一种化合物的晶型A及其制备纯化方法与流程
本发明涉及化学医药领域,有关布瓦西坦的关键中间体的晶型及其制备纯化方法。
背景技术:
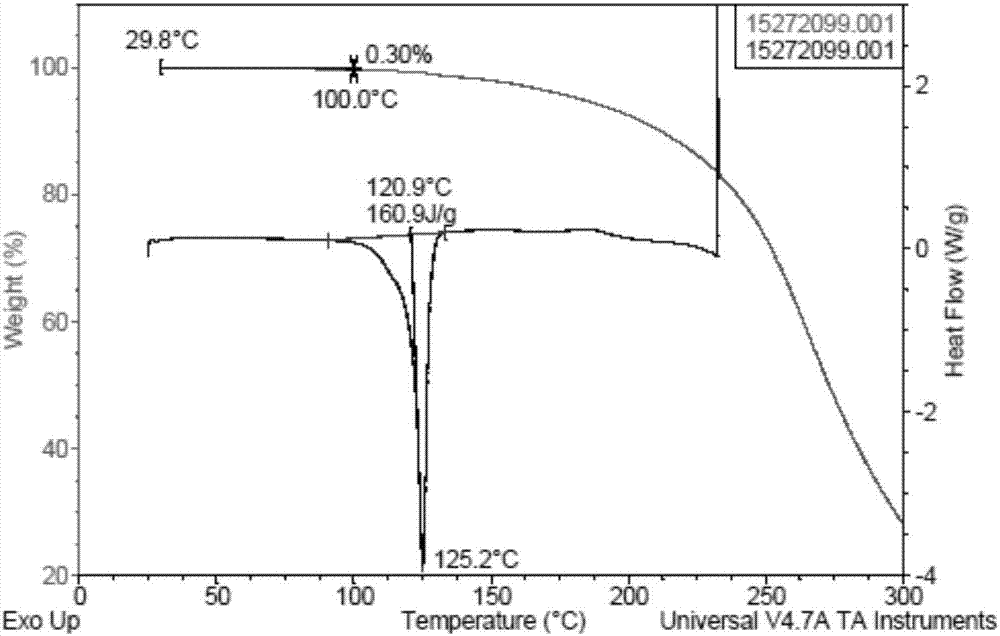
:癫痫是神经系统的常见病,在人群中的发病率为0.6%~1.1%,其中60%~70%的患者在服用抗癫痫药物时仍会发作,导致一部分患者自行停止药物治疗。目前我国有约600万以上的癫痫患者,每年新发癫痫患者65万~70万,大约25%为难治性癫痫。虽然目前癫痫的诊疗取得了很大的进展,但难治性癫痫患者的数量却在日益增多。广义难治性癫痫是指使用目前的抗癫痫药物(aeds)规范治疗,不能终止其发作或已被临床证实是难治的癫痫及癫痫综合征。布瓦西坦(brivaracetam)是一个新型的高亲和力的突触囊泡蛋白2a配体,可抑制神经元电压依赖性钠通道,用于治疗难治性癫痫部分性发作。布瓦西坦的ii期,iii期临床试验皆有较好的疗效。布瓦西坦的主要不良事件的发生率与安慰剂组的发生率相似,均为轻度至中度的疲劳、头痛、鼻咽炎、恶心、嗜睡和头晕。无患者因不良事件而中断治疗。结果表明布瓦西坦片在辅助治疗年龄为16~65岁的难治性癫痫部分性发作患者中是有效的且耐受性良好。总体而言,布瓦西坦是继左乙拉西坦之后的一个前景十分良好的第三代抗癫痫类药物。目前为止,关于布瓦西坦合成的国内专利还未见报道。国外相关专利报道也不多,诸如专利us6,784,197,us7,629,474,us8,957,226,us8,338,621,us8,076,493及其相关专利报道了布瓦西坦的合成,其中us6,784,197,us7,629,474和报道了一些合成路线,这些合成路线所存在的问题主要集中在,工艺路线不能选择性的合成目标手性分子,存在需要柱层析纯化、手性高效液相色谱纯化等不利于工业放大生产的操作方法。手性hplc分离需要昂贵的设备和耗材,不适合大规模生产。另外在手性hplc分离中,如果要提高收率,降低成本,必然会导致收集较多的异构体成分,品质会因此而降低,大量手性杂质的存在可能会导致药物不良反应,影响药物的安全性。另外,专利cn104892483a报道了不对称催化氢化的方法,但是其中使用了昂贵的手性配体,路线不经济。cn106279074a、cn106365986a和cn106432030a报道了一种通过低价易得的手性原料合成布瓦西坦,路线不涉及昂贵的手性催化剂或配体,也不需要手性hplc分离纯化最终产品,其中化合物i是合成布瓦西坦的一个中间体。较晚的另一篇专利cn105646319a也报道了类似的合成布瓦西坦的方法,其路线方案与cn106279074a、cn106365986a和cn106432030a中报道的部分路线类同。化合物的多晶型是指在化合物物中存在有两种或两种以上的不同晶型物质状态。多晶型现象的在有机化合物中广泛存在。同一化合物的不同晶型在溶解度、熔点、密度、稳定性等方面有显著的差异,从而不同程度的影响化合物的稳定性、均一性。不同的晶型在化合物的纯化过程中通过结晶对化合物的提纯能力有着明显的不同。因此,药物工艺研发中进行全面系统的多晶型筛选,选择最合适开发的晶型,是不可忽视的重要研究内容之一。药物中间体化合物的晶型与纯化工艺的研究有利于降低药物活性成分的生产成本,提高药物活性成分的品质,保证药物制剂的品质与安全性。技术实现要素:本发明提供一种布瓦西坦中间体化合物i的新晶型,本发明提供的新晶型有良好的稳定性、工艺可开发和易处理等有利性能,且制备方法简单,成本低廉,对该药物活性成分的生产、纯化和品质控制具有重要价值。具体的,本发明的一个目的是提供布瓦西坦中间体化合物i的一种新晶型,命名为晶型a。本发明提供的晶型a,其特征在于,其x射线粉末衍射图在2theta值为7.9°±0.2°,15.7°±0.2°,21.7°±0.2°,28.6°±0.2°处具有特征峰。更进一步的,所述的晶型a,其特征还在于,其x射线粉末衍射图在2theta值为2theta值为17.4°±0.2°,25.5°±0.2°,28.1°±0.2°处具有特征峰。更进一步的,所述的晶型a,其特征还在于,其x射线粉末衍射图在2theta值为14.0°±0.2°,19.1°±0.2°,36.5°±0.2°处具有特征峰。更进一步的,所述晶型a,其特征在于,其x射线粉末衍射图基本上与图1一致。本发明的另一个目的是提供化合物i的纯化方法,其特征在于,包括将化合物i粉末加入到一种或多种溶剂的混合体系中结晶得到。更进一步的,所述结晶方法包括混悬搅拌,加热降温,挥发或反溶剂添加。更进一步的,所述溶剂包括水,醇类,醚类,酮类,酯类,芳香烃,卤代烃,腈类,硝基烷烃,脂肪烃类溶剂的单一或混合体系。优选的,所述溶剂是四氢呋喃和正庚烷的混合溶剂。本发明的另一目的是提供一种包含有化合物i晶型a的化合物的用途,此化合物i通过一步简单的成环反应可以合成抗癫痫类的药物布瓦西坦。本发明的另一目的是提供一种高手性纯度化合物i,所述化合物i中,其对映及非对映异构体化合物的含量不大于2.5%。更进一步的,其对映及非对映异构体化合物的含量不大于1.0%。更进一步的,其对映及非对映异构体化合物的含量不大于0.5%。更进一步的,其对映及非对映异构体化合物的含量不大于0.15%。其特征还在于,对映及非对映异构体化合物的含量不大于2.5%的高手性纯度化合物i,通过晶型a,结晶纯化制得。本发明的有益效果为:本发明提供的晶型有较好的稳定性;本发明提供的结晶方法可以有效的将化合物i中对映和非对映异构体控制在2.5%以下,进一步的,1.0%以下,进一步的,0.5%以下,进一步的,0.15%以下;本发明提供的结晶方法得到的高手性纯度的化合物i可以用来生产高手性纯度的布瓦西坦,提高最终药物有效成分的手性纯度,降低因为手性杂质带来的潜在的不良反应,从而提高药物的安全性;本发明提供的新晶型的制备方法简单且重复性好,溶剂不易残留,且过程可控,适合直接用于工业化生产。附图说明图1为化合物i晶型a的xrpd图。图2为化合物i晶型a的tga图和dsc图叠图。具体的实施方式以下通过具体的实施例进一步的阐述本发明,但并不用于限制本发明的保护范围。本领域的技术人员可在权利要求范围内对制备方法和使用仪器做出改进,这些改进也应被视为本发明的保护范围。因此,本发明的保护范围应以权利要求为准。下述实施例中,所述的实验方法通常按照常规条件或制造厂商建议的条件实施;所述化合物i通过专利wo2016191435方法制备。本发明中所用到的缩写解释如下:xrpd:x射线粉末衍射dsc:差示扫描量热分析tga:热重分析本发明所述的x射线粉末衍射图在panalyticalempyreanx衍射粉末衍射仪上采集。xrpd扫描参数startposition[°2th.]:3.0056;endposition[°2th.]:39.9906;stepsize[°2th.]:0.0167;scansteptime[s]:17.8500;k-alpha1[å]:1.54060;k-alpha2[å]:1.54443;电压:40ma;电流:45kv。本发明所述差示扫描量热分析(dsc)图在taq200上采集。本发明所述差示扫描量热分析(dsc)的方法参数如下:扫描速率:10°c/min保护气体:氮气本发明所述热重分析(tga)图在taq500上采集。本发明所述热重分析(tga)的方法参数如下:扫描速率:10°c/min保护气体:氮气实施例1化合物i晶型a的制备方法:将30g化合物i加入180ml的四氢呋喃中,升温至60°c,搅拌0.5h,缓慢加入360ml正庚烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,所得固体经检测为晶型a,收率91%,hplc99.79%,手性hplc:化合物i为99.98%,化合物(r,s)-i未检测到,化合物(r,r)-i为0.01%,化合物(s,s)-i为0.01%。本实施例得到的晶型a的xrpd数据如表1所示。表12thetad间隔相对强度%7.8511.26100.0013.946.352.4615.685.6515.1517.395.102.7419.074.651.7121.734.096.5025.473.492.3028.043.183.3628.613.1220.7636.492.461.94实施例2化合物i晶型a的制备方法:将20g化合物i粉末加入120ml的四氢呋喃中,升温至60°c,搅拌0.5h,缓慢加入120ml正庚烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,所得固体经检测为晶型a,收率80%,hplc99.85%,手性hplc:化合物i为99.95%,化合物(r,s)-i未检测到,化合物(r,r)-i未检测到,化合物(s,s)-i为0.05%。本实施例得到的晶型a的xrpd数据如表2所示。表22thetad间隔相对强度%7.8611.24100.0013.966.346.6815.695.6422.2617.415.097.0319.104.646.8521.764.0825.6525.523.4910.5328.083.1813.4128.643.1150.9136.542.466.21实施例3化合物i晶型a的制备方法:将1.28kg化合物i粉末加入7.7l的四氢呋喃中,升温至60°c,搅拌0.5h,缓慢加入15.4l正庚烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,所得固体经检测为晶型a,收率82%,hplc99.00%,手性hplc:化合物i为99.95%,化合物(r,s)-i未检测到,化合物(r,r)-i为0.03%,化合物(s,s)-i为0.02%。本实施例得到的晶型a的xrpd数据如表3所示。表32thetad间隔相对强度%7.8611.24100.0013.956.344.9315.695.6417.5317.405.095.1319.084.644.2221.754.0816.2625.493.495.8828.063.178.2028.623.1135.4336.512.463.11实施例4化合物i重结晶方法:将500mg化合物i粉末加入4.5ml的丙酮中,升温至50°c,搅拌0.5h,缓慢加入6ml甲基环己烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率56.2%,hplc97.48%。实施例5化合物i重结晶方法:将500mg化合物i粉末加入4.5ml的丙酮中,升温至50°c,搅拌0.5h,缓慢加入6ml正庚烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率57.5%,hplc96.29%。实施例6化合物i重结晶方法:将500mg化合物i粉末加入4.5ml的丙酮中,升温至50°c,搅拌0.5h,缓慢加入4ml甲基叔丁基醚,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率51.2%,hplc95.73%。实施例7化合物i重结晶方法:将500mg化合物i粉末加入3ml的四氢呋喃中,升温至60°c,搅拌0.5h,缓慢加入5ml甲基环己烷,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率63.4%,hplc96.59%。实施例8化合物i重结晶方法:将500mg化合物i粉末加入3ml的四氢呋喃中,升温至60°c,搅拌0.5h,缓慢加入5ml甲基叔丁基醚,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率56.8%,hplc95.67%。实施例9化合物i重结晶方法:将500mg化合物i粉末加入4.5ml的乙腈中,升温至60°c,搅拌0.5h,缓慢加入6ml水,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率75.8%,hplc96.68%。实施例10化合物i重结晶方法:将500mg化合物i粉末加入4.5ml的丙酮中,升温至50°c,搅拌0.5h,缓慢加入6ml水,缓慢降温至0~10°c,过滤,所得滤饼置于50°c真空烘箱内干燥过夜,收率80.5%,hplc95.70%。本申请包括但不限于以上实施例,凡是在本申请精神的原则下进行的任何等同替代或局部改进,都将视为在本申请的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1