~2×1011cells/ml,电转后将Lm-inlB感受态细胞转入BHI培养基中振荡培养,离心收集菌体后涂布于含10μg/mL氯霉素的BHI平板于30℃培养,挑取菌落进行PCR鉴定,所述的引物为SEQ ID NO:1所示的引物actA-P1和SEQ ID NO:4所示的引物actA-P4,鉴定结果为阳性的菌株即为转化成功的菌株;
7)将转化成功的菌株转接到BHI液体培养基中41℃连续培养并转接8-10次,将末代细菌培养物划线于BHI平板上41℃过夜培养;挑取BHI平板上的单菌落于BHI液体培养基中30℃连续培养并转接6-8次,取末代培养物划线于BHI平板;
8)利用SEQ ID NO:3所示的引物actA-P3、SEQ ID NO:4所示的引物actA-P4、SEQ ID NO:5所示的引物actA-F、SEQ ID NO:6所示的引物actA-R及涂布无抗性平板对突变株进行鉴定,获得敲除了actA和inlB两个毒力因子的减毒单核细胞增生李斯特氏菌。
进一步的,所述的BHI液体培养基中含10μg/ml氯霉素。
本发明的一种双基因敲除的减毒单核细胞增生李斯特氏菌,其分类命名为:单核细胞增生李斯特氏菌EGD-e(actA-inlB)Listeria monocytogenes EGD-e(actA-inlB),该菌株保藏于中国典型培养物保藏中心(CCTCC)中,中国典型培养物保藏中心的地址为:中国、武汉、武汉大学,保藏日期为2016年5月26号,保藏号为CCTCC NO:M 2016282。
本发明利用同源重组技术对模式菌株EGDe单基因缺失菌株Lm-inlB进行二次减毒,构建了减毒后的疫苗载体----减毒李斯特氏菌Lm-actA/inlB。然后从生长状态、毒力基因表达水平和对HepG2细胞侵袭能力方面比较减毒菌株与野生型菌株之间的生物活性。
本发明和已有技术相比,其技术进步是显著的。本发明通过敲除actA和inlB两个毒力因子,使得减毒单核细胞增生李斯特氏菌可以作为疫苗载体,为食源性致病菌Lm致病机理研究提供基础。
附图说明:
图1是Lm-actA/inlB的构建原理图。
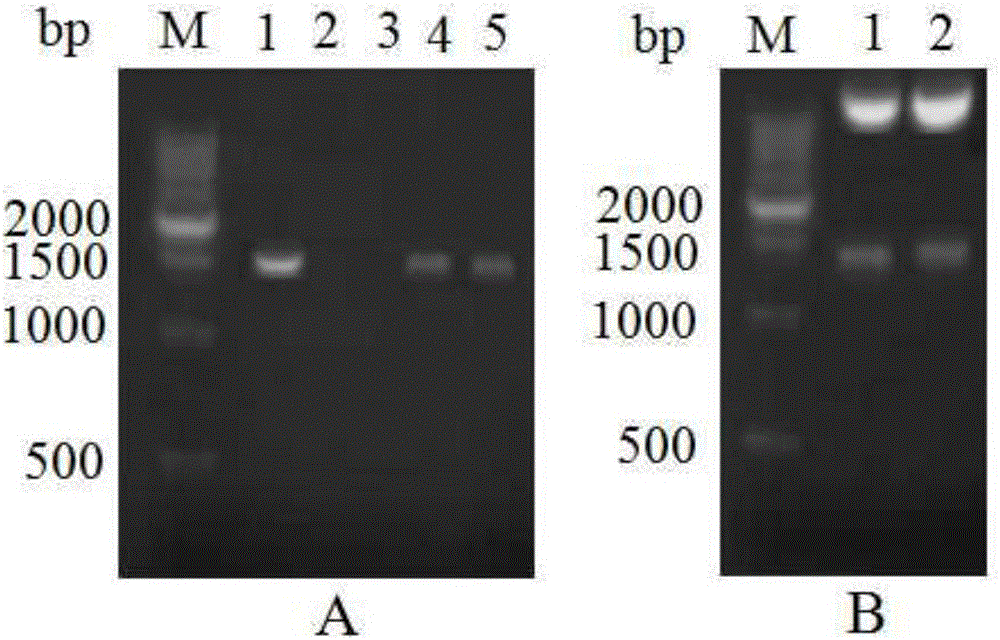
图2是重组质粒pKSV7-actA(P1/P4)鉴定结果;3-A PCR结果:M:DNA marker;1:actA(P1/P4);2:H2O;3:空载体;4-5:pKSV7-actA(P1/P4);3-B双酶切结果:M:DNA marker;1-2:pKSV7-actA(P1/P4)。
图3是减毒株Lm-actA/inlB的鉴定结果;3-A引物actA-F/actA-R的PCR结果;3-B引物actAP1/actAP4的PCR结果:1:H2O;4、5:阴性对照;2、3、6:Lm-actA/inlB;7:Lm(EGD-e)。
图4是Lm(EGD-e)和Lm-actA/inlB生长曲线。
图5是Lm-actA/inlB各毒力基因的表达水平。
图6是各菌株对HepG2细胞侵袭的菌落总数。
具体实施方式:
实施例1 双基因敲除的减毒单核细胞增生李斯特氏菌的构建方法
分别用引物actA-P1/actA-P2和actA-P3/actA-P4(表1)扩增EGDe基因组actA基因的上游片段P1/P2、下游片段P3/P4;
表1 Lm-actA/inlB构建及鉴定所需引物
限制性内切酶SmaI和XbaI、XbaI和SalI双酶切actA基因的上下游片段后使用T4DNA连接酶连接上下游片段,获得片段P1/P4;使用SmaI和SalI同时对P1/P4片段和穿梭质粒pKSV7进行双酶切;T4DNA连接酶连接双酶切后的产物,得到重组质粒;将获得的重组质粒导入DH5α中,同时导入空载体pKSV7作对照,PCR鉴定并使用SmaI和SalI对扩增得到的重组质粒进行双酶切鉴定。鉴定为阳性的克隆由上海华大基因科技有限公司测序,将测序正确的阳性克隆命名为重组质粒pKSV7-actA(P1/P4)。
感受态细胞DH5α的制备:从LB平板上挑取新鲜活化的E.coli单菌落接种于5ml LB液体培养基中,37℃,200rpm培养至稳定期;取1ml稳定期的菌液接种于100ml LB液体培养基中,37℃,200rpm培养3-4h至OD600=0.5左右,转入已经灭菌的50ml离心管中,冰浴10-20min后,于4℃,4000rpm,离心5min,弃上清,沉淀用10ml预冷的0.1M CaCl2悬浮,之后置冰上放置30min,4℃,4000rpm,离心5min,弃上清,沉淀用4ml预冷的含15%甘油的0.05MCaCl2轻轻重悬菌体至均匀,分装100μl置小离心管,冰浴2-7h后,即可用于转化;也可置于-80℃保存。
制备Lm-inlB感受态细胞,取1mL过夜培养的Lm-inlB菌液加入49mL新鲜BHI液体培养基中,37℃培养至OD600=0.4左右,加入青霉素至终浓度为5μg/ml,37℃培养至OD600=0.6左右,在4℃、6000rpm条件下离心10min,用5mL3.5×SMHEM(称取18.9g蔗糖;0.0967gHEPES;25mMMgcl2 5mL。双蒸水定容至50mL,0.2μm孔径的滤膜过滤除菌,置4℃冰箱预冷)溶液洗涤2次。最后用3.5×SMHEM溶液重悬,将感受态细胞浓度控制在1~2×1011cells/ml,分装1.5ml离心管中,每管200μl即为Lm-inlB感受态细胞,该细胞可立即用于后续试验,也可保存于-80℃。
然后用终浓度为5μg/ml的青霉素G和3.5×SMHEM溶液处理Lm-inlB感受态细胞菌体,将感受态细胞浓度控制在1.1×1011cells/ml左右。电转化法将重组质粒pKSV7-actA(P1/P4)导入Lm-inlB感受态细胞,电转条件为:使用0.2cm电击杯,每200μL感受态细胞加入约1000ng的质粒DNA,电转场强为11.25-12.5kV,电击时间为3-4ms。通过温度和氯霉素抗性压力进行同源重组。即将转化成功的菌株转接到BHI(含10μg/ml氯霉素)液体培养基中41℃连续培养并转接8-10次,将末代细菌培养物划线于BHI(含10μg/ml氯霉素)平板上41℃过夜培养;挑取BHI(含10μg/ml Cam)平板上的单菌落于BHI液体培养基中30℃连续培养并转接6-8次,取末代培养物划线于BHI平板。利用引物actA-F/actA-R、actAP1/actAP4(表1)及涂布无抗性平板对突变株进行鉴定,鉴定结果为阳性的菌株命名为Lm-actA/inlB。减毒株Lm-actA/inlB的构建如图1所示。
上述菌株命名为:单核细胞增生李斯特氏菌EGD-e(actA-inlB)Listeria monocytogenes EGD-e(actA-inlB),保藏编号为:CCTCC NO:M 2016282。
实施例2 本发明对菌株的鉴定方法包括如下步骤:
使用actA-P1/actA-P4引物PCR,鉴定actA(P1/P4)片段与质粒pKSV7的连接;使用SmaI和SalI,对扩增得到的重组质粒进行双酶切鉴定,鉴定为阳性的克隆由上海华大基因科技有限公司测序,将测序正确的阳性克隆命名为重组质粒pKSV7-actA(P1/P4)。电转化后利用引物actA-F/actA-R、actAP1/actAP4(表1)及涂布无抗性平板对突变株进行鉴定,鉴定结果为阳性的菌株命名为Lm-actA/inlB。
本发明菌株的鉴定结果如下:
1.重组质粒的鉴定结果:
使用actA-P1/actA-P4引物PCR鉴定。图2-A显示,泳道4、5表明actA上下游连接片段actA(P1/P4)已经成功插入质粒pKSV7中。使用SmaI和SalI对扩增得到的重组质粒进行双酶切鉴定,如图2-B所示,成功从重组质粒上酶切下和片段actA(P1/P4)一致大小(1500bp)的片段。进一步的DNA测序结果表明插入的片段与Genebank序列相似度为100%。
2.减毒株Lm-actA/inlB的鉴定结果:
如图3-A所示,使用引物actA-F/actA-R进行菌落鉴定时,预期突变株2、3、6泳道没有扩增条带,7泳道Lm(EGD-e)作为对照扩增出与预期大小(750bp)一致的条带;使用引物actAP1/actAP4对疑似突变株进行PCR鉴定,图3-B表明突变株与7泳道Lm(EGD-e)相比,2、3、6泳道有部分基因(750bp)缺失。结合3-A、3-B两图,突变株2、3、6为actA基因缺失的阳性转化子。
3.抗体平板鉴定结果:
阳性转化子分别涂布于抗性平板和非抗性平板,在含氯霉素的抗性平板上无菌落生长,在无氯霉素抗性的BHI平板上有菌落生长。
综合以上鉴定结果说明质粒携带的外源基因已经与自身基因组完成基因重组,并且质粒已经丢失,故将阳性转化子命名为Lm-actA/inlB。
本发明使用酶标仪测定12h内Lm(EGD-e)和Lm-actA/inlB在OD600下的生长曲线,以1%的接种量接种至200mL BHI培养基中,在37℃下200rpm摇床上培养,每隔1h取200μL菌液加入96孔板中测定OD600。比较actA和inlB基因缺失后的菌株与Lm(EGD-e)的活力是否存在明显的不同。
测定结果如下:图4表明Lm-actA/inlB在双基因缺失后菌落生长趋势与野生株Lm(EGD-e)完全一致,双基因的缺失并未对菌株的生长造成影响。
本发明为考察Lm(EGD-e)在缺失actA和inlB基因后其毒力基因的表达水平,提取Lm(EGD-e)、Lm-actA/inlB RNA后反转录成cDNA,以Lm(EGD-e)各毒力基因的表达水平作基准,RT-PCR分别检测Lm-actA/inlB中actA、inlA、inlB、plcB、hly基因的表达水平,各基因RT-PCR的引物序列如表2所示。
表2 RT-PCR所需引物
具体方法为:取5mL过夜培养的菌液中加入1mL预冷的苯酚乙醇混合液(苯酚:乙醇=1:9),冰浴30min;收集菌体后加入50μL变溶菌素(250U/mL)和50μL溶菌酶(25mg/mL)超声破碎30min;离心弃上清并加入1mL Trizol,离心后转移上清并加入200μL三氯甲烷,强烈震荡15s呈乳浊液后室温放置2min;离心转移上层水相并加入等体积异丙醇后缓慢摇动,室温放置5-10min;离心弃上清后加入1mL 75%乙醇清洗沉淀,晾干乙醇,用10-30μL DEPC水溶解沉淀。提取获得的RNA根据Takara反转录试剂盒(Takara Code:DRR047A)进行反转录成cDNA(其中包括基因组DNA的去除反应),以cDNA为模板进行SYBR Green荧光染料法RealTime-PCR(具体步骤见表3和表4)。
表3 反转录反应
1.基因组DNA的去除反应。
2.反转录反应
表4 RT-PCR反应
计算方法为:各毒力基因的CT值减去内参基因16S的CT,消减不平衡误差,扩增效率统一为2,各基因的计算数值表示Lm-actA/inlB相比与Lm(EGD-e)的表达倍数。
检测结果为:在actA和inlB基因缺失后,两者的表达水平几乎为零,plcB和hly基因的表达水平分别上升至原来的8倍和3倍,inlA基因的表达水平变化较小(图5)。
实施例3
本发明为考察Lm(EGD-e)在缺失actA和inlB基因后对人肝癌细胞株HepG2侵袭能力的变化,对细菌和细胞进行计数,调整浓度使得细菌和细胞分别为108CFU/mL和106个/mL,调整比例为细菌数:细胞数=100:1后进行细菌侵袭细胞实验。侵袭之前将细胞培养板中换成不含血清的不完全培养基,排除血清对侵袭实验的干扰。放入细胞培养箱分别侵袭5h,侵袭结束后加入0.5mL700μg/mL的庆大霉素,放置于细胞培养箱处理30min充分灭活胞外细菌。使用PBS清洗庆大霉素后加入0.5mL 1%Triton-X-100,室温处理30min,充分破碎细胞后稀释涂布BHI平板,37℃培养进行平板计数。
计算结果为:Lm(EGD-e)在缺失actA和inlB基因后对HepG2细胞侵袭能力显著降低(图6)。
- 还没有人留言评论。精彩留言会获得点赞!