mPEG3‑替诺福韦酯及其制备方法与流程
本发明属于有机化合物合成技术领域,涉及一种药物化合物mPEG3-替诺福韦酯及其合成工艺,具体地,涉及一种mPEG3-替诺福韦酯的制备方法。
背景技术:
富马酸替诺福韦酯(Tenofovir Disoproxil Fumarate,TDF),化学名为(R)-[[2-(6-氨基-9H-嘌呤-9-基)-1-甲基乙氧基]甲基]膦酸二异丙氧羰氧基甲酯富马酸盐,是美国Gilead Sciences公司研发的一种新型核苷酸类逆转录酶抑制剂,主要通过抑制HIV-1逆转录酶的活性而抑制HIV病毒的复制。该制剂2001年首次在美国上市,目前已在加拿大、欧洲等多个国家和地区上市,其作为治疗HIV的一线药物,具有很好的应用前景。
替诺福韦的活性成分替诺福韦双磷酸盐可通过直接竞争性地与天然脱氧核糖底物相结合而抑制病毒聚合酶,及通过插入DNA中终止链。替诺福韦几乎不经胃肠道吸收,因此进行酯化、成盐,成为替诺福韦酯富马酸盐。替诺福韦酯具有水溶性,可被迅速吸收并降解成活性物质替诺福韦,替诺福韦然后被转变为活性代谢产物替诺福韦双磷酸盐。
富马酸替诺福韦酯现有生产工艺是从替诺福韦一水合物或者无水物作为起始原料与氯甲基碳酸异丙酯经酯化反应后合成替诺福韦酯,然后与富马酸成盐得富马酸替诺福韦酯,但现有合成技术中反应转化率偏低,其主要原因是由于替诺福韦在酯化时先进行单酯化,然后继续反应成双酯,但是单酯化后因为位阻增大,反应活性降低,双酯化难度加大,导致反应中一直有单酯存在而影响反应收率。且因为单取代产物大量存在,反应后处理也比较困难,产品质量较难保证。虽然通过增加原料投料量来促进反应的正向进行,但是这会大大提高生产成本,并且当原料增加到一定量时,反应正向进行会达到一个平衡,不在继续转化。此外,现有合成技术中酯化反应均使用高沸点的溶剂,而替诺福韦酯在高温下不稳定,无法直接回收,所以后处理时一般都采用大量的冰水进行盐析或者大量的水洗涤,导致大量溶剂进入污水处理系统,不仅增加了环保的压力,而且由于溶剂无法回收利用造成产品成本太高。
此外,目前已知的富马酸替诺福韦酯可能引起的不良反应有:(1)全身无力;(2)胃肠道反应:轻至中度的胃肠道不适,常见的有腹泻、腹痛、食欲减退、恶心、呕吐和胃肠胀气、胰腺炎;(3)代谢系统:低磷酸盐血症(1%发生率);脂肪蓄积和重新分布,包括向心性肥胖、水牛背、末梢消瘦、乳房增大、库兴综合征;(4)可能引起乳酸中毒、与脂肪变性相关的肝肿大等;(5)神经系统:头晕、头痛;(6)呼吸系统:呼吸困难;(7)皮肤:药疹。
技术实现要素:
本申请发明人发现,采用三甘醇单甲醚对甲苯磺酸酯修饰后的替诺福韦酯,与现有技术及对照品四甘醇单甲醚对甲苯磺酸酯修饰后的替诺福韦酯相比,对PBMC细胞具有减轻的毒性,对HIV-1TC-1感染的PBMC有很好的抑制HIV-1TC-1在PBMC中复制的作用。有鉴于此,本发明的目的是克服现有技术的不足,提供一种安全、质量好的mPEG3-替诺福韦酯产品。
为实现上述目的,本发明的技术方案为:
mPEG3-替诺福韦酯,其结构式如式I所示;
本发明还提供了一种mPEG3-替诺福韦酯的制备方法,其制备方法成本低、工艺安全、适合工业化生产。
为实现上述目的,本发明的技术方案为:
mPEG3-替诺福韦酯的制备方法,包括如下进行的步骤:
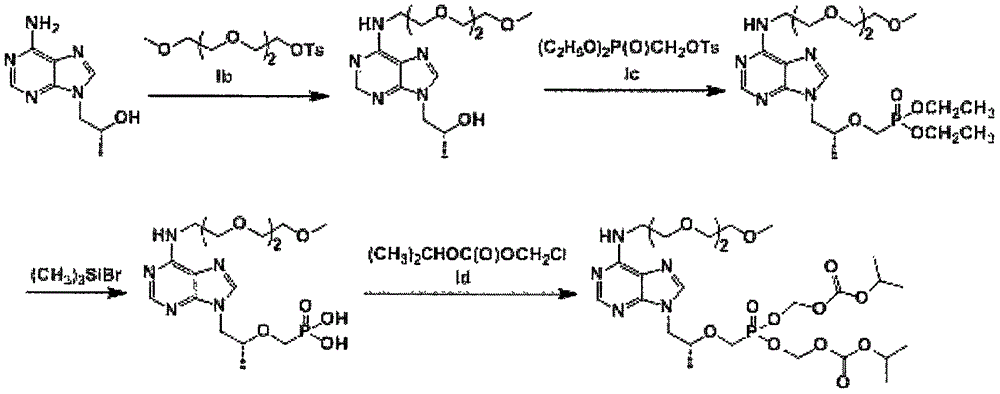
(1)以(R)-9-(2-羟丙基)腺嘌呤为原料,在碳酸盐的作用下,与三甘醇单甲醚对甲苯磺酸酯反应得式II所示的mPEG3-(R)-9-(2-羟丙基)腺嘌呤;
(2)步骤(1)所得式II化合物与对甲苯磺酰氧甲基磷酸二乙酯在碱的作用下反应制得式III所示的mPEG3-替诺福韦二乙酯;
(3)步骤(2)所得式III化合物在水解试剂作用下经水解反应制得式IV所示的mPEG3-替诺福韦;
(4)步骤(3)所得式IV化合物与氯甲基碳酸异丙酯在碱作用下经缩合制得式I所示的mPEG3-替诺福韦酯;
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(1)中,所述碳酸盐为碱金属碳酸盐;所用溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、乙腈和N-甲基吡咯烷酮中的一种或多种。
优选的,所述碳酸盐为碳酸钾;所用溶剂为N,N-二甲基甲酰胺。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(1)中,(R)-9-(2-羟丙基)腺嘌呤与三甘醇单甲醚对甲苯磺酸酯的摩尔比为1∶1.5-2。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(1)中,所述(R)-9-(2-羟丙基)腺嘌呤与碳酸盐的摩尔比为1∶2-3。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(1)中,反应温度为90-110℃。
优选的,所述反应温度为100℃。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(2)中,所述碱为叔丁氧基钠、叔丁氧基钾和叔丁氧基镁中的一种或多种。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(2)中,先将mPEG3-(R)-9-(2-羟丙基)腺嘌呤溶于溶剂中,所用溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、乙腈和N-甲基吡咯烷酮中的一种或多种。
优选的,所用溶剂为N,N-二甲基甲酰胺。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(2)中,所述mPEG3-(R)-9-(2-羟丙基)腺嘌呤与对甲苯磺酰氧甲基磷酸二乙酯的摩尔比为1∶1-2。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(2)中,所述mPEG3-(R)-9-(2-羟丙基)腺嘌呤与碱的摩尔比为1∶8-10。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(2)中,反应温度为30-40℃。
优选的,所述反应温度为35℃。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(3)中,所述水解试剂为三甲基溴硅烷和/或三甲基氯硅烷;所用溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、乙腈和N-甲基吡咯烷酮中的一种或多种。
优选的,所述水解试剂为三甲基溴硅烷。
优选的,所用溶剂为乙腈。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(3)中,水解在室温条件下进行,水解时间为20-40小时。
优选的,水解时间为24小时。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(4)中,所述碱为吡啶、4-二甲氨基吡啶(DMAP)、N,N-二异丙基乙胺(DIPEA)、2,6-二甲基吡啶、碳酸钠、碳酸钾、三乙胺和二乙胺中的一种或多种;所用溶剂为乙醇、乙腈、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜和N-甲基吡咯烷酮中的一种或多种。
优选的,所述碱为三乙胺。
优选的,所用溶剂为无水乙醇与N-甲基吡咯烷酮混合物。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(4)中,所述mPEG3-替诺福韦与氯甲基碳酸异丙酯的摩尔比为1∶5-8。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(4)中,所述mPEG3-替诺福韦与碱的摩尔比为1∶5-8。
进一步,所述的mPEG3-替诺福韦酯的制备方法,步骤(4)中,反应温度为45-60℃。
优选的,反应温度为55℃。
本发明还提供了mPEG3-替诺福韦酯的应用。所述的mPEG3-替诺福韦酯在制备治疗HIV、HBV药物中的应用。
本发明的有益效果在于:
本发明提供的mPEG3-替诺福韦酯,弥补了现有技术的空缺,与现有技术相比,对PBMC细胞具有减轻的毒性,对HIV-1TC-1感染的PBMC有很好的抑制HIV-1TC-1在PBMC中复制的作用,具有安全、质量好的优点,具有良好的应用开发前景。该化合物的开发,将为艾滋病患者、乙肝患者带来福音。
本发明提供的mPEG3-替诺福韦酯的制备方法,方法简洁,原料来源广,反应条件温和,生产成本低,并且反应整体收率高,产品质量也相对稳定。此外,用乙醇与N-甲基吡咯烷酮做混合溶剂进行酯化反应,使用低沸点的乙醇作为主要溶剂,可以在温度较低的条件下将溶剂回收利用,在不影响产品质量的前提下解决因溶剂损失导致的成本偏高问题,也减轻了环保的压力。
附图说明
图1为本发明所述mPEG3-替诺福韦酯的制备方法的合成路线图。
具体实施方式
所举实施例是为了更好地对本发明的内容进行说明,但并不是本发明的内容仅限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,还包括各具体实施方式间的任意组合,仍属于本发明的保护范围。
三甘醇单甲醚对甲苯磺酸酯、四甘醇单甲醚对甲苯磺酸酯购自北京键凯科技有限公司;(R)-9-(2-羟丙基)腺嘌呤购自武汉远成赛创科技有限公司;对甲苯磺酰氧甲基磷酸二乙酯购自天津威赛诺科技发展有限公司;其余均购自国药集团化学试剂有限公司
实施例1 式II所示的mPEG3-(R)-9-(2-羟丙基)腺嘌呤的制备
将(R)-9-(2-羟丙基)腺嘌呤(Ia,22.8g,117mmol)、三甘醇单甲醚对甲苯磺酸酯(Ib,67.2g,211mmol)和K2CO3(32.4g,234mmol)溶于DMF(900mL)中,加热升温至100℃保温反应7h。反应完毕后,冷却至室温,加入蒸馏水(1200mL),搅拌混合均匀。用乙酸乙酯(720mL×4)提取,混合有机层用无水硫酸钠干燥,过滤、浓缩,残留物用硅胶柱分离得油状固体产品19.6g,收率49.4%,HPLC纯度98.0%。
实施例2 式III所示的mPEG3-替诺福韦二乙酯的制备
将mPEG3-(R)-9-(2-羟丙基)腺嘌呤(式II,25.4g,74.8mmol)溶于DMF(150mL)中,缓慢加入叔丁氧基锂(53.9g,673mmol)的2.0M四氢呋喃溶液,然后再缓慢加入对甲苯磺酰氧甲基磷酸二乙酯(Ic,44.8g,89.8mmol),升温至35℃搅拌反应8h。反应完毕后,冷却至20℃后,加入冰醋酸(6.0mL),减压蒸馏除去挥发物,加入蒸馏水(200mL)。用CH2Cl2分次提取,合并提取液,用无水硫酸钠干燥。过滤、浓缩得油状固体产品12.8g,收率34.9%,HPLC纯度95.8%。
实施例3 式IV所示的mPEG3-替诺福韦的制备
将mPEG3-替诺福韦二乙酯(式III,19.2g,28.8mmol)溶于乙腈(250mL)和三甲基溴硅烷(25mL)中,隔绝空气室温搅拌过夜。减压蒸馏除去挥发物,加入蒸馏水(500mL),用CH2Cl2洗涤。水相用NaOH水溶液调节pH至3.2,冷却搅拌6h。过滤收集固体,依次用冰水和丙酮洗涤,真空干燥得固体6.7g,收率53.7%,HPLC纯度98.0%。
实施例4 式I所示的mPEG3-替诺福韦酯的制备
在1L反应瓶中加入无水乙醇(80mL)、N-甲基吡咯烷酮(8mL)和mPEG3-替诺福韦(式IV,11.4g,26.2mmol),开启搅拌,控制温度在20℃下滴加三乙胺(13.4g,132mmol),滴完后继续滴加氯甲基碳酸异丙酯(Id,20g,132mmol),滴完后将反应体系升温至55℃保温反应6h,将反应液在40℃以下减压浓缩至干,向浓缩残液中加入乙酸乙酯(100mL)和6%的碳酸氢钠溶液(50mL),搅拌混匀后分层萃取,有机相用蒸馏水(50mL)洗涤两次后,用无水硫酸钠干燥。过滤、浓缩得油状固体产品14.9g,收率85.4%,HPLC纯度98.5%。ESI-MS:666[M+H]+。
对比例1:mPEG4-替诺福韦酯的制备
将(R)-9-(2-羟丙基)腺嘌呤(Ia,22.8g,117mmol)、四甘醇单甲醚对甲苯磺酸酯(215mmol)和K2CO3(32.4g,234mmol)溶于DMF(900mL)中,加热升温至100℃保温反应7h。反应完毕后,冷却至室温,加入蒸馏水(1200mL),搅拌混合均匀。用乙酸乙酯(720mL×4)提取,混合有机层用无水硫酸钠干燥,过滤、浓缩,残留物用硅胶柱分离得油状固体产品18.6g。将上述固体产品溶于DMF(150mL)中,缓慢加入叔丁氧基锂(667mmol)的2.0M四氢呋喃溶液,然后再缓慢加入对甲苯磺酰氧甲基磷酸二乙酯(89.8mmol),升温至35℃搅拌反应8h。反应完毕后,冷却至20℃后,加入冰醋酸(6.0mL),减压蒸馏除去挥发物,加入蒸馏水(200mL)。用CH2Cl2分次提取,合并提取液,用无水硫酸钠干燥。过滤、浓缩得油状固体产品11.8g。将上述油状固体产品溶于乙腈(250mL)和三甲基溴硅烷(25mL)中,隔绝空气室温搅拌过夜。减压蒸馏除去挥发物,加入蒸馏水(500mL),用CH2Cl2洗涤。水相用NaOH水溶液调节pH至3.2,冷却搅拌6h。过滤收集固体,依次用冰水和丙酮洗涤,真空干燥得固体6.3g。在1L反应瓶中加入无水乙醇(40mL)、N-甲基吡咯烷酮(4mL)和mPEG4-替诺福韦(12.2mmol),开启搅拌,控制温度在20℃下滴加三乙胺(66mmol),滴完后继续滴加氯甲基碳酸异丙酯(66mmol),滴完后将反应体系升温至55℃保温反应6h,将反应液在40℃以下减压浓缩至干,向浓缩残液中加入乙酸乙酯(100mL)和6%的碳酸氢钠溶液(50mL),搅拌混匀后分层萃取,有机相用蒸馏水(50mL)洗涤两次后,用无水硫酸钠干燥。过滤、浓缩得油状固体产品5.8g。ESI-MS:710[M+H]+。
实施例5 mPEG3-替诺福韦酯在细胞培养内的抗HIV-1活性
本实施例所需主要试剂与主要仪器:
主要试剂:
RPMI-1640、胎牛血清为Invitrogen、硫酸链霉素(Streptomycin sulfate)、氨苄青霉素、硫酸卡那霉素、HEPES(N-2,2-Hydroxyothyl piperazine-N’-2-ethanesufonic acid)、MTT(3,(4,5-dimethylthiazol-2-yl,噻唑蓝)-2,5-diphenyl tetrazolium bromide)、DMF(N,N’-Dimethyl formamine,N,N-二甲基甲酰胺)、Triton X-100、青霉素(Penicillin)、谷氨酰胺(Glutamine)、碳酸氢钠、碳酸钠、氯化钾、磷酸二氢钠、磷酸氢二钠、磷酸二氢钾、磷酸氢二钾、二甲基亚砜(DMSO)。
主要仪器:
酶标仪(Biotek ELX800)、离心机(Thermo IEC CL40)、二级生物安全柜(Thermo Forma Class II,A2)、CO2培养箱(Thermo)、加样枪(Thermo Finnpipeette F2)。
(1)待试药物:本发明的mPEG3-替诺福韦酯(命名为:化合物GQ-80604),生产批号:080604,水不溶,DMSO可溶解,试验时用DMSO溶解配成适当浓度后用培养基稀释,立即加入细胞培养。
对照药物A:mPEG4-替诺福韦酯。
(2)阳性对照药:齐多夫定(AZT),购自东北制药集团市售药品,片剂,规格0.1g,避光、密闭、干燥处保存。
(3)病毒
HIV-1临床分离株选用HIV-1TC-1,由天津第二人民医院惠赠。按常规方法培养制备HIV-1病毒。HIV-1感染性滴定按常规方法进行。病毒小量分装于冻存管,-80℃保存。
(4)细胞
人外周血单个核细胞(PBMC)自健康人血液浓缩白细胞中分离,由天津第二人民医院惠赠。细胞培养基:RPMI-1640完全培养基,含有10%胎牛血清,2mM L-谷氨酰胺,10mM HEPES,50μM 2-巯基乙醇,100IU/ml青霉素,100μg/ml 链霉素。
细胞按常规方法进行复苏,传代:从液氮中复苏细胞,用RPMI-164培养基洗去冻存液,加入含10%FCS的完全培养基10ml,混匀,37℃ 5%CO2培养。每隔3天对细胞传一次代。实验前1天传一次代,使所用细胞处于对数生长期。
(5)GQ-80604对PBMC的毒性试验
在96孔细胞培养板上,100μl PHA-P刺激转化的PBMC(5×105个细胞)与100μl不同稀释度浓度的待测药物溶液混合,每个稀释度3个重复孔,同时设置不含药物的对照孔。37℃,5%CO2培养箱培养。第4天每孔补充100μl含有IL-2(终浓度50U/ml)的新鲜完全细胞培养基,第7天采用MTT比色法检测细胞毒性。ELx800酶标仪测定OD值,测定波长为570nm参考波长为630nm。计算得到CC50值(50%Cytotoxic concentration),即对50%的PBMC产生毒性时的药物浓度。
在人体内,HIV感染宿主细胞主要是CD4+T淋巴细胞和单核巨噬细胞。从外周血分离获得的PBMC主要细胞成分是淋巴细胞(包括T、B淋巴细胞)和单核巨噬细胞。体外HIV感染PBMC实验时,HIV-1需要在PHA活化后的PBMC中复制。本实验以MTT比色法检测了GQ-80604对活化PBMC的毒性作用。结果如表1所示。
表1 GQ-80604和AZT对PBMC细胞的毒性作用
结果表明:GQ-80604对PBMC细胞毒性相对于替诺福韦及mPEG4-替诺福韦酯显著降低,而同样进行修饰的对照品A对PBMC细胞的毒性与AZT没有显著性差异,即并非所有对替诺福韦进行的聚乙二醇结构修饰都会带来细胞毒性的显著改变。
(6)GQ-80604对HIV-1TC-1感染PBMC中病毒复制的抑制试验
收集PHA刺激72小时的PBMC,用含有IL-2(终浓度为50U/ml)的1640完全培养基重悬细胞,染色计数,存活率>98%。调细胞浓度5×106/ml,加入HIV-1TC-1贮存液(M.O.I.=0.06),37℃感染4小时后,离心、PBS洗涤3次,去除游离的病毒。用含有IL-2(终浓度为50U/ml)的培养基重悬细胞,调细胞浓度为5×106/ml。每孔滴加100μl细胞悬液至含有100μl不同浓度待测药物的96孔板中。37℃,5%CO2培养箱培养。第4天每孔补加100μl含有IL-2的细胞培养基,第7天收集培养上清,用终浓度0.5%Triton X-100裂解灭活。采用捕捉p24抗原ELISA方法,计算出药物对HIV-1TC-1在PBMC中复制表达p24抗原的抑制率和EC50。
为了检测GQ-80604抗HIV-1临床分离株活性作用,我们以从天津市HIV/AIDS患者体内分离的HIV-1临床分离株HIV-1TC-1感染PBMC,此株临床分离株是07BC亚型,X4嗜性。结果如表2所示。
表2 GQ-80604和AZT对HIV-1TC-1感染PBMC的复制的抑制试验
结果表明:GQ-80604对对HIV-1TC-1感染的PBMC有很好的抑制HIV-1TC-1在PBMC中复制的作用,阳性对照化合物AZT三批次实验的EC50与对照品A作用相当。
由表1和表2可知,GQ-80604对PBMC细胞的毒性较小,同时对HIV-1TC-1感染的PBMC有很好抑制HIV-1TC-1在PBMC中复制的作用,具有良好的应用开发前景。该化合物的开发,将为艾滋病患者、乙肝患者带来福音。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!