一种基于稀土元素的温敏性复合物及其制备方法和应用与流程
本发明涉及稀土材料,具体涉及一种基于稀土元素的温敏性复合物及其制备方法和应用。
背景技术:
稀土高分子体系既包括稀土化合物均匀地掺杂于高分子聚合物或单体中所形成的掺杂型稀土高分子(doping-type rare earth polymers),也包括稀土离子以化学键合配位方式键合于高分子或单体中形成的键合型稀土高分子(bonding-type rare earth polymers),以高分子聚合物作为基质所制得的稀土高分子材料,一方面具备高分子化合物机械强度好、易加工等优点,另一方面具有稀土元素特殊的光致发光性能,因此引起了世界各国科学家的广泛关注。
稀土元素独特的4f电子构型使得它的化合物表现出优良的电、光、磁性能,与其他元素相比,稀土元素无法比拟的光谱学性质使得含有稀土元素的发光材料格外引人关注。目前,有关固体发光领域的研究总会不可避免的涉及到稀土发光材料。因此,研究稀土配合物以求实现它的功能化就显得十分有意义。
聚N-异丙基丙烯酰胺,因其水溶液或水凝胶的温度在临界溶解温度(LCST)附近时,它的溶胀性能和其他相关性能会发生改变,因而使它在药物控释体系、生物物质的分离、免疫分析、生物酶的固定等领域有潜在的应用。
综上,通过RAFT方法将聚N-异丙基丙烯酰胺末端羧基化,制备出一种温度响应型高分子聚合物,并将聚合物与具有荧光标记作用的稀土离子相结合,检测其荧光特性、温敏特性,以探求在生物工程领域应用的可行性正成为研究的方向。
技术实现要素:
本发明设计开发了一种基于稀土元素的温敏性复合物。
本发明的第二发明目的是提供上述基于稀土元素的温敏性复合物的制备方法。
本发明的第三发明目的是提供上述基于稀土元素的温敏性复合物在荧光材料领域的应用以及温敏材料领域的应用。
本发明提供的技术方案为:
一种基于稀土元素的温敏性复合物,结构式如下:
其中,M为Eu3+或者Tb3+,n为65;以及
所述温敏性复合物的数均分子量为Mn=7700g/mol,分子量分布为1.03,由GPC测定。
一种基于稀土元素的温敏性复合物的制备方法,包括如下步骤:
步骤一、由α-噻吩甲酰三氟丙酮、三价稀土金属盐酸盐以及5-氨基-1,10-邻菲罗啉进行配位,得到稀土金属配合物;
步骤二、通过1-十二硫醇、丙酮以及四丁基溴化铵作为原料,在碱性条件下,加入丙酮以及二硫化碳溶液继续反应后,在加入氯仿和氢氧化钠继续作用后,酸化体系,得到S-S′-双(1-十二烷基)三硫代醋酸酯;
步骤三、通过所述步骤二中得到S-S′-双(1-十二烷基)三硫代醋酸酯、N-异丙基丙烯酰胺、1,3,5-三噁烷以及偶氮二异丁腈进行聚合反应,得到末端含有羧基的聚(N-异丙基丙烯酰胺);
步骤四、通过所述稀土金属配合物与所述聚(N-异丙基丙烯酰胺)在乙醇溶液中搅拌反应,得到所述温敏性复合物。
优选的是,在所述步骤一包括:将α-噻吩甲酰三氟丙酮溶解在乙醇中,用的氢氧化钠调节溶液pH为6.5,将三价稀土金属盐酸盐的乙醇溶液滴加到上述反应,搅拌,再加二次蒸馏水继续恒温搅拌,过滤,复合物在冷却至室温的过程中沉淀,滤出沉淀物,用水洗涤,并在真空下干燥;所得复合物为三价稀土金属:α-噻吩甲酰三氟丙酮的摩尔数比例为1:3的二元产物,将5-氨基-1,10-邻菲罗啉在乙醇中与所述二元产物络合在一起,同时搅拌将溶液加热回流,24小时后过滤,用乙醇和氯仿将过量的无界络合物冲走,得到铕配合物。
优选的是,所述三价稀土金属盐酸盐为EuCl3或者TbCl3。
优选的是,在所述步骤二中包括:将1-十二烷硫醇,丙酮与四丁基溴化铵放在三口烧瓶中反应,冷却,在氮气下进行混合;溶液中加入氢氧化钠溶液混合,将反应物搅拌,再将丙酮以及二硫化碳溶液加入,要超过20分钟,溶液颜色变成红色,将氯仿溶液加入,然后滴加氢氧化钠溶液超过30分钟,将反应物搅拌过夜;溶液中加入水,然后由浓盐酸的水溶液酸化,通过鼓动反应器,以帮助氮气蒸发掉丙酮,收集固体,然后加入搅拌的2-丙醇,未溶解的固体滤出,得到S-S′-双(1-十二烷基)三硫代醋酸酯。
优选的是,在所述步骤三中包括:将所述S-S′-双(1-十二烷基)三硫代醋酸酯、N-异丙基丙烯酰胺、1,3,5-三噁烷偶氮二异丁腈放入聚合管中,加入无水N,N-二甲基甲酰胺作溶剂,放在磁力搅拌器上,进行多次通氮气后再抽真空的操作,每次用时10~15分钟,然后将反应混合物在氮气保护下,放入油浴中反应,接着升温反应,取出后冷却,进行旋蒸,旋蒸冷却后,加入无水乙醚沉淀,用抽滤瓶过滤,得到末端含有羧基的聚(N-异丙基丙烯酰胺)。
优选的是,在所述步骤四中包括:将所述末端含有羧基的聚(N-异丙基丙烯酰胺)和所述稀土金属配合物放入乙醇溶液中搅拌,反应结束后将产物旋转蒸发,除去乙醇溶液,然后将所得液体置于恒温干燥箱中干燥后得到所述温敏性复合物。
优选的是,所述步骤一中,合成所述5-氨基-1,10-邻菲罗啉包括如下步骤:
步骤a、在1,10-邻菲罗啉和浓硫酸中加入浓硝酸,放入70℃油浴锅中,接着将温度升到120℃恒温搅拌,将反应混合物用冰水稀释逐渐冷却,同时将温度保持在低于5℃,用碱中和到成中性,所形成的微黄色的沉淀物通过过滤收集并用冰冷的水充分洗涤,残留物进一步用乙醇重结晶,在真空烘箱中干燥过夜后,得到5-硝基-1,10-邻菲罗啉;
步骤b、将5-硝基-1,10-邻菲罗啉和钯碳催化剂的乙醇悬浮液加热,然后通入氮气,加入水合肼溶液,将反应混合物回流,趁热过滤后,将滤液在旋转式蒸发器上蒸干,将得到的复合物粗品用乙醇重结晶进一步纯化,干燥后,在真空烘箱中于室温下过夜,得到5-氨基-1,10-邻菲罗啉。
所述的复合物在温敏材料中的应用。
所述的复合物在荧光材料中的应用。
本发明与现有技术相比较所具有的有益效果:
1、通过本发明的合成方法所合成的DMP-PNIPAM/Eu(III)复合物最低临界溶解温度为34.1℃,相对于DMP-PNIPAM的最低临界溶解温度的33.9℃以及PNIPAM的最低临界溶解温度的32℃均有升高,XPS结果显示,Eu(Ш)与配体中的O、N原子以配位键结合。复合物具有优异的荧光性能,荧光发射谱中580、591、614、621nm处发射峰分别对应5D0—>7fj(j=0,1,2,3,4)的电子跃迁,其中614nm处发射峰强度最大。同时测定,DMP-PNIPAM/Eu(Ш)的荧光寿命为11.78ms,使DMP-PNIPAM/Eu(III)复合物能够作为温敏材料以及荧光材料应用;
2、通过本发明的合成方法所合成的DMP-PNIPAM/Tb(III)复合物最低临界溶解温度为34.0℃,相对于DMP-PNIPAM的最低临界溶解温度的33.9℃以及PNIPAM的最低临界溶解温度的32℃均有升高,XPS结果显示,Tb(Ш)与配体中的O、N原子以配位键结合。复合物具有优异的荧光性能,荧光发射谱中487、545、583、616nm处发射峰分别对应5D4—>7fj(j=6,5,4,3)的电子跃迁,其中545nm处发射峰强度最大。同时测定,DMP-PNIPAM/Eu(Ш)的荧光寿命为10.67ms,使DMP-PNIPAM/Tb(III)复合物能够作为温敏材料以及荧光材料应用
附图说明
图1为本发明所述的DMP的红外光谱图。
图2为本发明所述的DMP的核磁共振氢谱图。
图3为本发明所述的DMP-PNIPAM的红外光谱图。
图4为本发明所述的DMP-PNIPAM的核磁共振氢谱图。
图5为本发明所述的5-硝基-1,10-邻菲罗啉的红外光谱图。
图6为本发明所述的5-硝基-1,10-邻菲罗啉的核磁共振氢谱图。
图7为本发明所述的5-氨基-1,10-邻菲罗啉的红外光谱图。
图8为本发明所述的5-氨基-1,10-邻菲罗啉的核磁共振氢谱图。
图9为本发明所述的DMP-PNIPAM/Eu(III)复合物的红外光谱图。
图10为本发明所述的DMP-PNIPAM/Eu(III)复合物的XPS元素分析图。
图11为本发明所述的DMP-PNIPAM/Tb(III)复合物的红外光谱图。
图12为本发明所述的DMP-PNIPAM/Tb(III)复合物的XPS元素分析图。
图13为本发明所述的DMP-PNIPAM/Eu(III)复合物的荧光激发光谱图。
图14为本发明所述的DMP-PNIPAM/Eu(III)复合物的荧光发射光谱图。
图15为本发明所述的DMP-PNIPAM/Eu(III)复合物的荧光寿命图。
图16为本发明所述的DMP-PNIPAM/Tb(III)复合物的荧光激发光谱图。
图17为本发明所述的DMP-PNIPAM/Tb(III)复合物的荧光发射光谱图。
图18为本发明所述的DMP-PNIPAM/Tb(III)复合物的荧光寿命图。
具体实施方式
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
实施例1
1、合成三硫代末端羧基化合物DMP
将20mmol的1-十二烷硫醇,165.5mmol的丙酮与8mmol的四丁基溴化铵放在三口烧瓶中反应,冷却至10℃,在氮气下进行混合,溶液中加入21mmol的质量分数为50%的氢氧化钠溶液,将反应物搅拌30分钟,再将34.5mmol的丙酮和20mmol的二硫化碳混合溶液加入,要超过20分钟,在该时间内溶液的颜色变成红色,将30mmol的氯仿溶液加入,然后滴加100mmol的质量分数为50%氢氧化钠溶液超过30分钟,将反应物搅拌过夜;过夜后溶液中加入30ml水,然后加入5ml浓盐酸水溶液酸化,通过鼓动反应器帮助氮气蒸发掉丙酮,用布氏漏斗收集固体,将固体加入搅拌的50ml的2-丙醇中,未溶解的固体滤出,并鉴定为S-S′-双(1-十二烷基)三硫代醋酸酯(DMP),再将2-丙醇的溶液浓缩至干,将得到的固体用正己烷中重结晶,得到4.625g黄色结晶固体,熔点62.3℃;DMP合成路线反应式如下:
2、RAFT法制备末端羧基化合物DMP-PNIPAM
将0.0508g的DMP、1.1298g的N-异丙基丙烯酰胺(PNIPAM)、0.0465g的1,3,5-三噁烷以及0.0017g的偶氮二异丁腈放入聚合管中,加入无水N,N-二甲基甲酰胺(DMF)作溶剂,放在磁力搅拌器上,循环3次通氮气再抽真空,每次用时10分钟,然后将反应混合物在氮气保护下,放入60℃油浴中反应,接着将温度升到80℃恒温反应5h,取出冷却,进行旋蒸除去溶剂,旋蒸完冷却,加入无水乙醚开始沉淀,用抽滤瓶过滤,即得目标产物末端含有羧基的聚(N-异丙基丙烯酰胺)(DMP-PNIPAM);DMP-PNIPAM(A)合成路线反应式如下:
3、合成5-氨基-1,10-邻菲罗啉
首先,在配有冷凝器和磁力搅拌棒的250mL三口烧瓶中,装入0.017mmol的1,10-邻菲罗啉和16mL的浓硫酸,然后加入8mL的浓硝酸,放入70℃油浴锅中,接着将温度升到120℃恒温搅拌3小时后,将反应混合物用冰水稀释逐渐冷却至0℃,同时将温度保持在低于5℃,用氢氧化钠水溶液中和到成中性,所形成的微黄色的沉淀物通过过滤收集并用冰冷的水充分洗涤,残留物进一步用体积分数为95%的乙醇重结晶,在50℃真空烘箱中干燥过夜后,得到1.3513g浅黄色固体5-硝基-1,10-邻菲罗啉;
然后,在250ml三口烧瓶中加入19.98mmol的5-硝基-1,10-邻菲罗啉,0.8g的质量分数为10%的Pd/C催化剂和100mL的乙醇,将该悬浮液加热至70℃,然后通入氮气30分钟,加入61.5mmol的水合肼溶液,将反应混合物回流5小时,在60~70℃过滤后,将滤液在旋转式蒸发器上蒸干,得到明亮黄色粉末的化合物粗品,将该化合物粗品用乙醇重结晶进一步纯化,干燥后,在真空烘箱中于室温下过夜,得到3.0g的黄色晶体5-氨基-1,10-邻菲罗啉;5-氨基-1,10-邻菲罗啉合成路线反应式如下:
4、合成稀土噻吩甲酰三氟丙酮配合物
将6mmol的α-噻吩甲酰三氟丙酮(TTA)溶解在30ml的乙醇中,用1mol·L-1的氢氧化钠溶液调节PH≈6.5,将2mmol的EuCl3·6H2O的乙醇溶液滴加到上述反应中,60℃下搅拌20min,再加200ml二次蒸馏水继续恒温搅拌1h,过滤,复合物在冷却至室温的过程中沉淀,滤出沉淀物,用水洗涤,并在真空下干燥;所得复合物为摩尔比Eu:TTA=1:3的二元产物;
将0.1025g的5-氨基-1,10-邻菲罗啉在乙醇中与0.1034g的Eu(TTA)3(H2O)2复合物络合在一起,同时搅拌将溶液加热回流,24小时后过滤,用乙醇和氯仿将过量的无界络合物冲走,得到浅棕色产物;稀土噻吩甲酰三氟丙酮配合物(B)合成路线反应式如下:
5、合成DMP-PNIPAM/Eu(III)复合物
将DMP-PNIPAM(A)和稀土噻吩甲酰三氟丙酮配合物(B)放入乙醇溶液中搅拌,反应结束后透析(MWCO=4000),除去乙醇溶液及小分子物质,然后将所得液体置于恒温干燥箱中24h后得到目标产物;DMP-PNIPAM/Eu(III)复合物合成路线反应式如下:
实施例2
1、合成三硫代末端羧基化合物DMP
将20mmol的1-十二烷硫醇,165.5mmol的丙酮与8mmol的四丁基溴化铵放在三口烧瓶中反应,冷却至10℃,在氮气下进行混合,溶液中加入21mmol的质量分数为50%的氢氧化钠溶液,将反应物搅拌30分钟,再将34.5mmol的丙酮和20mmol的二硫化碳混合溶液加入,要超过20分钟,在该时间内溶液的颜色变成红色,将30mmol的氯仿溶液加入,然后滴加100mmol的质量分数为50%氢氧化钠溶液超过30分钟,将反应物搅拌过夜;过夜后溶液中加入30ml水,然后加入5ml浓盐酸水溶液酸化,通过鼓动反应器,以帮助氮气蒸发掉丙酮,用布氏漏斗收集固体,将固体加入搅拌的50ml的2-丙醇中,未溶解的固体滤出,并鉴定为S-S′-双(1-十二烷基)三硫代醋酸酯(DMP),再将2-丙醇的溶液浓缩至干,将得到的固体用正己烷中重结晶,得到4.625g黄色结晶固体,熔点62.3℃;DMP合成路线反应式如下:
2、RAFT法制备末端羧基化合物DMP-PNIPAM
将0.0508g的DMP、1.1298g的N-异丙基丙烯酰胺(PNIPAM)、0.0465g的1,3,5-三噁烷以及0.0017g的偶氮二异丁腈放入聚合管中,加入无水N,N-二甲基甲酰胺(DMF)作溶剂,放在磁力搅拌器上,循环3次通氮气再抽真空,每次用时10分钟,然后将反应混合物在氮气保护下,放入60℃油浴中反应,接着将温度升到80℃恒温反应5h,取出冷却,进行旋蒸除去溶剂,旋蒸完冷却,加入无水乙醚开始沉淀,用抽滤瓶过滤,即得目标产物末端含有羧基的聚(N-异丙基丙烯酰胺)(DMP-PNIPAM);DMP-PNIPAM(A)合成路线反应式如下:
3、合成5-氨基-1,10-邻菲罗啉
首先,在配有冷凝器和磁力搅拌棒的250mL三口烧瓶中,装入0.017mmol的1,10-邻菲罗啉和16mL的浓硫酸,然后加入8mL的浓硝酸,放入70℃油浴锅中,接着将温度升到120℃恒温搅拌3小时后,将反应混合物用冰水稀释逐渐冷却至0℃,同时将温度保持在低于5℃,用氢氧化钠水溶液中和到成中性,所形成的微黄色的沉淀物通过过滤收集并用冰冷的水充分洗涤,残留物进一步用体积分数为95%的乙醇重结晶,在50℃真空烘箱中干燥过夜后,得到1.3513g浅黄色固体5-硝基-1,10-邻菲罗啉;
然后,在250ml三口烧瓶中加入19.98mmol的5-硝基-1,10-邻菲罗啉,0.8g的质量分数为10%的Pd/C催化剂和100mL的乙醇,将该悬浮液加热至70℃,然后通入氮气30分钟,加入61.5mmol的水合肼溶液,将反应混合物回流5小时,在60~70℃过滤后,将滤液在旋转式蒸发器上蒸干,得到明亮黄色粉末的化合物粗品,将该化合物粗品用乙醇重结晶进一步纯化,干燥后,在真空烘箱中于室温下过夜,得到3.0g的黄色晶体5-氨基-1,10-邻菲罗啉;5-氨基-1,10-邻菲罗啉合成路线反应式如下:
4、合成稀土噻吩甲酰三氟丙酮配合物
将6mmol的α-噻吩甲酰三氟丙酮(TTA)溶解在30ml的乙醇中,用1mol·L-1的氢氧化钠溶液调节PH≈6.5,将2mmol的TbCl3·6H2O的乙醇溶液滴加到上述反应中,60℃下搅拌20min,再加200ml二次蒸馏水继续恒温搅拌1h,过滤,复合物在冷却至室温的过程中沉淀,滤出沉淀物,用水洗涤,并在真空下干燥,所得复合物为摩尔比Tb:TTA=1:3的二元产物;
将0.1025g的5-氨基-1,10-邻菲罗啉在乙醇中与0.1034g的Tb(TTA)3(H2O)2复合物络合在一起,同时搅拌将溶液加热回流,24小时后过滤,用乙醇和氯仿将过量的无界络合物冲走,得到浅棕色产物;稀土噻吩甲酰三氟丙酮配合物(B)合成路线反应式如下:
5、合成DMP-PNIPAM/Tb(III)复合物
将DMP-PNIPAM(A)和稀土噻吩甲酰三氟丙酮配合物(B)放入乙醇溶液中搅拌,反应结束后透析(MWCO=4000),除去乙醇溶液及小分子物质,然后将所得液体置于恒温干燥箱中24h后得到目标产物;DMP-PNIPAM/Tb(III)复合物合成路线反应式如下:
对实施例1以及实施例2中的复合物的结构进行表征确认。
表征方法及仪器
(1)傅里叶红外光谱(FT-IR)测试
采用FT-IR-8400型傅里叶红外光谱仪对合成的产物进行测试,将干燥后的样品粉末采用KBr压片法制样,扫描范围4000~500cm-1。
(2)核磁共振光谱(NMR)的测定
采用JEOL JNM-400型核磁共振仪,以CDCl3作为溶剂,以四甲基碘硅烷做为内标进行测试。
(3)紫外光谱测试
浊度法测试聚合物温敏性是在UV mini 1240(Shimadzu)型紫外-可见分光光度计上进行的。先配制一定浓度的聚合物水溶液(2mg/mL),通过恒温循环水槽(SDR-30,Hitachi)调节升温速率(0.5℃/min),紫外-可见分光光度计上测定λ=500nm处聚合物水溶液的透过率,绘制透过率对不同温度的曲线,从而确定其LCST值。
(4)荧光光谱测试
日本岛津RF-5301PC型荧光光度计,激发光源为150W氙灯,分别取相同质量高分子配合物粉末置于固体样品池中,激发及发射单色器狭缝根据样品荧光强度均确定为3nm。
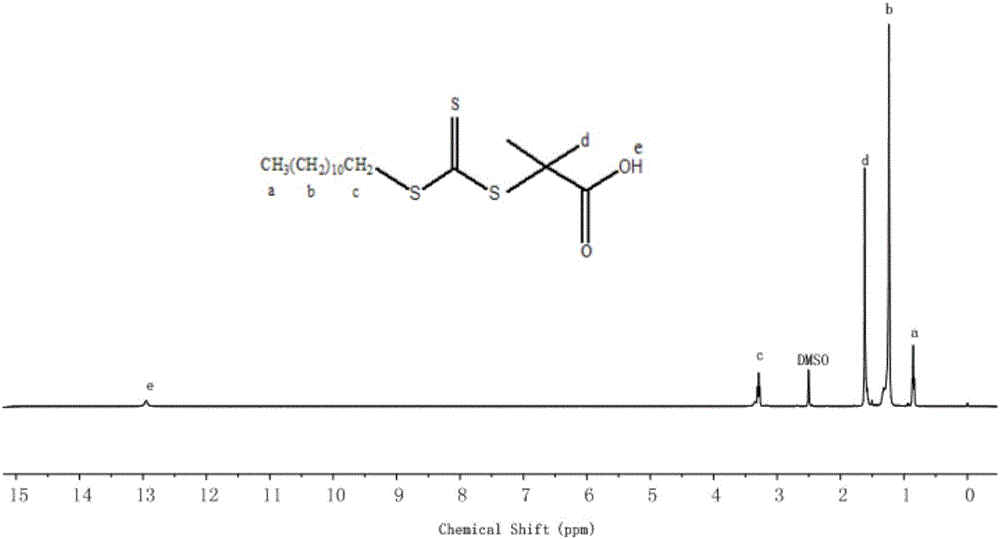
如图1所示,3441cm-1处,为羟基的特征吸收O-H的伸缩振动特征吸收,2920cm-1、2851cm-1处分别为-CH3和-CH2-的对称和不对称伸缩振动峰,1719cm-1处的吸收峰为DMP中羧基C=O双键伸缩振动。在吸收峰为1070cm-1处C=S键的吸收振动,904cm-1为甲基的C-H变形振动,812cm-1处对应于—CH2(CH2)10-CH3的面内摇摆振动;如图2所示,图标a处为-CH3的特征峰,图标b处为亚甲基-CH2-的特征峰,图标c处为与S连接的-CH2-的特征峰,图标d处为-CH3的特征峰,图标e为末端-COOH的特征峰;因此,综合图1和图2可以判断DMP已经生成。
如图3所示,3304cm-1处,对应是仲氨基的N-H键的伸缩振动,1652cm-1为酰胺基的C=O很强的吸收谱带,为Ⅰ谱带,1547cm-1为N-H的弯曲振动强吸收峰,为Ⅱ谱带;如图4所示,分析可知,在图标a处为十二烷基的特征峰,图标b处为与硫连接的-CH2-的特征峰,图标c处为是PNIPAM上重复单元次甲基上H的特征位移,图标d处为断裂链处的甲基特征峰,图标e处为酰胺基-NH-上H的特征位移,图标f处为聚合物PNIPAM链末端-CH3-上H的特征位移,图标g处为与羧基连接碳上的甲基的特征位移;因此,综合图3和图4可以判断DMP-PNIPAM已经生成。
如图5所示,在波数1434cm-1和1530cm-1处出现的吸收峰分别为硝基的对称和不对称伸缩振动特征峰,说明有-NO2的存在,在波数1600cm-1处出现的吸收峰为-C-N-的伸缩振动峰,表明邻菲罗啉已发生硝化反应;如图6所示,图标a处为4C上质子H的特征峰,图标b处为8C上质子H的特征峰,图标c处为9C上质子H的特征峰,图标d处为2C上质子H的特征峰,图标e处为3C上质子H的特征峰,图标f处为7C上质子H的特征峰,图标g处为6C上质子H的特征峰;因此,综合图5和图6可以判断5-硝基-1,10-邻菲罗啉已经生成。
如图7所示,3401cm-1、3300cm-1处为氨基N-H键的反对称和对称伸缩振动特征双峰,1640cm-1处的吸收峰对应的是C=N键的伸缩振动,说明有氨基存在;如图8所示,图标a处为4C上质子H的特征峰,图标b处为8C上质子H的特征峰,图标c处为9C上质子H的特征峰,图标d处为2C上质子H的特征峰,图标e处为3C上质子H的特征峰,图标f处为7C上质子H的特征峰,图标g处为6C上质子H的特征峰,图标h处为-NH2的特征峰;因此,综合图7和图8可以判断5-氨基-1,10-邻菲啰啉已经生成。
如图9所示,3285cm-1处,对应是仲氨基的N-H键的伸缩振动,1640cm-1为酰胺基的C=O很强的吸收谱带,为Ⅰ谱带、1533cm-1为N-H的弯曲振动强吸收峰,为Ⅱ谱带。相比DMP-PNIPAM红外光谱图对应的相应吸收峰,均发生了红移现象,说明Eu3+可能与PNIPAM侧链的氮原子或羰基氧发生配位,吸收电子的诱导效应分别使NH,-C=O键间电子云密度下降,键力常数变小,致使振动频率降低,导致红位移现象;如图10、表1所示,曲线a为DMP-PNIPAM的XPS全图,曲线b为DMP-PNIPAM/Eu(III)的XPS全图,结合能在530.3eV、399.1eV、284.2eV处的峰值表明了复合物中的O、N、C元素的存在,曲线b与a相比,多了Eu的峰值,从曲线c上,141.5eV处的峰值代表了Eu元素的存在,DMP-PNIPAM/Eu(Ш)与配体DMP-PNIPAM相比,它的O1s、N1s结合能都有所增加,而DMP-PNIPAM/Eu(Ш)配合物中Eu(Ш)的4d的结合能比EuCl3中Eu(Ш)的4d的结合能低。由于内层电子的屏蔽作用受核外电子的电荷分布影响,因此当外层电荷密度降低时,内层电子的屏蔽作用就会减弱,内层电子的结合能随之增加,与此相反结合能便会减小。因此可以推断DMP-PNIPAM/Eu(Ш)中配体DMP-PNIPAM羧基上的氧原子在形成配位键后电子转移到稀土Eu(III)的外层空轨道上,使得Eu(Ш)外层电子密度增加,屏蔽作用增强,Eu(Ш)电子结合能下降,在诱导效应的作用下氮原子将电子传递给氧原子,造成氮原子外层的电子云密度减小,屏蔽作用减弱,而内层电子结合能升高。XPS测试结果反映了配体与Eu(Ш)的配位成键情况,表明已经成功合成了Eu(III)与DMP-PNIPAM的复合物。
表1EuCl3、DMP-PNIPAM、DMP-PNIPAM/Eu(Ш)的O1s、N1s和Tb4d的结合能
如图11所示,在3286cm-1处为N-H键的伸缩振动峰,在1641cm-1处为NH-C=O的C=O很强的吸收谱带,在1532cm-1处为N-H的弯曲振动强吸收峰。相比DMP-PNIPAM红外光谱图对应的相应吸收峰,均发生了红移现象。这种红移现象可能是由于稀土噻吩甲酰三氟丙酮配合物中-NH2基与DMP-PNIPAM中的-COOH基发生酰化反应引起的,也有可能稀土噻吩甲酰三氟丙酮配合物并不稳定,分解出的Tb3+与PNIPAM侧链的氮原子或羰基氧发生配位,吸收电子的诱导效应分别使N-H,C=O键间电子云密度下降,键力常数变小,致使振动频率降低,导致红移;如图12、表2所示,DMP-PNIPAM/Tb(Ш)与配体DMP-PNIPAM相比,它的O1s结合能有增加了1.74eV,同时配合物中的N1s结合能增加了2.57eV,而DMP-PNIPAM/Tb配合物中Tb(Ш)的4d的结合能比TbCl3中Tb(Ш)的4d的结合能低4.29eV,表明DMP-PNIPAM与Tb(Ш)反应成功。由于内层电子的屏蔽作用受核外电子的电荷分布影响,因此当外层电荷密度降低时,内层电子的屏蔽作用就会减弱,内层电子的结合能随之增加,与此相反结合能便会减小。因此可以推断DMP-PNIPAM/Tb(Ш)中配体DMP-PNIPAM羧基上的氧原子在形成配位键后电子转移到稀土Tb(III)的外层空轨道上,使得Tb(Ш)外层电子密度增加,屏蔽作用增强,Tb(Ш)电子结合能下降,在诱导效应的作用下氮原子将电子传递给氧原子,造成氮原子外层的电子云密度减小,屏蔽作用减弱,而内层电子结合能升高。
表2TbCl3、DMP-PNIPAM、DMP-PNIPAM/Tb(Ш)的O1s、N1s和Tb4d的结合能
对实施例1中的DMP-PNIPAM/Eu(III)复合物以及实施例2中的DMP-PNIPAM/Tb(III)复合物进行GPC分析;
凝胶渗透色谱通过具有分子筛性质的固定相,用来分离相对分子质量较小的物质,并且还可以分析分子体积不同、具有相同化学性质的高分子同系物。
GPC的色谱条件为:流速为1.0ml/min,柱温15.0℃,进样量20μL,视差折光检测器。流动相为四氢呋喃,用Agilent HP1100专用GPC软件采集和分析数据。用凝胶渗透色谱法测定分子量时,由于非体积效应等原因无法进行直接测量,需要找到适合的色谱条件,制定正确的校正曲线才能有效克服非体积效应。
GPC测定DMP-PNIPAM/Eu(III)复合物中聚合物的Mn=7700g/mol,多分散性指数PDI(Mw重均/Mn数均)为1.03,DMP的分子量为364,N-异丙基丙烯酰胺分子量为113.18,经计算可知,聚合度n=65,转化率46.3%(产量/反应物的量)。
GPC测试DMP-PNIPAM/Tb(III)复合物中聚合物的Mn=7700g/mol,多分散性指数PDI(Mw重均/Mn数均)为1.03,DMP的分子量为364,N-异丙基丙烯酰胺分子量为113.18,经计算可知,聚合度n=65,转化率为46.3%(产量/反应物的量)。
试验例
1、荧光性能检测
如图13、图14、图15所示,首先,以620nm的发射波长测铕复合物的激发光谱,在以275nm光激发,测得铕复合物的发射光谱,图13在270—370nm出现了3个与激发光谱峰重叠的半频峰,图14在620nm附近出现了两个倍频峰,这两项干扰峰均与次配合物的荧光光谱性无关;在275nm紫外光激发下,位于580和590nm处出现两组强度不同的发射峰;由图14可知:(1)三元配合物体系发射谱中仅出现Eu3+离子5D0—>7fj(j=0,1,2,3,4)跃迁对应的特征发射峰,分别位于580nm,591nm,614nm,621nm,其中614nm处5D0—>7f2电偶极跃迁发射峰发射强度最强,为配体微扰的中心离子的发光;(2)DMP-PNIPAM/Eu(III)三元配合物只发射Eu3+的特征荧光,单色性好,色纯度高;(3)PNIPAM在325nm处的发射峰在三元配合物中几乎完全消失,可能是高分子的激发三重态与Eu3+的激发态能级之间差别较大,不能进行有效的能量传递,使高分子吸收的能量以非辐射方式损耗;如图15所示,经过荧光测定,测得铕复合物的荧光寿命达到11.78ms,其量子产率为Φ=0.176,DMP-PNIPAM聚合物本身不具有荧光性,但是加入很少部分Eu(Ш)形成铕的复合物后,发出红色荧光,寿命相对来说很长,因此,本发明所述的DMP-PNIPAM/Eu(III)复合物可以作为荧光材料的应用。
如图16、图17、图18所示,DMP-PNIPAM/Tb(III)复合物的荧光激发光谱特征峰强度在350nm处最大,这一结果说明在高分子聚合物中加入稀土铽(III)和小分子配体TTA,复合物同样具有荧光特性,TTA的加入与Tb(III)粒子发生了协同配位作用,更大程度地满足Tb(III)的配位数,这种方式也大大提高了Tb(III)离子的发射效率,使复合物荧光性能增强,从图17可知,复合物体系发射谱中仅出现Tb3+离子5D4—>7Fj(j=6,5,4,3)跃迁对应的特征发射峰,分别位于487nm,545nm,583nm,616nm,其中545nm处5D4—>7F5电偶极跃迁发射峰发射强度最强,为配体微扰的中心离子的发光,DMP-PNIPAM/Tb(III)复合物只发射Tb3+的特征荧光,单色性好,色纯度高,PNIPAM在325nm处的发射峰在复合物中几乎完全消失,可能是高分子的激发三重态与Tb3+的激发态能级之间差别较大,不能进行有效的能量传递,使高分子吸收的能量以非辐射方式损耗;图16、图17充分的证明了配位成功,即聚合物具有荧光性;由图18可知,DMP-PNIPAM/Tb(III)复合物的荧光寿命达到10.67ms,因为DMP-PNIPAM聚合物本身不具有荧光性,但是加入很少部分Tb(Ш)的配合物后,发出绿色荧光,并且能达到10.67ms,荧光寿命较长,由此可以推断,合成出荧光性能较好的DMP-PNIPAM/Tb(Ш)复合物;因此,本发明所述的DMP-PNIPAM/Tb(III)复合物可以作为荧光材料的应用。
2、温敏性检测
如表3所示,聚N-异丙基丙烯酰胺的低临界溶解温度(LCST)为32℃左右,而DMP-PNIPAM的地临界溶解温度(LCST)为33.9℃,相对于聚N-异丙基丙烯酰胺的临界溶解温度有所提高,这是因为邻苯二甲酸二甲酯与聚N-异丙基丙烯酰胺聚合后,分子上有极性基团羧基的缘故。DMP-PNIPAM/Eu(III)复合物的地临界溶解温度为34.1℃,少量Eu(Ш)的加入可使DMP-PNIPAM的LCST略微升高。这是由于DMP-PNIPAM/Eu(III)聚合物溶液中Eu(Ш)离子配位数较高还可以与水分了配位,稀土离子与水分子羟基配位键的键能高于水分子本身氢键的键能,因此当温度升高至LCST附近时,需要消耗更多的能量才能破坏稀土离子与水分子间的配位键,使配合物发生构象转变,从而使高分子配合物的相转变略微滞后;因此,本发明所述的DMP-PNIPAM/Eu(III)复合物可以作为温敏材料的应用。
表3DMP-PNIPAM和DMP-PNIPAM/Eu(III)复合物的临界溶解温度(LCST)
如表4所示,将制备好的DMP-PNIPAM和DMP-PNIPAM/Tb(III)复合物用紫外-可见光光谱分析,由于在聚合过程中PNIPAM末端引入了亲水性的羧基,使得DMP-PNIPAM的LCST较PNIPAM(32℃)温度略微升高。少量Tb(Ш)的加入可使DMP-PNIPAM的LCST温度略微升高。这是由于DMP-PNIPAM/Tb(III)聚合物溶液中Tb(Ш)离子配位数较高还可以与水分子配位,稀土离子与水分子羟基配位键的键能高于水分子本身氢键的键能,因此当温度升高至LCST温度附近时,需要消耗更多的能量才能破坏稀土离子与水分子间的配位键,使配合物发生构象转变,从而使高分子配合物的相转变略微滞后;因此,本发明所述的DMP-PNIPAM/Tb(III)复合物可以作为温敏材料的应用。
表4DMP-PNIPAM和DMP-PNIPAM/Tb(III)复合物的临界溶解温度(LCST)
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
- 还没有人留言评论。精彩留言会获得点赞!