一种可以裂解大肠杆菌及沙门氏菌的噬菌体裂解酶的制备方法与流程
本发明涉及动物生物医药工程技术领域,更具体地,涉及一种可以裂解大肠杆菌及沙门氏菌的噬菌体裂解酶的制备方法。
背景技术:
大肠杆菌(Escherichia coli)既是人和动物肠道中的一种正常栖息菌,同时也是一种重要的致病菌。大肠杆菌大致可分为肠道非致病性共生菌株、肠道致病性菌株和肠道外致病性菌株三大类。作为一种人和动物常见的致病菌,它可导致肠道感染、尿道感染、全身性感染和其它临床感染等。其中肠道外致病性菌株可引起严重的肠道外感染,尤其是尿道感染,并且在抗生素耐药性传播中扮演重要角色。在大肠杆菌多重耐药性菌株中,肠道外致病性菌株比例居多。多重耐药性问题的出现,给大肠杆菌感染的防控增添了不少难题。
沙门氏菌(Salmonella)是革兰氏阴性肠道菌,是一种重要的食源性疾病的病原,已经在世界范围内造成重大的经济损失并严重威胁到公共卫生安全。由沙门氏菌感染导致的沙门氏菌病是一种食源性人畜共患病,临床症状包括肠胃炎、菌血症、伤寒症和病灶感染等;肠胃炎症状可从温和型腹部不适到呕吐脱水,甚至出现死亡。人经常因为摄入被污染的动物源食品而感染沙门氏菌,如摄入被沙门氏菌污染的鸡蛋。在全球范围内,每年由沙门氏菌感染导致的肠胃炎案例估计可达93,800,000例,死亡人数达155,000人之多,其中由食物传播引起的案例占85%左右。
在大肠杆菌耐药现状中,β-内酰胺类抗生素耐药问题尤为严重。这类菌株的耐药机制主要是因为β-内酰胺酶的存在,该酶能够降解β-内酰胺类抗生素。近年来,在肠道外致病性菌株中出现了新的β-内酰胺酶,如质粒介导的AmpC β-内酰胺酶、广谱β-内酰胺酶和碳青霉烯酶等。最出名的新型β-内酰胺酶于1983年首次被报道,并被命名为广谱β-内酰胺酶。这类酶能够降解青霉素类、头孢菌素类和单环β-内酰胺类抗生素,但不能降解头霉素和碳青霉烯类抗生素,其活性可被典型的β-内酰胺酶抑制剂所抑制。碳青霉烯抗性是最近出现的新问题,主要是由质粒编码的碳青霉烯酶所导致的,且目前碳青霉烯抗性主要出现在医院的大肠杆菌菌株,给大肠杆菌感染的防治造成不小的难题。
沙门氏菌耐药性问题也日益严重,多重耐药菌株层出不穷,对红霉素、氨苄青霉素、复方新诺明、环丙沙星等的耐药性均见报道。规模化猪场的沙门氏菌耐药性问题也比较严峻,某研究者对分离自猪场粪样的沙门氏菌进行耐药性分析,发现分离菌株对四环素、环丙沙星、恩诺沙星、阿莫西林、卡那霉素、氟苯尼考和氨苄西林等表现出高度耐药。目前,多重耐药菌株的广泛流行,以及流行菌株不断产生的对喹诺酮类、第三代头孢菌素类等临床重要抗生素的耐药性,成为世界范围内新兴的棘手问题。因此,新型抗菌制剂的研发显得极为迫切。
噬菌体广泛存在于环境中,是以真菌、细菌等微生物为宿主的病毒,烈性噬菌体具有高效的杀菌能力。噬菌体在侵染周期的末期合成一种噬菌体裂解酶,裂解酶破坏宿主的细胞壁,使得宿主细胞渗透压失衡并最终破裂而达到释放子代噬菌体的目的。噬菌体及其裂解酶对抗生素耐药菌同样具有杀灭作用,通过基因工程技术,可以大量地人工合成噬菌体裂解酶。因此,噬菌体及其裂解酶具有巨大的应用前景。
技术实现要素:
本发明所要解决的技术问题是寻求新的可同时抑制大肠杆菌和沙门氏菌的抗菌制剂,以期减少和替代抗生素的使用,提供表达能裂解大肠杆菌及沙门氏菌的噬菌体重组裂解酶TrxA-lysin的重组大肠杆菌BL21 /pET-32a-lysin的制备方法。
本发明的第二个目的是提供上述方法制备得到的重组裂解酶TrxA-lysin。
本发明的第三个目的是提供所述重组裂解酶TrxA-lysin用于裂解大肠杆菌和沙门氏菌。
本发明的目的是通过以下技术方案予以实现的:
一种可以裂解大肠杆菌及沙门氏菌的噬菌体裂解酶的制备方法,包括以下步骤:
S1. 扩增噬菌体ECGD1的裂解酶lysin基因;与质粒相连,并转入基因工程菌获得含lysin重组质粒的基因工程菌;
S2. 培养所述含lysin重组质粒的基因工程菌至菌液的OD值为0.6~0.8,用IPTG诱导含lysin重组质粒的基因工程菌,获得可溶性重组蛋白TrxA-lysin;所述裂解酶lysin基因的序列如SEQ ID NO:1所示。
优选地,S1所述裂解酶lysin基因由引物P1和P2扩增获得,所述引物P1和P2的序列如SEQ ID NO:2和SEQ ID NO:3所示。
pET-32a表达系统具有以下优点:属于原核表达系统,适用于噬菌体裂解酶基因的表达,自带硫氧还原蛋白TrxA基因,可增加外源蛋白的二硫键形成,增加目的蛋白的可溶性表达。
作为一种具体的实施方式,本发明设计引物,采用PCR的方法获得可裂解大肠杆菌及沙门氏菌的噬菌体裂解酶lysin基因。将该基因连接于pET-32a表达质粒中构建重组表达质粒pET-32a-lysin,以ITPG诱导表达重组蛋白TrxA-lysin。
优选地,S2所述IPTG诱导的条件为:IPTG的终浓度为1mM,诱导时间为2h,诱导温度为37℃。
作为一种更优选地实施方式,本发明所述噬菌体裂解酶的制备方法的S1,包括以下步骤:
(1)设计引物,利用PCR技术对能裂解大肠杆菌及沙门氏菌的噬菌体ECGD1的裂解酶lysin基因进行扩增。
(2)将获得的lysin基因连接至pET-32a并筛选得到重组质粒pET-32a-lysin;采用热激转化法将pET-32a-lysin重组质粒导入大肠杆菌BL21中,获得重组大肠杆菌BL21 /pET-32a-lysin。
优选地,(1)所述PCR的反应程序为:98℃预变性3 min;98℃变性10 s,55℃退火10 s,72℃延伸10 s,完成30循环;72℃后延伸5 min。
优选地,(2)所述热激转化法为:取大肠杆菌BL21感受态细胞,加入pET-32a-lysin重组质粒,混匀冰浴30 min,于42℃热激转化90 s后,冰浴2 min,加入LB液体培养基复苏30 min,平板筛选阳性转化子。
优选地,本发明所述噬菌体裂解酶的制备方法还包括纯化可溶性重组蛋白TrxA-lysin的步骤。
更优选地,所述纯化可溶性重组蛋白TrxA-lysin的步骤为:离心收集诱导表达后的BL21/pET-32a-lysin菌体细胞,洗涤,重悬后过滤,用Ni NTA Beads装入层析柱纯化重组蛋白TrxA-lysin。
本发明还提供上述方法获得的噬菌体裂解酶重组蛋白TrxA-lysin。
本发明还提供所述噬菌体裂解酶重组蛋白TrxA-lysin在裂解细菌方面的应用。
本发明还提供所述噬菌体裂解酶重组蛋白TrxA-lysin在制备预防和/或治疗细菌感染的药物方面的应用。
优选地,上述应用中,所述细菌为大肠杆菌和/或沙门氏菌。
与现有技术相比,本发明具有以下有益效果:
本发明提供了一种表达噬菌体ECGD1裂解酶lysin基因的重组大肠杆菌的制备方法。即采用E.coli BL21/pET-32a大肠杆菌表达系统,对噬菌体ECGD1裂解酶lysin基因进行了重组表达,通过硫铁还原蛋白TrxA增加目的蛋白二硫键的形成和表达条件的优化,成功获得高活性的可溶性蛋白,蛋白纯化无需经过复杂的变性复性过程。同时,将诱导后大肠杆菌BL21/pET-32a-lysin重悬液于-80℃和室温之间冻融3次后,细菌重悬液变得澄清、粘稠,即通过简单的冻融,可使细胞破裂并将大部分目的蛋白释放出来。本发明中重组裂解酶粗提物的制备可通过简单的冻融获得,不仅操作简便、成本低廉,而且不易导致目的蛋白变性,适合用于裂解酶的大量制备。
将制备得到的重组蛋白TrxA-lysin用于裂解氯仿预处理过的大肠杆菌和沙门氏菌菌株,发现TrxA-lysin对多株大肠杆菌和沙门氏菌菌株具有裂解作用,即证实了TrxA-lysin可同时裂解大肠杆菌和沙门氏菌。同时TrxA-lysin的宽裂解谱揭示其在大肠杆菌和沙门氏菌感染的防控上具有潜在的应用价值。
附图说明
图1为噬菌体ECGD1电镜照片(放大49,000倍)。
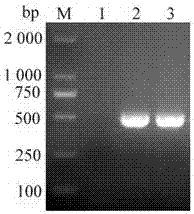
图2为噬菌体ECGD1裂解酶lysin基因的PCR扩增结果:M为DL 2 000 DNA Marker;1为阴性对照;2和3为裂解酶lysin基因的PCR扩增产物。
图3为重组质粒pET-32a-lysin的双酶切鉴定结果:M1为DL10000 DNA Marker;M2为DL 5 000 DNA Marker;1为重组质粒pET-32a-lysin;2为重组质粒pET-32a-lysin经过Nco I、Hind III进行双酶切鉴定结果。
图4为裂解酶重组蛋白TrxA-lysin的纯化结果:M为蛋白标准分子量;1为BL21/pET-32a诱导4 h的细菌总蛋白;2和3分别为BL21/pET-32a-lysin诱导2 h的细菌总蛋白及其菌体冻融上清经0.22 μm过滤后的样品;4为镍柱纯化后的裂解酶重组蛋白。
图5为裂解酶重组蛋白TrxA-lysin对氯仿预处理过的DH5α细胞的裂解效果。
具体实施方式
下面结合说明书附图和具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件。除非另行定义,文中所使用的所有专业与科学用语与本领域技术人员熟悉的意义相同。
实施例1 含lysin基因的大肠杆菌BL21表达菌株的构建
包括以下步骤:
S1. 重组质粒pET-32a-lysin的构建
S11. lysin基因的获得:从生活污水中分离得到了一株可同时裂解大肠杆菌及沙门氏菌的噬菌体(命名为ECGD1,其电镜照片见图1),大量培养噬菌体后提取该噬菌体的基因组,并进行高通量测序和测序结果的拼接和分析。
噬菌体ECGD1基因组的提取:取600 μl过滤除菌后的噬菌体浓缩液,加入DNase I与RNase A至终浓度均为1 μg/ml,37℃消化过夜(彻底去除宿主菌核酸),80℃灭活15 min(注意:该温度不能灭活RNase A);向浓缩液中分别加入24 μl的0.5 mol/L EDTA(终浓度20 mmol/L),1.5 μl的20 mg/mL蛋白酶K(终浓度50 μg/mL),30 μl的10%SDS(终浓度0.5%),56℃水浴1 h;加等体积平衡酚,温和震荡1 min,12 000 rpm离心5 min,转移上层水相于新的离心管中;加等体积酚—氯仿—异戊醇(25:24:1),温和震荡1 min,12 000 rpm离心5 min,转移上层水相于新离心管中;加等体积异丙醇,-70℃放置3 h;4℃,13 000 rpm离心20 min,缓慢倒掉上清;加入等体积75%冰乙醇,静置1 min,7 000 g离心1 min,缓慢倒掉乙醇,室温开盖放置10 min,使乙醇完全挥发,用适量的去离子水溶解DNA,-20℃保存。
将噬菌体ECGD1全基因序列上传至GenBank(序列号为:KU522583.1),通过序列注释和比对获得了lysin基因,大小为501 bp。根据该基因序列设计引物P1、P2,在上游引物P1中插入限制性内切酶Nco I酶切位点,Nco I为含有起始密码子ATG的融合表达酶切位点,在其5’端加上4个保护碱基CATG。在下游引物P2中插入限制性内切酶Hind III的酶切位点,在下游引物5’端加入3个保护碱基CCC。lysin基因序列如SEQ ID NO:1所示;合成的P1、P2引物序列如SEQ ID NO:2~3所示;以1 μL噬菌体ECGD1的基因组为模板,加入高保真DNA聚合酶Prime STAR Max(2×) 25 μL,10 μM的引物P1、P2各1 μL,用ddH2O补至50 μL,反应程序为98℃预变性3 min;98℃变性10 s,55℃退火10 s,72℃延伸10 s,完成30循环;72℃后延伸5 min。PCR反应完成,将产物进行1%琼脂糖凝胶观察与回收,可见大小约500 bp的扩增条带,与预期结果一致(如图2)。
S12. 重组质粒pET-32a-lysin的构建:将S11中回收的PCR产物用Nco I、Hind III进行双酶切处理,胶回收大小约500 bp的条带;以同样的方法对pET-32a空质粒进行双酶切,胶回收大小约5900 bp的条带。分别取4 μL双酶切后胶回收的lysin基因完整修饰片段和1 μL双酶切后胶回收的pET-32a空质粒,加入1 μL的T4 DNA连接酶和1 μL 的10 × buffer,用ddH2O补至10 μL,混匀后置于16℃条件下连接过夜,将连接产物转化E.coli DH5α感受态细胞,在含有100 μg/mL 氨苄青霉素(Ampicillin)的LB琼脂培养板中,37℃培养过夜,然后挑取单个菌落。将菌落接种于含100 μg/mL 氨苄青霉素(Ampicillin)的LB液体培养基中过夜培养。PCR鉴定以待检菌液为模板,加入DNA聚合酶2×EsTaq Master Mix 10 μL,引物P1、P2各0.5 μL,模板0.5 μL,用ddH2O补至20 μL,PCR反应程序为95℃预变性5 min,95℃变性30 s,55℃退火30 s,72℃延伸1 min,30个循环;72℃后延伸10 min。PCR产物用1%琼脂糖凝胶电泳进行检测,可见大小约500 bp的扩增条带,与预期结果一致(如图1)。对检测阳性的菌液用质粒DNA抽提试剂盒进行质粒抽提,并进行双酶切鉴定和序列测定,双酶切后电泳出现约500 bp和5900 bp两条目的条带(如图3),并且测序鉴定结果与预期一致,即获得了重组质粒pET-32a-lysin。
S2. 含lysin基因的大肠杆菌BL21表达菌株的构建
S21. 大肠杆菌BL21感受态细胞的制备:将冻存的E.coli BL21于LB平板培养基上划线复苏,挑取单菌落于LB液体培养基中过夜扩大培养。按照1:100的比例将过夜培养的BL21接种于25 ml新鲜LB液体培养基中,培养至对数期,OD600nm值约为0.6。将培养液转入离心管中,冰上放置10 min,于4℃下3000 g离心10 min。弃去上清,用预冷的0.05 mol/L的CaCl2 溶液10 mL轻轻悬浮细胞,冰上放置15-30 min后,4℃下3000 g离心10 min。弃去上清,加入4 mL预冷含15%甘油的0.05 mol/L的CaCl2 溶液,轻轻悬浮细胞,冰上放置几分钟,即成感受态细胞悬液。
S22. 大肠杆菌BL21的转化及转化子的PCR鉴定:取50 µL大肠杆菌BL21感受态细胞,冰浴上融解,加入1 µL pET-32a-lysin重组质粒,轻轻混匀;将以上混合物冰浴30 min后放入42℃热激90 s,迅速将混合物冰浴3 min,加入100µL的LB液体培养基,于37℃,140 rpm的培养条件培养45 min。将培养后的菌液均匀涂布于含100 μg/mL 氨苄青霉素(Ampicillin)的LB固体培养基中,于37℃培养过夜,挑取单菌落。将菌落接种于含100 μg/mL 氨苄青霉素(Ampicillin)的LB液体培养基中过夜培养。PCR鉴定以待检菌液为模板,加入DNA聚合酶2×EsTaq Master Mix 10 μL,引物P1、P2各0.5 μL,模板0.5 μL,用ddH2O补至20 μL,PCR反应程序为95℃预变性5 min,95℃变性30 s,55℃退火30 s,72℃延伸1 min,30个循环;72℃后延伸10 min。。PCR产物用1%琼脂糖凝胶电泳进行检测,可见大小约500 bp的扩增条带。
S3. 含lysin基因的E.coli BL21/pET-32a-lysin重组表达菌诱导表达过程:
首先是最佳诱导时间的确定,将PCR鉴定为阳性的BL21/pET-32a-lysin划线培养过夜;挑取单菌落接种于3 mL LA液体培养基中,37℃,220 rpm培养10 h,取出菌液,置4℃冰箱过夜;按1%体积比例将上述菌液接种于3 mL LA液体培养基,37℃,220 rpm振摇培养;当OD600为0.6~0.8时,加入终浓度为1 mmol/L IPTG进行诱导表达。取诱导不同时间的菌液,12,000 rpm离心2 min,去上清,细菌沉淀中加100 µL 1×SDS上样缓冲液,沸水中煮5 min左右,12,000 rpm离心2 min,取上清进行SDS-PAGE电泳检测最佳诱导时间。同时,设立pET-32a空质粒在BL21中的诱导表达菌液作为对照。上样前,将凝胶放进垂直电泳槽,将电压设置为80 V进行预电泳30 min以消除凝胶中未聚合物质的影响;上样后设定电压为80 V,待溴酚蓝染料压成一条水平的直线,即将进入分离胶时将电压升高至160 V;染料接近凝胶底部时停止电泳。取下凝胶,于水平摇床上用考马斯亮蓝染色液染色1 h左右,再用脱色液进行脱色,期间更换脱色液,脱色至蛋白区带清晰为止,用凝胶成像系统对凝胶进行照相。
结果显示最佳诱导时间为2 h。利用上述确定的最佳诱导时间,进行目的蛋白的诱导表达。离心收集诱导后的菌体,用1 mL PBS重悬后,在-80℃和室温之间反复冻融3次,4℃条件下10,000 rpm离心2 min,取上清液,加入等体积的2×SDS上样缓冲液,沸水中煮5 min左右,12,000 rpm离心2 min,取上清进行SDS-PAGE电泳检测重组蛋白的表达形式,结果显示目的蛋白主要为可溶性表达。
S4.重组蛋白TrxA-lysin纯化过程:离心收集诱导表达后的BL21/pET-32a-lysin菌体细胞,用PBS洗涤两次。按照菌体:Lysis buffer = 1:10(W/V)将菌体悬浮起来,于-80℃和室温之间反复冻融3次,4℃条件下10,000 rpm离心20 min,取上清液,并用0.22 μm滤膜过滤,收集滤液,用于目的蛋白的纯化。
将Ni NTA Beads装入合适的层析柱,用5倍柱体积的Lysis buffer平衡柱子,使填料与目的蛋白处于相同的缓冲体系下,起到保护蛋白的作用。将样品加到平衡好的Ni NTA Beads中(保证目的蛋白与Ni2+充分接触,提高目的蛋白的回收率),收集流出液,用于SDS-PAGE分析蛋白质的结合情况。用10~15倍柱体积的Wash buffer进行清洗,去除非特异性吸附的杂蛋白,收集洗杂液。使用5~10倍柱体积的Elution buffer洗脱柱子,收集洗脱液,即目的蛋白组分。利用10 kDa超滤离心管对纯化的蛋白进行后续脱盐及浓缩处理。纯化结果如图4所示。
实施例2 重组蛋白TrxA-lysin的活性检测
包括以下步骤:
S1. 用重组蛋白TrxA-lysin裂解氯仿预处理过的DH5α:按1%比例将待测菌株接种于LB培养基中,过夜培养,菌液中加入终浓度为5%的氯仿,振荡培养15 min,离心收集菌体,并用去离子水清洗两次,-80℃保存备用。测定活性之前,细菌沉淀用50 mmol/L的Tris-HCl(PH 8.2,含0.1% Triton X-100)重悬。将10 µL的裂解酶溶液(重组蛋白TrxA-lysin溶液)加入到90 µL细菌重悬液中,室温静置至裂解效果明显,测定OD450。试验设置三个重复,且将Tris-HCl缓冲液处理作为阴性对照。由于噬菌体ECGD1能裂解大肠杆菌基因工程菌株DH5α,因此先用DH5α作为待测菌株检测重组裂解酶TrxA-lysin的裂解活性。利用氯仿预处理的浊度法检测重组裂解酶TrxA-lysin的裂解活性,发现菌液在TrxA-lysin的作用下迅速变得澄清,OD450在10 min内降低了0.79(图5),说明本试验表达的TrxA-lysin具有裂解活性。
S2. 多粘菌素B与重组蛋白TrxA-lysin联用裂解活菌:该试验分为两组,包括多粘菌素B与重组蛋白协同作用组和多粘菌素B对照组。协同作用组试验处理大致为,用2倍倍比稀释法制备含多粘菌素B终浓度为210、29、28、27……2-4、2-5 µg/mL的LB培养基(每个梯度3 mL),往每个稀释梯度添加100 µL重组蛋白TrxA-lysin溶液,再按1%比例接种大肠杆菌DH5α,37℃振荡培养过夜。多粘菌素B对照组处理同上,但是不添加重组蛋白TrxA-lysin溶液。结果显示,多粘菌素B对照组中,多粘菌素B对DH5α最低抑菌浓度为2 µg/mL;裂解酶TrxA-lysin与多粘菌素B协同作用组中,多粘菌素B对DH5α最低抑菌浓度为1 µg/mL。即裂解酶TrxA-lysin的存在降低了多粘菌素B的最低抑菌浓度,从而证明这两者存在协同抑菌效果。
S3. 重组蛋白TrxA-lysin裂解谱的测定:利用S1所述的氯仿预处理的方法检测裂解酶重组蛋白对测试菌株的裂解活性,观察细菌裂解效果及其OD450值变化。测试菌株包括大肠杆菌和沙门氏菌临床分离菌株,以及大肠杆菌基因工程菌株。裂解酶对待测菌株的裂解值=Δ450nm(试验组降低值)—Δ450nm(Tris-HCl对照组降低值)。相对裂解活性=裂解酶对待测菌株的裂解值/裂解酶对DH5α的裂解值。以大肠杆菌DH5α为裂解阳性标准,利用氯仿预处理的浊度法检测裂解酶对大肠杆菌及沙门氏菌的裂解活性,结果显示,裂解酶对不同来源的大肠杆菌及沙门氏菌均有裂解活性,表现出对大肠杆菌及沙门氏菌的广谱杀菌活性。由相对裂解活性测定结果得知,裂解酶对大肠杆菌的裂解活性普遍高于沙门氏菌。
SEQUENCE LISTING
<110> 华南农业大学
<120> 一种可以裂解大肠杆菌及沙门氏菌的噬菌体裂解酶的制备方法
<130>
<160> 3
<170> PatentIn version 3.3
<210> 1
<211> 501
<212> DNA
<213> 裂解酶lysin基因序列
<400> 1
atggctaaag tagttgatgt gtttgatatg ttacgttttg atgaaggact aaagctaact 60
gtatatactg ataccgaagg gtattggacg gttggtattg gacaccttct gacaaaactt 120
aaagataaat cagaagctat acgcattctt gataacttag taggtagaaa aactaatggg 180
gttattactg aagcggaagc tagaaaaatc tttgagggtg acgtaaagaa agcgatacaa 240
caaatccatt caagtaccat attatctcct atttacgata aagtaagtcc taatcgtaaa 300
atggctatta tcaatatggt atttcaaatg ggattgaaag gtgcagaatc tttcaaaaat 360
agcttgactt tagtgagtaa ttcatattat actcaagcct ctataaattt acgtaaaagt 420
aaatggtatc gccaaacgcc taatcgcgca gagcgtgtaa ttcaggtgct taaaactgga 480
acattagacg cttataacta a 501
<210> 2
<211> 26
<212> DNA
<213> 引物P1序列
<400> 2
catgccatgg ctaaagtagt tgatgt 26
<210> 3
<211> 29
<212> DNA
<213> 引物P2序列
<400> 3
cccaagcttt tagttataag cgtctaatg 29
- 还没有人留言评论。精彩留言会获得点赞!